

Abstract
Phosphatidic acid generated by the activation of phospholipase D (PLD)
functions as a second messenger and plays a vital role in cell signaling. Here
we demonstrate that PLD-dependent generation of phosphatidic acid is critical
for Rac1/IQGAP1 signal transduction, translocation of
p47phox to cell periphery, and ROS production. Exposure of
[32P]orthophosphate-labeled human pulmonary artery endothelial
cells (HPAECs) to hyperoxia (95% O2 and 5% CO2) in the
presence of 0.05% 1-butanol, but not tertiary-butanol, stimulated PLD as
evidenced by accumulation of [32P]phosphatidylbutanol. Infection of
HPAECs with adenoviral constructs of PLD1 and PLD2 wild-type potentiated
hyperoxia-induced PLD activation and accumulation of
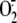 /reactive oxygen species
(ROS). Conversely, overexpression of catalytically inactive mutants of PLD
(hPLD1-K898R or mPLD2-K758R) or down-regulation of expression of PLD with PLD1
or PLD2 siRNA did not augment hyperoxia-induced
[32P]phosphatidylbutanol accumulation and ROS generation. Hyperoxia
caused rapid activation and redistribution of Rac1, and IQGAP1 to cell
periphery, and down-regulation of Rac1, and IQGAP1 attenuated
hyperoxia-induced tyrosine phosphorylation of Src and cortactin and ROS
generation. Further, hyperoxia-mediated redistribution of Rac1, and IQGAP1 to
membrane ruffles, was attenuated by PLD1 or PLD2 small interference RNA,
suggesting that PLD is upstream of the Rac1/IQGAP1 signaling cascade. Finally,
small interference RNA for PLD1 or PLD2 attenuated hyperoxia-induced cortactin
tyrosine phosphorylation and abolished Src, cortactin, and
p47phox redistribution to cell periphery. These results
demonstrate a role of PLD in hyperoxia-mediated IQGAP1 activation through Rac1
in tyrosine phosphorylation of Src and cortactin, as well as in
p47phox translocation and ROS formation in human lung
endothelial cells.
/reactive oxygen species
(ROS). Conversely, overexpression of catalytically inactive mutants of PLD
(hPLD1-K898R or mPLD2-K758R) or down-regulation of expression of PLD with PLD1
or PLD2 siRNA did not augment hyperoxia-induced
[32P]phosphatidylbutanol accumulation and ROS generation. Hyperoxia
caused rapid activation and redistribution of Rac1, and IQGAP1 to cell
periphery, and down-regulation of Rac1, and IQGAP1 attenuated
hyperoxia-induced tyrosine phosphorylation of Src and cortactin and ROS
generation. Further, hyperoxia-mediated redistribution of Rac1, and IQGAP1 to
membrane ruffles, was attenuated by PLD1 or PLD2 small interference RNA,
suggesting that PLD is upstream of the Rac1/IQGAP1 signaling cascade. Finally,
small interference RNA for PLD1 or PLD2 attenuated hyperoxia-induced cortactin
tyrosine phosphorylation and abolished Src, cortactin, and
p47phox redistribution to cell periphery. These results
demonstrate a role of PLD in hyperoxia-mediated IQGAP1 activation through Rac1
in tyrosine phosphorylation of Src and cortactin, as well as in
p47phox translocation and ROS formation in human lung
endothelial cells.
Phagocytic cells of the immune system (neutrophils, eosinophils, monocytes,
and macrophages) generate superoxide
(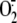 )2
instrumental in the killing of invading pathogens solely by NADPH oxidase
(1-3).
Deficiency of
)2
instrumental in the killing of invading pathogens solely by NADPH oxidase
(1-3).
Deficiency of 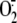 results in
the genetically inherited disorder chronic granulomatous disease, a condition
in which the affected individuals are susceptible to infection
(4). Phagocytic NADPH oxidase
is activated when cytosolic p47phox,
p67phox, and Rac2 translocate to the phagosomes and plasma
membrane and form a complex with integral membrane cytochrome
b558, which, in turn, is a Nox2
(gp91phox)/p22phox heterodimer
(5,
6). Assembly of phagocytic
NADPH oxidase is initiated by two signals. The first is the phosphorylation of
multiple serine and tyrosine residues in the p47phox
domain, which leads to unmasking of p47phox SH3 domains
that bind to a proline-rich target in the C terminus of
p22phox
(7-10).
The interaction between p47phox and
p22phox seems to be an essential requirement for the
translocation of other cytosolic components of the oxidase. The second signal
is the binding of GTP to Rac2, which leads to the dissociation of Rac from
Rho-GDI and binding to p67phox, followed by translocation
of p67phox/GTP-Rac2 to the membrane
(11). Nonphagocytic cells
express predominantly Rac1, Tiam1 (a GEF involved in Rac1 activation), Nox1-5,
and most of the other cytosolic phagocytic oxidase components
(12); however, the oxidative
output of non-phagocytes is much smaller compared with the phagocytes. A
recent study indicates that IQGAP1, an effector of Rac1, may link Nox2 to
actin, thereby enhancing ROS production and contributing to cell motility in
ECs (13). The one or more
mechanisms responsible for differences in the oxidative burst between the
phagocytic and non-phagocytic cells are yet to be defined.
results in
the genetically inherited disorder chronic granulomatous disease, a condition
in which the affected individuals are susceptible to infection
(4). Phagocytic NADPH oxidase
is activated when cytosolic p47phox,
p67phox, and Rac2 translocate to the phagosomes and plasma
membrane and form a complex with integral membrane cytochrome
b558, which, in turn, is a Nox2
(gp91phox)/p22phox heterodimer
(5,
6). Assembly of phagocytic
NADPH oxidase is initiated by two signals. The first is the phosphorylation of
multiple serine and tyrosine residues in the p47phox
domain, which leads to unmasking of p47phox SH3 domains
that bind to a proline-rich target in the C terminus of
p22phox
(7-10).
The interaction between p47phox and
p22phox seems to be an essential requirement for the
translocation of other cytosolic components of the oxidase. The second signal
is the binding of GTP to Rac2, which leads to the dissociation of Rac from
Rho-GDI and binding to p67phox, followed by translocation
of p67phox/GTP-Rac2 to the membrane
(11). Nonphagocytic cells
express predominantly Rac1, Tiam1 (a GEF involved in Rac1 activation), Nox1-5,
and most of the other cytosolic phagocytic oxidase components
(12); however, the oxidative
output of non-phagocytes is much smaller compared with the phagocytes. A
recent study indicates that IQGAP1, an effector of Rac1, may link Nox2 to
actin, thereby enhancing ROS production and contributing to cell motility in
ECs (13). The one or more
mechanisms responsible for differences in the oxidative burst between the
phagocytic and non-phagocytic cells are yet to be defined.
We have demonstrated previously that hyperoxia activates lung endothelial
NADPH oxidase, which in part is mediated by ERK, p38 MAPK
(14,
15), and Src
(16), and hyperoxia-induced
p47phox tyrosine phosphorylation and translocation to cell
periphery is dependent on Src
(16). Further, tyrosine
phosphorylation of cortactin mediated by Src is essential for
hyperoxia-induced p47phox translocation and
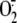 /ROS generation in HPAECs
(17). In addition to Src,
phosphatidic acid (PA) or diacylglycerol also stimulated phosphorylation of
p47phox and p22phox in neutrophils
both in vivo and in vitro
(18-20).
PA is generated in mammalian cells via de novo biosynthesis or
hydrolysis of membrane phospholipids catalyzed by phospholipase D (PLD)
(21-25).
Activation of polymorphonuclear leukocytes with formyl-Met-Leu-Phe enhanced
the oxidative burst that correlated with PA accumulation, and inclusion of
short-chain primary alcohols attenuated the NADPH oxidase mediated
/ROS generation in HPAECs
(17). In addition to Src,
phosphatidic acid (PA) or diacylglycerol also stimulated phosphorylation of
p47phox and p22phox in neutrophils
both in vivo and in vitro
(18-20).
PA is generated in mammalian cells via de novo biosynthesis or
hydrolysis of membrane phospholipids catalyzed by phospholipase D (PLD)
(21-25).
Activation of polymorphonuclear leukocytes with formyl-Met-Leu-Phe enhanced
the oxidative burst that correlated with PA accumulation, and inclusion of
short-chain primary alcohols attenuated the NADPH oxidase mediated
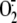 /ROS generation,
suggesting a potential role for PLD in the regulation of NADPH oxidase
(12,
26,
27). However, the downstream
targets of PLD that signal NADPH oxidase activation have not been fully
characterized.
/ROS generation,
suggesting a potential role for PLD in the regulation of NADPH oxidase
(12,
26,
27). However, the downstream
targets of PLD that signal NADPH oxidase activation have not been fully
characterized.
Here, we identify for the first time that activation of IQGAP1 by Rac1 is downstream of PLD in hyperoxia-induced ROS generation. In addition, we show that activation of Rac1/IQGAP1 by PLD also regulates Src-dependent tyrosine phosphorylation of cortactin and p47phox translocation to cell periphery. Thus, our results define a novel molecular mechanism for hyperoxia-induced NADPH oxidase activation by PLD/PA-mediated p47phox membrane translocation via Rac1/IQGAP1/Src/cortactin signaling cascade.
EXPERIMENTAL PROCEDURES
Materials—Human pulmonary artery endothelial cells (HPAECs), endothelial basal media (EBM-2), and a bullet kit were obtained from Lonza (San Diego, CA). Phosphate-buffered saline (PBS) was obtained from Biofluids Inc. (Rockville, MD). DCFDA (6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate), Alexa Fluor 488, 568, or 594 mouse, rabbit, donkey, chicken, or goat secondary antibodies, Prolong Gold maintain media, and precast Tris-glycine polyacrylamide gel were purchased from Invitrogen-Molecular Probes (Eugene, OR). The enhanced chemiluminescence (ECL) kit was from Thermo Scientific (Rockford, IL). Polyclonal goat anti-p47phox antibody was provided by Dr. Leto (National Institutes of Health, Bethesda, MD). Adenoviral constructs, wild type (Wt) and mutant (Mn) for hPLD1, mPLD2, and dominant Rac1 were generated at the services of the University of Iowa Gene Transfer Vector Core (Iowa City, IA). Antibody for PLD1 was provided by Dr. Bourgoin (Laval University, Canada), and antibody for PLD2 (28) was from Drs. Nozawa and Banno (Gifu International Institute of Biotechnology, Japan). IQGAP1-Myc (Wt and Mn) were provided by Dr. Sacks (Harvard University, MA). siRNA for PLD1, PLD2, Tiam1, Rac1, IQGAP1, antibodies for cortactin, Src, ERK1 and ERK2, Tiam1, IQGAP1, protein A/G plus agarose, rabbit IgG, and bovine serum albumin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Gene silencer was from Genlantis (San Diego, CA), and FuGENE HD transfection reagent was from Roche Applied Science. Phosphatase inhibitor mixture, anti-actin, and anti-phospho-Src (Y418) antibodies were from Sigma. Anti-phospho-cortactin (Y486) antibody was obtained from Chemicon (Boronia, Australia). c-Myc antibody was from Biomol (Plymouth Meeting, PA). Rac1 inhibitor (NSC23766) was purchased from Calbiochem. Rac1 antibody was from BD Biosciences (San Jose, CA). Rac1 activation kit was from Upstate Biotechnology, Inc. (Temecula, CA). Anti-phospho-tyrosine, -phospho-serine, and -phospho-threonine were from Zymed Laboratories Inc. (San Francisco, CA). TLC plates were from AnalTech (Newark, DE). Microscopy Lab-Tek slides chambers were from Electron Microscopy Sciences (Hatfield, PA). Incubator chamber for hyperoxia exposure was from Billups-Rothenberg (Del Mar, CA). Cell lyses buffer was from Cell Signaling Technology (Danvers, MA). Polyvinylidene difluoride and nitrocellulose membranes were from Millipore (Billerica, MA). Protein standard and secondary IgG (H+L)-horseradish peroxidase-conjugated antibodies were from Bio-Rad.
Endothelial Cell Culture—HPAECs at passages 5-8 in EGM-2 complete medium with 10% fetal bovine serum, 100 units/ml penicillin, and streptomycin were grown to contact-inhibited monolayers with a typical cobblestone morphology in a 37 °C incubator under a 5% CO2-95% air atmosphere. Cells were detached from T-75 flasks with 0.05% trypsin and resuspended in fresh complete medium, then cultured in 35-mm, 60-mm, or 100-mm dishes or on slide chambers for immunofluorescence studies. All cells were starved overnight in EGM-2 medium containing 1% fetal bovine serum prior to exposure to normoxia or hyperoxia.
Exposure of Cells to Hyperoxia—HPAECs were placed in a humidity-controlled airtight modulator incubator chamber and flushed continuously with 95% O2-5% CO2 for 30 min until the oxygen level inside the chamber reached ∼95%. Chamber was then placed in a cell culture incubator at 37 °C for the desired length of time. The concentration of O2 inside the chamber was monitored with a digital oxygen monitor. The buffering capacity of the cell culture medium did not change significantly during the period of hyperoxic exposure and was maintained at pH ∼ 7.4.
RNA Isolation and Real-time Reverse Transcription-PCR—Total RNA was isolated from HPAECs grown on 35-mm dishes using TRIzol® reagent according to the manufacturer's instruction. iQ SYBR Green Supermix was used to do the real-time measurements using iCycler by Bio-Rad. 18 S (sense, 5′-GTAACCCGTTGAACCCCATT-3′, and antisense, 5′-CCATCCAATCGGTAGTAGCG-3′) was used as a housekeeping gene to normalize expression. The reaction mixture consisted of 0.3 μg of total RNA (target gene) or 0.03 μg of total RNA (18 S rRNA), 12.5 μl of iQ SYBR Green, 2 μl of cDNA, 1.5 μm target primers, or 1 μm 18 S rRNA primers, in a total volume of 25 μl. For all samples, reverse transcription was carried out at 25 °C for 5 min, followed by cycling to 42 °C for 30 min and 85 °C for 5 min with iScript cDNA synthesis kit. Amplicon expression in each sample was normalized to its 18 S rRNA content. The relative abundance of target mRNA in each sample was calculated as 2 raised to the negative of its threshold cycle value times 106 after being normalized to the abundance of its corresponding 18 S rRNA (housekeeping gene), (2-(primer Threshold Cycle)/2-(18 S Threshold Cycle) × 106). All primers were designed by inspection of the genes of interest using Primer 3 software. Negative controls, consisting of reaction mixtures containing all components except target RNA, were included with each of the reverse transcription-PCR runs. To verify that amplified products were derived from mRNA and did not represent genomic DNA contamination, representative PCR mixtures for each gene were run in the absence of the reverse transcription enzyme after first being cycled to 95 °C for 15 min. In the absence of reverse transcription, no PCR products were observed.
Transfection and Infection of HPAECs—For siRNA experiments HPAECs were transfected with Fl-luciferase GL2 duplex siRNA (target sequence: 5′-CGTACGCGGAATACTTCGA-3′, Dharmacon, CO) as a positive control (scrambled RNA). HPAECs grown to ∼60-70% confluence were transfected with Gene Silencer transfection agent plus scrambled RNA or PLD1, PLD2, Tiam1, Rac1, or IQGAP1 siRNA (50 nm) in serum-free EBM-2 medium according to the manufacturer's recommendations. At 3 h post-transfection, fresh complete EGM-2 medium was added, and the cells were cultured for an additional 72 h prior to experiments.
For cDNA experiments HPAECs grown to ∼50% confluence were transfected with 1 μg/ml Vector-control or plasmids DNA of IQGAP1-Myc (Wt and Mn) or p47phox-GFP using FuGene HD (3 μg/ml) transfection reagent in serum-free EGM-2 medium according to the manufacturer's recommendation. After 3 h the medium was replaced by complete EGM-2, and the cells were incubated for 72 h post-transfection.
For transient infection adenoviral constructs (5 plaqueforming units/cell) of Vector-control, PLD1 Wt, PLD1 Mn, PLD2 Wt, PLD2 Mn, or Rac1 dominant-negative were added to HPAECs grown to ∼80% confluence in complete EGM containing 10% fetal bovine serum. After overnight culture, the virus-containing medium was replaced with fresh complete medium, exposed to normoxia or hyperoxia, and treated as indicated.
Determination of Hyperoxia-induced ROS Formation—ROS production in HPAECs exposed to either normoxia or hyperoxia was determined by the DCFDA fluorescence measured by spectrofluorometer or fluorescence microscopy (14, 16, 17). HPAECs were loaded with 10 μm DCFDA for 30 min in serum-free medium at 37 °C in a 95% air-5% CO2 environment. At the end of incubation, the medium containing DCFDA was aspirated, cells were washed once with complete medium, complete medium was added, and cells were exposed to normoxia and hyperoxia. For spectrofluorometer measurements cells were scraped, and the medium containing cells was transferred to 1.5-ml microcentrifuge tubes and centrifuged at 8,000 × g for 10 min at 4 °C. The medium was aspirated, and the cell pellet was washed twice with ice-cold PBS and sonicated on ice with a probe sonicator for 15 s in 500 μl of ice-cold PBS to prepare cell lysates. Fluorescence of oxidized DCFDA in cell lysates, an index of formation of ROS, was measured on an Aminco Bowman series 2 spectrofluorometer with excitation and emission set at 490 and 530 nm, respectively, using appropriate blanks. All above steps were performed in the dark. The extent of ROS formation was expressed as a percentage of normoxic control. For fluorescent microscopy measurements cells were washed twice with Phenol Red-free basal EBM-2, and fluorescence of oxidized DCFDA was examined under a Nikon Eclipse TE 2000-S fluorescence microscopy with a Hamamatsu digital charge-coupled device camera (Japan) using a 20× objective lens. Statistics of entire images were calculated using MetaVue software (Universal Imaging Corp., PA) and expressed as % of Control.
PLD Activation in Intact ECs—HPAECs were labeled with [32P]orthophosphate (5 μCi/ml) in phosphate-free medium containing 2% fetal bovine serum for 18-24 h (29-31). Cells were washed in minimal essential medium and exposed to either normoxia or hyperoxia for varying time periods in the presence of 0.05% 1-butanol or tertiary butanol as indicated in the figures. The incubations were terminated by addition of methanol-concentrated HCl (100:1, v/v). [32P]PBt formed as a result of PLD activation and trans-phosphatidylation reaction, an index of in vivo PLD stimulation (31), was separated by TLC in 1% potassium oxalate-impregnated silica gel H plates using the upper phase of ethyl acetate-2,2,4-trimethyl pentane-glacial acetic acid-water (65:10:15:50, v/v) as the developing solvent system (31). Unlabeled PBt was added as a carrier during the lipid separation by TLC and was visualized under iodine vapors. Radioactivity associated with PBt was quantified by liquid scintillation counting, and data are expressed as dpm normalized to 106 counts in total lipid extract or as a percentage of control.
Rac1 Activation Assay—HPAECs were cultured in 100-mm dishes, exposed to normoxia or hyperoxia and Rac1 activation was evaluated using the Rac1 Activation Assay Kit as per the manufacturer's protocol. Briefly, after exposure to hyperoxia, cells were washed twice with ice-cold PBS and lysed with lyses buffer. Cell lysates (0.5-1 mg/ml) were loaded with 10 μg of PAK-1 p21-binding domain fusion-protein conjugated to agarose for 1 h to bind Rac1-GTP, centrifuged, and washed three times with lyses buffer. The proteins were separated by SDS-PAGE, transferred to nitrocellulose membrane, and probed with antibodies as indicated. Quantification of the bands was performed by ImageJ software (NIH) and expressed in pixels (% Control). Total cell lysates were also probed separately with anti-actin antibody to confirm equal loading.
Immunofluorescence Microscopy—HPAECs grown on slide chambers were exposed to either normoxia or hyperoxia then immediately fixed with 3.7% paraformaldehyde in PBS for 10 min, permeabilized for 4 min in 3.7% paraformaldehyde containing 0.25% Triton X-100. In some experiments for Rac1, Tiam1, IQGAP1, and p-Src staining, permeabilization was performed by methanol treatment for 4 min at -20 °C. Then cells were rinsed three times with PBS and incubated for 30 min at room temperature in TBST blocking buffer containing 1% bovine serum albumin followed by incubation with primary antibodies (1:200 dilution in blocking buffer, 1 h). Thoroughly rinsed with TBST, cells were then stained with Alexa Fluor secondary antibodies (1:200 dilution in blocking buffer, 1 h). After washing slides were prepared with maintain media and examined with a Nikon Eclipse TE 2000-S fluorescence microscope and Hamamatsu digital camera (Japan) using a 60× oil immersion objective lens. Protein redistribution to cell periphery was estimated by statistics of equivalent square of cell periphery from normoxic and hyperoxic samples and MetaVue software (Universal Imaging Corp.).
Preparation of Cell Lysates, Immunoprecipitation, and Western Blotting—HPAECs were serum-deprived for ∼18 h in EBM-2 containing 1% fetal bovine serum. After exposure to normoxia or hyperoxia, cells were washed with ice-cold PBS containing 1 mm vanadate, scraped into 1 ml of lysis buffer (50 mm Tris-HCl, pH 7.4; 150 mm NaCl; 1% Nonidet P-40; 0.25% sodium deoxycholate; 1 mm EDTA; 1 mm phenylmethylsulfonyl fluoride; 1 mm Na3VO4; 1 mm NaF; 10 μg/ml aprotinin; 10 μg/ml leupeptin; and 1 μg/ml pepstatin) containing 1% phosphatases inhibitor mixture, sonicated on ice with a probe sonicator (15 s), and centrifuged at 5000 × g in a microcentrifuge (4 °C) for 5 min. Protein concentrations of the supernatants were determined using a Pierce protein assay kit. Equal volumes of the supernatants, adjusted to 1 mg of protein/ml, were denatured by boiling in 6× SDS sample buffer for 5 min, and samples were separated on SDS-PAGE gels and analyzed by Western blotting. For immunoprecipitation, cell lysates (0.5-1 mg of protein) were incubated overnight with 2 μg/ml appropriate antibodies, conjugated to protein A/G PLUS-agarose (50 μl) for 2 h at 4 °C, and then centrifuged at 5000 × g in a microcentrifuge. Pellets were washed in lyses buffer, dissociated by boiling in 2× SDS sample buffer for 5 min and separated on SDS-PAGE. Protein bands were transferred overnight (25 V, 4 °C) on polyvinylidene difluoride or nitrocellulose membranes, probed with primary and secondary antibodies according to the manufacturer's protocol, and detected by the ECL kit. The blots were scanned (UMAX Power Lock II) and quantified by an ImageJ software (NIH). To verify unspecific protein co-immunoprecipitation cell lysates (1 mg of protein) were incubated overnight with 2 μg of rabbit IgG and IQGAP1 or Cortactin, conjugated to protein A/G PLUS-agarose (50 μl) for 2 h at 4 °C and Western blotted as described above.
Statistics—Analysis of variance and Student-Newman-Keul's test were used to compare means of two or more different treatment groups. The level of significance was set to p < 0.05 unless otherwise stated. Data are expressed as mean ± S.E.
RESULTS
Role of PLD in Hyperoxia-induced ROS Formation in
HPAECs—Previous studies have shown that hyperoxia stimulates ROS
generation (14,
16,
17,
32,
33) and activates PLD in ECs
(22,
34); however, the role of PLD
in hyperoxia-induced NADPH oxidase activation and ROS generation is not well
defined. Therefore, we investigated the role of PLD1 and PLD2, the two
isoforms of PLD (21,
23,
25,
35,
36), on hyperoxiamediated
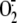 /ROS generation. HPAECs
overexpressing PLD1/PLD2 wild-type or catalytically inactive mutants were
labeled with [32P]orthophosphate and exposed to hyperoxia for 2 h.
As shown in Fig. 1,
overexpression of hPLD1/mPLD2 wild type increased both basal and
hyperoxia-induced [32P]PBt production and ROS formation, whereas
the hPLD1-K898R/mPLD2-K758R mutants attenuated the formation of labeled PBt
and ROS production. Consistent with the data using adenoviral hPLD1/mPLD2
vectors, down-regulation of PLD1 or PLD2 with siRNA attenuated ROS generation
mediated by hyperoxia (supplemental Fig. S1). Further, accumulation of
[32P]PBt was significantly higher at 2 and 3 h as compared with
cells exposed to normoxia (supplemental Fig. S2A), and 1-butanol, but
not tertiary-butanol, attenuated hyperoxia-mediated ROS production, indicating
a role for PLD in NADPH oxidase activation (supplemental Fig. S2B).
Importantly, either PLD1 or PLD2 siRNA affected the mRNA level of PLD2 or
PLD1, respectively (supplemental Fig. S1A). These results show a role
for PLD1 and PLD2 in hyperoxia-induced ROS production in HPAECs.
/ROS generation. HPAECs
overexpressing PLD1/PLD2 wild-type or catalytically inactive mutants were
labeled with [32P]orthophosphate and exposed to hyperoxia for 2 h.
As shown in Fig. 1,
overexpression of hPLD1/mPLD2 wild type increased both basal and
hyperoxia-induced [32P]PBt production and ROS formation, whereas
the hPLD1-K898R/mPLD2-K758R mutants attenuated the formation of labeled PBt
and ROS production. Consistent with the data using adenoviral hPLD1/mPLD2
vectors, down-regulation of PLD1 or PLD2 with siRNA attenuated ROS generation
mediated by hyperoxia (supplemental Fig. S1). Further, accumulation of
[32P]PBt was significantly higher at 2 and 3 h as compared with
cells exposed to normoxia (supplemental Fig. S2A), and 1-butanol, but
not tertiary-butanol, attenuated hyperoxia-mediated ROS production, indicating
a role for PLD in NADPH oxidase activation (supplemental Fig. S2B).
Importantly, either PLD1 or PLD2 siRNA affected the mRNA level of PLD2 or
PLD1, respectively (supplemental Fig. S1A). These results show a role
for PLD1 and PLD2 in hyperoxia-induced ROS production in HPAECs.
FIGURE 1.

Effects of overexpression of wild-type and catalytically inactive mutants of hPLD1 and mPLD2 on hyperoxia-induced [32P]PBt formation and ROS production in HPAECs. HPAECs were infected with vector, wild-type, or mutant hPLD1 or mPLD2 adenoviral constructs and subsequently labeled with [32P]Pi, exposed to normoxia and hyperoxia as indicated. A, accumulation of [32P]PBt, an index of PLD activation, was quantified as described under “Experimental Procedures.” Values are mean ± S.D. of three independent experiments in triplicate. *, p < 0.05 compared with vector control/normoxia; **, p < 0.01 compared with vector control/normoxia; ***, p < 0.05 compared with vector control/hyperoxia; ****, p < 0.001 compared with PLD1 or PLD2 wild-type. B, ROS formation, under normoxia or hyperoxia, was determined by DCFDA oxidation as described under “Experimental Procedures.” Values are mean ± S.D. from three independent experiments in triplicate. *, p < 0.05 compared with vector control/normoxia; **, p < 0.01 compared with vector control/normoxia; ***p < 0.001 compared with vector control/normoxia; ****, p < 0.05 compared with PLD1 or PLD2 wild-type infected cells. In separate experiment, HPAECs were collected with lyses buffer, separated by 10% SDS-PAGE, and Western blotted with PLD1 or PLD2 antibodies as indicated (insets).
siRNA for PLD1 and PLD2 Attenuate Hyperoxia-induced p47phox Translocation to Cell Periphery, Activation, and Association of p47phox with Cortactin and Src—We have previously demonstrated that hyperoxia enhances Src-dependent tyrosine phosphorylation of p47phox and translocation and association of p47phox with Src and cortactin in HPAECs (16, 17). Based on our observation that PLD is involved in hyperoxia-mediated ROS production, we investigated the role of PLD on p47phox translocation and its enhanced association with Src and cortactin. As shown in Fig. 2A, most of the native p47phox was distributed in the cytoplasm and perinuclear region in normoxic cells, and hyperoxia enhanced the translocation of p47phox from the cytoplasm toward cell periphery within lamellipodia structures. Furthermore, down-regulation of PLD1 or PLD2 with siRNA blocked hyperoxia-mediated redistribution of p47phox to cell periphery and localization in lamellipodia (scrambled siRNA: normoxia, 100 ± 8%; hyperoxia, 178 ± 12%; PLD1 siRNA: normoxia, 100 ± 6%; hyperoxia, 105 ± 8%; PLD2 siRNA: normoxia, 100 ± 4%; hyperoxia, 96 ± 4%) (Fig. 2A). Next, we investigated the role of PLD2 in enhancing the association between p47phox, Src, and cortactin mediated by hyperoxia. As shown in Fig. 2B, overexpression of PLD2 mutant attenuated hyperoxia-induced tyrosine phosphorylation of p47phox, Src, and cortactin as well as hyperoxia-induced association of p47phox with Src and cortactin. Interestingly, overexpression of PLD2 mutant protein increased the association of p47phox with Src and cortactin even in normoxia. These results demonstrate that PLD regulates hyperoxia-induced p47phox phosphorylation and redistribution to cell periphery and enhances association with Src and cortactin.
FIGURE 2.

Effects of PLD1 or PLD2 siRNA and catalytically inactive mPLD2 mutant on hyperoxia-induced translocation of p47phox to cell periphery and association of p47phox with Src and cortactin. A, HPAECs were transfected with scrambled, PLD1, or PLD2 siRNA prior to exposure to either normoxia or hyperoxia (3 h) probed with anti-p47phox antibody, and examined by immunofluorescence microscopy using a ×60 oil objective. Exposure of cells to hyperoxia resulted in redistribution of p47phox to cell periphery, whereas siRNA for PLD1 or PLD2 blocked p47phox redistribution. A representative image from several experiments is shown. B, HPAECs were infected with vector control or mPLD2 K758R mutant in adenoviral construct, exposed to either normoxia or hyperoxia (3 h), and cell lysates were subjected to immunoprecipitation with anti-p47phox antibody under non-denaturing conditions as described under “Experimental Procedures.” Immunoprecipitates were analyzed by 10% SDS-PAGE and probed with antibodies as indicated. Representative blots from three independent experiments are shown.
Hyperoxia Induces Translocation of Tiam1 and Rac1 to Cell Periphery in HPAECs—Earlier studies have demonstrated that Rac1 is a key cytosolic component that regulates assembly and activation of phagocytic NADPH oxidase (8, 33, 37-41). To characterize the role of Rac1 in hyperoxia-induced ROS production, we determined if Tiam1, a GEF involved in Rac1 activation by hyperoxia. Exposure of HPAECs to hyperoxia for 3-h induced redistribution of Tiam1 and Rac1 to cell periphery as evidenced by immunofluorescence microscopy (normoxia: Tiam1, 100 ± 4; Rac1, 100 ± 5; hyperoxia: Tiam1, 172 ± 14; Rac1 193 ± 8) (Fig. 3A). Activation of Rac1 by hyperoxia was also verified by precipitation of Rac1 bound to GTP with PAK-1 PBD. Hyperoxia increased activation of Rac1 in a time-dependent fashion as evidenced by Western blotting of PAK-1 PBD precipitates (normoxia: 100 ± 6; hyperoxia: 1 h, 146 ± 5; 2 h, 214 ± 8; 3 h, 270 ± 9 (Fig. 3B). Knocking down Tiam1 expression with Tiam1 siRNA decreased hyperoxia-induced Rac1 activation (data not shown). Further, down-regulation of Rac1 with Rac1 siRNA had no effect on the expression of IQGAP1 and cortactin proteins (supplemental Fig. S7). These results demonstrate that hyperoxia activates Tiam1 and Rac1 in HPAECs.
FIGURE 3.

Involvement of Tiam1 and Rac1 in hyperoxia-induced ROS formation and p47phox redistribution to cell periphery. A, HPAECs were exposed to either normoxia or hyperoxia (3 h), probed with anti-Tiam1 or anti-Rac1 antibodies, and redistribution examined by immunofluorescence microscopy using a ×60 oil objective. Exposure of cells to hyperoxia resulted in translocation of Tiam1 and Rac1 from the cytoplasm to cell periphery. A representative image from three independent experiments is shown. B, HPAECs were exposed to either normoxia or hyperoxia (1, 2, and 3 h). Cell lysates were prepared, and activated Rac1 was precipitated using PAK-1 PBD-agarose beads as described under “Experimental Procedures.” Precipitated Rac1 beads were separated by 4-20% gradient SDS-PAGE and Western blotted with anti-Rac1 antibody. Shown is a representative blot from three independent experiments. C, HPAECs were transfected with scrambled or Rac1 siRNA, and cells were loaded with 10 μm DCFDA for 30 min prior to exposure to either normoxia or hyperoxia (3 h). Oxidation of DCFDA was monitored by DCFDA fluorescence as described under “Experimental Procedures.” Western blot insets reflect Rac1 expression by scrambled or Rac1 siRNA. Values are mean ± S.D. of three independent experiments in triplicate. *, p < 0.05 compared with scrambled siRNA/normoxia; **, p < 0.01 compared with scrambled siRNA/hyperoxia. D, HPAECs were transfected with scrambled, Rac1 siRNA, or Rac1 siRNA for 72 h plus p47phox-GFP (1 μg/ml plasmid DNA) for 48 h. Cells were exposed to normoxia or hyperoxia (3 h) and probed with anti-p47phox antibody and examined by immunofluorescence microscopy using a ×60 oil objective. A representative result from three independent experiments is shown.
Rac1 Depletion Attenuates Hyperoxia-induced p47phox Translocation and ROS Production—We next examined the functional role of Rac1 in hyperoxia-induced ROS production using molecular strategies to down-regulate or block Rac1 action. HPAECs were pretreated with Rac1 siRNA for 72 h, which reduced >90% Rac1 protein expression, respectively (Fig. 3C). Further, down-regulation of Rac1 attenuated ROS generation mediated by hyperoxia. In parallel experiments, Rac1 siRNA attenuated hyperoxia-induced translocation of native p47phox or p47phox-GFP to cell periphery (Fig. 3D). Semiquantitation of the redistribution of p47phox-GFP to cell periphery, using an image analyzer, showed an ∼1.9-fold increase in the intensity during hyperoxia as compared with cells exposed to normoxia while transfection of cells with Rac1 siRNA attenuated hyperoxia-induced p47phox translocation to cell periphery (scrambled siRNA: normoxia, 100 ± 6%; hyperoxia, 189 ± 15%; Rac1 siRNA: normoxia, 100 ± 7%; hyperoxia, 117 ± 9%). Taken together, these results suggest that Tiam1 and Rac1 regulate hyperoxia-dependent p47phox redistribution to cell periphery and ROS formation in HPAECs.
PLD2 siRNA Attenuates Hyperoxia-induced Tiam1 and Rac1 Activation in HPAECs—As Tiam1/Rac1 regulates NADPH oxidase activation and ROS generation in HPAECs, we next studied the role of PLD in hyperoxia-induced Tiam1/Rac1 activation and redistribution to cell periphery. HPAECs were transfected with scrambled or PLD2 siRNA (50 nm) for 72 h, and cells were challenged with normoxia or hyperoxia. Exposure to hyperoxia stimulated translocation of Tiam1 and Rac1 to the plasma membrane and knock down of PLD2 with PLD2 siRNA attenuated hyperoxia-induced translocation of Tiam1 and Rac1 (scrambled siRNA: normoxia-Tiam1, 100 ± 8%; Rac1, 100 ± 6%; hyperoxia-Tiam1, 136 ± 12%; Rac1, 198 ± 15%; PLD2 siRNA: normoxia-Tiam1, 100 ± 4%; Rac1, 100 ± 8%; hyperoxia-Tiam1, 109 ± 6%; Rac1, 119 ± 10%) (Fig. 4A). The immunofluorescence results were confirmed by precipitation of activated Rac1 using PAK-1 PBD-agarose beads followed by Western blotting. HPAECs were infected with a catalytically inactive PLD2-K758R mutant for 24 h, followed by exposure to either normoxia or hyperoxia for 2 h. Hyperoxia-increased activation of Rac1, which was attenuated by PLD2-K758R mutant (Vector Control: normoxia, 100 ± 6%; hyperoxia, 625 ± 56%; PLD2-K758R: normoxia, 170 ± 28%; hyperoxia, 310 ± 46%) (Fig. 4B). These results show that PLD2 regulates hyperoxia-induced Tiam1/Rac1 activation in HPAECs.
FIGURE 4.

PLD2 siRNA attenuates hyperoxia-induced activation of Tiam1 and Rac1. A, HPAECs were transfected with scrambled or PLD2 siRNA prior to exposure to normoxia or hyperoxia (3 h). Cells were probed with anti-Tiam1 (green) or anti-Rac1 (red) antibodies and redistribution was examined by immunofluorescence microscopy using a ×60 oil objective. Shown is a representative immunofluorescence micrograph from three independent experiments. B, HPAECs were infected with vector control or catalytically inactive mutant of mPLD2, exposed to either normoxia or hyperoxia (3 h), and activated Rac1 was precipitated from total cell lysates using PAK-1 PBD-agarose beads as described under “Experimental Procedures.” Shown is a representative blot from three independent experiments.
Hyperoxia-induced ROS Generation Is Dependent on IQGAP1 Activation in HPAECs—IQGAP1 is an IQ domain protein with a region containing sequence that has homology to RasGAP (42-45), and a recent study indicated that IQGAP1 may link Nox2 with actin (13) at the leading edge, thereby enhancing ROS production and ROS-dependent endothelial migration and proliferation (13, 46). However, very little information is available concerning hyperoxia-mediated activation of IQGAP1 and endothelial NADPH oxidase assembly and activation. HPAECs were exposed to normoxia or hyperoxia (3 h), and activation of IQGAP1 was determined by immunohistochemistry. As shown in Fig. 5 (A and B), hyperoxia enhanced translocation of IQGAP1 to plasma membrane (normoxia, 100 ± 12%; hyperoxia, 153 ± 15%), and time-dependent phosphorylation of IQGAP1 at serine/threonine and tyrosine residues indicating activation of IQGAP1. Overexpression of Myc-tagged IQGAP1 mutant attenuated hyperoxia-induced ROS formation; however, overexpression of IQGAP1 wild type had no effect on ROS production under hyperoxia (Fig. 5C). These results show a role for activated IQGAP1 in hyperoxiamediated ROS production in HPAECs.
FIGURE 5.

Involvement of IQGAP1 in hyperoxia-induced ROS formation. A, HPAECs were exposed to either normoxia or hyperoxia (3 h), probed with anti-IQGAP1 antibody, and redistribution was examined by immunofluorescence microscopy using a ×60 oil objective as described under “Experimental Procedures.” Hyperoxia induced the translocation of IQGAP1 to the cell periphery and in structures resembling membrane ruffles. Shown is an immunofluorescence micrograph from three independent experiments. B, HPAECs were exposed to either normoxia or hyperoxia (1, 2, and 3 h). Cell lysates were immunoprecipitated using anti-IQGAP1 antibody, separated by 10% SDS-PAGE, and Western blotted with antibodies as indicated. Shown is a representative blot from three independent experiments. C, HPAECs were transfected with vector control or Myc-tagged IQGAP1 wild-type (Wt) or mutant (Mn), loaded with 10 μm DCFDA, exposed to either normoxia or hyperoxia (3 h), and ROS formation was quantified by DCFDA fluorescence as described under “Experimental Procedures.” Values are mean ± S.D. from three independent experiments in triplicate. *, p < 0.01 compared with vector control/normoxia; **, p > 0.05 compared with vector control/hyperoxia; ***, p < 0.05 compared with vector control/hyperoxia. In the inset, overexpression of IQGAP1 Wt or Mn was verified by Western blotting of cell lysates (20 μg of protein) and probed with anti-Myc antibody.
IQGAP1 siRNA Attenuates Association of IQGAP1 with Src, Cortactin, and p47phox—We further investigated the potential interaction between IQGAP1 and components of NADPH oxidase and cytoskeletal proteins. HPAECs were exposed to either normoxia or hyperoxia for varying time periods, and cell lysates were subjected to immunoprecipitation with anti-IQGAP1 antibody and analyzed for NADPH oxidase activating proteins. As shown in Fig. 6 (A and B), IQGAP1 is constitutively associated with PLD2, Tiam1, Rac1, Src, cortactin, and p47phox under normoxia, and exposure to hyperoxia (1-3 h) further increased this association with the exception of total Rac1. Additionally, the cortactin immunoprecipitates also revealed the presence of immunodetectable IQGAP1, Tiam1, and p47phox under basal conditions, which was further increased after hyperoxia (Fig. 6C). Next, we investigated by immunofluorescence microscope the association between IQGAP1 and p47phox in normoxia and hyperoxia. As shown in Fig. 6D, IQGAP1 and GFP-p47phox seemed to be dispersed mainly in the cytoplasm; however, exposure of HPAECs to hyperoxia (3 h) enhanced translocation of both IQGAP1 and GFP-p47phox to the cell periphery and merging of the immunofluorescence stains of IQGAP1 (red) and GFP-p47phox (green) showed co-localization (yellow) at the plasma membrane of the cell. Transfection of HPAECs with IQGAP1 siRNA blocked translocation of GFP-p47phox (Fig. 6D) without affecting the expression of Rac1 and cortactin (supplemental Fig. S7). Similarly, down-regulation of IQGAP1 with siRNA attenuated translocation of cortactin to cell periphery (Fig. 7A) and decreased hyperoxia-induced tyrosine phosphorylation of Src (Fig. 7B). These results suggest the involvement of IQGAP1 in hyperoxia-induced p47phox and cortactin redistribution to cell periphery and association of IQGAP1 with PLD2, Tiam1, Src, cortactin, and p47phox in HPAECs.
FIGURE 6.

Hyperoxia enhances association of IQGAP1 with PLD2, Tiam1, Src, cortactin, and p47phox. HPAECs were exposed to normoxia or hyperoxia for varying time periods, cell lysates (500 μg of proteins) were subjected to immunoprecipitation with anti-IQGAP1 or anti-cortactin. A, IQGAP1 immunoprecipitates were separated by SDS-PAGE and Western blotted with anti-IQGAP1, -Tiam1, -PLD2, -Src, and -Rac1 antibodies as described under “Experimental Procedures.” B, cells were exposed to normoxia or hyperoxia for 2 h, and cell lysates (500 μg of proteins) were subjected to immunoprecipitation with IgG or anti-IQGAP1 antibody. Immunoprecipitates were subjected to SDS-PAGE and Western blotting with anti-IQGAP1, -Tiam1, -cortactin, and -p47phox antibodies. C, cell lysates (500 μg of proteins) from B were subjected to immunoprecipitation with IgG or anti-cortactin antibody and immunoprecipitates were separated by SDS-PAGE and Western blotted with anti-cortactin, -IQGAP1, -Tiam1, and -p47phox antibodies. Shown are representative blots from three independent experiments. D, HPAECs were transfected with scrambled or IQGAP1 siRNA and p47phox-GFP plasmid, cells were exposed to either normoxia or hyperoxia (3 h), and prepared for immunofluorescence analysis of IQGAP1 (red) and p47phox-GFP (green) localization. In normoxic conditions, IQGAP1 and p47phox-GFP were distributed diffusely in the cytoplasm and perinuclear areas, respectively. Hyperoxia induced redistribution of both proteins to the cell periphery where they appear to co-localize (yellow in merged images), and down-regulation of IQGAP1 attenuated p47phox-GFP redistribution to cell periphery. Shown are representative images from three independent experiments.
FIGURE 7.

Down-regulation of IQGAP1 with IQGAP1 siRNA blunts of hyperoxia-induced translocation of cortactin and phosphorylation of Src. A, HPAECs was transfected with scrambled or IQGAP1 siRNA prior to exposure to either normoxia or hyperoxia (3 h), probed with anti-cortactin antibody, and examined by immunofluorescence microscopy. Shown is a representative immunofluorescence micrograph from three independent experiments. The redistribution of cortactin to cell periphery was quantified using an image analyzer as described under “Experimental Procedures.” Values are mean± S.D. from three independent experiments and expressed as relative pixels. *, p < 0.01 compared with scrambled siRNA-transfected cells/normoxia; **, p < 0.05 compared with scrambled siRNA transfected cells/hyperoxia. B, in parallel experiments ECs were transfected with scrambled or IQGAP1 siRNA, cells exposed to either normoxia or hyperoxia (3 h), total cell lysates separated by SDS-PAGE 10% gels, and Western blotted with total Src, IQGAP1, and phospho-Src antibodies as described under “Experimental Procedures.” Shown is a representative blot from three independent experiments.
PLD2 and Rac1 Regulate Hyperoxia-induced IQGAP1 Activation—Having established roles for PLD2 in Rac1 activation, and in the assembly and activation of NADPH oxidase and ROS generation, we next studied the role of PLD2 and Rac1 in IQGAP1 activation by hyperoxia. HPAECs were transfected with scrambled, PLD2 (50 nm), or Rac1 siRNA (50 nm) for 72 h prior to exposure to normoxia or hyperoxia. As shown in Fig. 8A, PLD2 siRNA, but not scrambled siRNA, attenuated hyperoxia-induced translocation of IQGAP1 to cell periphery. Similarly, overexpression of the catalytically inactive mPLD2-K758R mutant did not stimulate hyperoxia-induced serine/threonine and tyrosine phosphorylation of IQGAP1; however, enhanced basal tyrosine phosphorylation, but not serine/threonine phosphorylation, of IQGAP1 (Fig. 8B). Additionally, overexpression of catalytically inactive mPLD2-K758R mutant attenuated association of IQGAP1 with activated Rac1 as verified by precipitation of activated Rac1 bound to GTP with PAK-1 PBD (Vector Control: normoxia, 100 ± 12%; hyperoxia, 270 ± 22%; adenoviral PLD2-K758R mutant: 20 ± 6%; hyperoxia, 70 ± 14%) (Fig. 8C). Down-regulation of PLD1 with PLD1 siRNA and exposure of cells to either normoxia or hyperoxia resulted in a similar attenuation of IQGAP1 activation (data not shown). These data suggest the involvement of PLD in hyperoxia-induced IQGAP1 activation. As Rac1 activation is downstream to hyperoxia-induced PLD stimulation, the role of Rac1 in IQGAP1 activation and association with p47phox was investigated. Exposure of HPAECs to hyperoxia (3 h) revealed translocation of both IQGAP1 and GFP-p47phox to the cell periphery and merging of the immunofluorescence stains of IQGAP1 (red) and GFP-p47phox (green) showed co-localization (yellow) at the plasma membrane of the cell. Furthermore, down-regulation of Rac1 expression with Rac1 siRNA blocked hyperoxia-induced translocation of IQGAP1 and GFP-p47phox to cell periphery (Fig. 9A). Further, the increased association of IQGAP1 with phospho-Src, cortactin, and phospho-p47phox was diminished in Rac1 siRNA-transfected cells after exposure to hyperoxia (Fig. 9B). These results show a role for PLD2 in Rac1-dependent activation of IQGAP1 by hyperoxia.
FIGURE 8.

PLD2 regulates hyperoxia-induced IQGAP1 activation and association with Rac1. A, HPAECs were transfected with scrambled or PLD2 siRNA, exposed to either normoxia or hyperoxia (3 h), and probed with anti-IQGAP1 antibody. Redistribution of IQGAP1 was examined by immunofluorescence microscopy using a ×60 oil objective. Shown is a representative immunofluorescence micrograph from three independent experiments. B, HPAECs were infected with vector control or adenoviral construct of mPLD2 mutant (Mn), exposed to either normoxia or hyperoxia (3 h), cell lysates were subjected to immunoprecipitation with anti-IQGAP1 antibody, and immunoprecipitates were assayed for phosphorylation of IQGAP1 with anti-phosphoserine, anti-phosphothreonine, or anti-phosphotyrosine antibodies as described under “Experimental Procedures.” Shown is a representative Western blot from three independent experiments. C, cell lysates from B were subjected to immunoprecipitation with PAK-1 PBD-agarose beads to bring down activated Rac1 bound to GTP as described under “Experimental Procedures.” Rac1-GTP bound to PAK-1 PBD was separated by 4-20% SDS-PAGE and probed with anti-Rac1 antibody. Shown is a representative blot from three independent experiments.
FIGURE 9.

Rac1 siRNA attenuates hyperoxia-induced IQGAP1 redistribution to cell periphery and co-localization with p47phox. A, HPAECs were transfected with scrambled or Rac1 siRNA, and additionally transfected with plasmid DNA of p47phox-GFP. 24 h later cells were exposed to either normoxia or hyperoxia (3 h) and analyzed by immunofluorescence microscopy for IQGAP1 (red) and p47phox-GFP (green) localization. In normoxic condition, IQGAP1 and p47phox-GFP were distributed diffusely in the cytosol and hyperoxia induced redistribution of both the proteins to cell periphery, where they appear to co-localize (yellow in merged image). Rac1 siRNA blocked the redistribution and co-localization of IQGAP1 and p47phox (less yellow visible in merged image). A representative image from three independent experiments is shown. B, in parallel experiments, HPAECs were transfected with scrambled or Rac1 siRNA, cells exposed to either normoxia or hyperoxia (3 h), and total cell lysates were subjected to immunoprecipitation with anti-IQGAP1 antibody as described under “Experimental Procedures.” Immunoprecipitates were analyzed after separation by SDS-PAGE for total IQGAP1, cortactin, Src, and p47phox with anti-IQGAP1, -cortactin, -Src, and -p47phox antibodies, respectively. Shown is a representative blot from three independent experiments.
Role of Rac1 in Hyperoxia-induced Tyrosine Phosphorylation of Src and Cortactin in HPAECs—As it was determined from the earlier experiments of this study that Rac1 via IQGAP1-regulated hyperoxia-induced p47phox translocation to cell periphery and ROS production; we also investigated the role of Rac1 in the activation of Src and cortactin. HPAECs were transfected with scrambled or Rac1 siRNA (50 nm) for 72 h prior to exposure to hyperoxia. As shown in Fig. 10 (A and B), Rac1 siRNA attenuated hyperoxia-mediated tyrosine phosphorylation of Src as evidenced by immunocytochemistry (scrambled siRNA: normoxia, 100 ± 12%; hyperoxia, 303 ± 24%; Rac1 siRNA: normoxia, 100 ± 4%; hyperoxia, 143 ± 16%), and Western blotting with phospho-Src and total Src antibodies as compared with scrambled cells exposed to hyperoxia. Similarly, treatment of cells with dominant-negative Rac1 or NSC23766, an inhibitor of Rac1, blocked hyperoxia-induced tyrosine phosphorylation of Src (data not shown). In our recent studies, we have shown that tyrosine phosphorylation of cortactin is essential for hyperoxia-mediated assembly of NADPH oxidase components with the actin cytoskeleton during ROS generation in HPAECs (17). We next examined the effect of Rac1 siRNA or dominant-negative Rac1 on hyperoxia-induced tyrosine phosphorylation of cortactin. Cells under normoxia showed basal tyrosine phosphorylation of cortactin as determined by immunohistochemistry or Western blotting with phospho-specific cortactin antibodies. Exposure to hyperoxia enhanced tyrosine phosphorylation of cortactin, which was partially blocked by Rac1 siRNA (Fig. 11A) or dominant-negative Rac1 (Fig. 11B). These results show that Rac1 plays a critical role in hyperoxia-mediated enhanced tyrosine phosphorylation of Src and cortactin in HPAECs.
FIGURE 10.

Effect of Rac1 siRNA on hyperoxia-induced Src activation. A, HPAECs were transfected with scrambled or Rac1 siRNA prior to exposure to either normoxia or hyperoxia (3 h) and probed with anti-phospho-Src antibody. Shown is a representative immunofluorescence micrograph from three independent experiments. Hyperoxia enhanced immunostaining of phospho-Src near cell periphery, which was blocked by Rac1 siRNA. B, HPAECs were transfected with scrambled or Rac1 siRNA, and total cell lysates were separated by 4-20% SDS-PAGE and Western blotted with anti-Src, anti-phospho-Src, or anti-Rac1 antibodies as described under “Experimental Procedures.” Shown is a representative blot from three independent experiments.
FIGURE 11.

Down-regulation of Rac1 prevents hyperoxia-induced tyrosine phosphorylation of cortactin. A, HPAECs were transfected with scrambled siRNA or Rac1 siRNA, exposed to either normoxia or hyperoxia (3 h) and probed with anti-phospho-cortactin antibody as described under “Experimental Procedures.” Hyperoxia stimulated tyrosine phosphorylation and translocation of phospho-cortactin to cell periphery, which was attenuated by Rac1 siRNA. Shown is a representative immunofluorescence micrograph from three independent experiments. The redistribution of p-cortactin to cell periphery was quantified using an image analyzer as described under “Experimental Procedures.” The values are the mean ± S.D. from three independent experiments as relative pixels. *, p < 0.01 compared with scrambled siRNA/normoxia; **, p < 0.05 compared with scrambled siRNA/hyperoxia. B, HPAECs were infected with vector-control or adenoviral dominant-negative Rac1 prior to exposure to normoxia or hyperoxia (3 h), cell lysates (20 μg of protein) were separated by 4-20% SDS-PAGE and Western blotted with anti-cortactin, anti-phospho-cortactin, or anti-Rac1 antibodies. Overexpressed dominant-negative Rac1 ran slightly ahead of native Rac1 on SDS-PAGE. Shown is a representative Western blot from three independent experiments, and hyperoxia-induced phosphorylation of cortactin was calculated by densitometry and normalized to total cortactin. The values are mean ± S.D. from three independent experiments. *, p < 0.05 compared with vector control/normoxia; **, p < 0.001 compared with vector control/hyperoxia.
PLD2 siRNA Attenuates Hyperoxia-induced Tyrosine Phosphorylation of Src and Cortactin in HPAECs—Having established the role of PLD2 in Rac1 and IQGAP1 activation as well as ROS generation by hyperoxia, we next investigated the role of PLD2 in modulation of tyrosine phosphorylation of Src and cortactin. Down-regulation of PLD2 with PLD2 siRNA attenuated tyrosine phosphorylation of Src as evidenced by immunofluorescence microscopy (scrambled siRNA: normoxia, 100 ± 8%; hyperoxia, 249 ± 16%; PLD2 siRNA: normoxia, 100 ± 8%; hyperoxia, 97 ± 18%) (Fig. 12A). Because tyrosine phosphorylation of p47phox is dependent on Src (16), and PLD2 is essential for Src phosphorylation (Fig. 12A), it raises the possibility that PLD2 plays a role in hyperoxia-mediated enhanced association of cortactin and p47phox with Src. This was explored by down-regulation of cells with PLD2 siRNA, Src was immunoprecipitated for co-immunoprecipitation studies under normoxia and hyperoxia. Down-regulation of PLD2 expression partly abolished hyperoxia-mediated co-immunoprecipitation of cortactin, phospho-cortactin, phospho-Src, and p47phox with Src (Fig. 12B). Further, PLD2 siRNA prevented cortactin redistribution to lamellipodia and tyrosine phosphorylation of cortactin (Fig. 13, A and B). Furthermore, additional experiments were carried out in the presence of 1-butanol or tertiary butanol to demonstrate the involvement of PLD pathway as PA is channeled to PBt by 1-butanol and not by tertiary-butanol. Addition of 1-butanol, but not tertiary-butanol, attenuated hyperoxia-mediated redistribution of Rac1, IQGAP1, cortactin, and p47phox to cell periphery, suggesting a role for PA or DAG generated by the PLD pathway in activation of the various components of NADPH oxidase (supplemental Figs. S3-S6). Although the PLD1, PLD2 siRNA, or 1-butanol experiments suggest a role for PLD signaling in hyperoxia-induced tyrosine phosphorylation of Src and cortactin, the involvement of PA or DAG derived from PLD pathway is not conclusive as PA could be converted to DAG by lipid phosphate phosphatases or lipins (22, 23, 47).
FIGURE 12.

PLD mediates hyperoxia-induced Src and cortactin activation. A, HPAECs were transfected with scrambled siRNA or PLD2 siRNA prior to exposure to either normoxia or hyperoxia (3 h) and probed with anti-phospho-Src antibody. Translocation of phospho-Src was quantified by image analyzer using MetaVue software. Shown is a representative immunofluorescence micrograph from three independent experiments. Hyperoxia enhanced tyrosine phosphorylation of Src in cell periphery, which was attenuated by PLD2 siRNA. The values are mean ± S.D. from three independent experiments. *, p < 0.01 compared with scrambled siRNA/normoxia; **, p < 0.05 compared with scrambled siRNA/hyperoxia. B, HPAECs were infected with adenoviral vector control or mPLD2 mutant prior to exposure to normoxia or hyperoxia (3 h). Cell lysates were subjected to immunoprecipitation with anti-Src antibody as described under “Experimental Procedures,” and immunoprecipitates were subjected to SDS-PAGE and Western blotted with anti-Src, -cortactin, -p47phox, and -phospho-Src antibodies as described under “Experimental Procedures.” Hyperoxia increased co-immunoprecipitation of cortactin, p47phox, phospho-Src, and phospho-cortactin with Src, which was attenuated by PLD2 siRNA. Shown is a representative blot from three independent experiments.
FIGURE 13.

PLD2 siRNA attenuates hyperoxia-induced redistribution and tyrosine phosphorylation of cortactin. A, HPAECs were transfected with scrambled or PLD2 siRNA prior to exposure to either normoxia or hyperoxia (3 h), probed with anti-cortactin antibody, and examined by immunofluorescence microscopy using a ×60 oil objective. Exposure to hyperoxia resulted in redistribution of cortactin to cell periphery, which was attenuated by PLD2 siRNA. Shown is a representative immunofluorescence micrograph from several independent experiments. B, HPAECs were transfected with scrambled or PLD2 siRNA prior to exposure to either normoxia or hyperoxia. Cell lysates were subjected to SDS-PAGE and Western blotted with anti-cortactin, -phospho-cortactin, -actin, and -PLD2 antibodies. PLD2 siRNA attenuated hyperoxia-induced phosphorylation of cortactin. Shown is a representative blot from three independent experiments.
DISCUSSION
Vascular NADPH oxidase has emerged as an important generator of lung
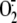 /ROS that regulates
endothelial signaling, motility, proliferation, and barrier function
(1-3,
12,
48). The mechanisms of
vascular NADPH oxidase activation, akin to phagocytic oxidase activation, are
complex, requiring phosphorylation of the cytosolic components and assembly of
the phosphorylated cytosolic subunits with Rac1 to the cytochrome
b558. In the present study, we show that (i) hyperoxia
activated PLD; (ii) transient overexpression of adenoviral constructs of PLD1
and PLD2 wild type potentiated hyperoxia-induced PLD activation,
p47phox translocation, and accumulation of ROS, while the
catalytically inactive mutants of PLD1 and PLD2 attenuated hyperoxia-mediated
activation of NADPH-oxidase and ROS generation; (iii) hyperoxia caused a rapid
activation and redistribution to cell periphery of Tiam1/Rac1 as well as
IQGAP1; (iv) down-regulation of Rac1/IQGAP1 blocked hyperoxia-induced Src and
cortactin tyrosine phosphorylation, p47phox translocation,
and ROS production; and (v) down-regulation of PLD2 either with siRNA or
dominant-negative PLD2 attenuated activation of Rac1, IQGAP1, Src, cortactin,
and p47phox redistribution to cell periphery.
/ROS that regulates
endothelial signaling, motility, proliferation, and barrier function
(1-3,
12,
48). The mechanisms of
vascular NADPH oxidase activation, akin to phagocytic oxidase activation, are
complex, requiring phosphorylation of the cytosolic components and assembly of
the phosphorylated cytosolic subunits with Rac1 to the cytochrome
b558. In the present study, we show that (i) hyperoxia
activated PLD; (ii) transient overexpression of adenoviral constructs of PLD1
and PLD2 wild type potentiated hyperoxia-induced PLD activation,
p47phox translocation, and accumulation of ROS, while the
catalytically inactive mutants of PLD1 and PLD2 attenuated hyperoxia-mediated
activation of NADPH-oxidase and ROS generation; (iii) hyperoxia caused a rapid
activation and redistribution to cell periphery of Tiam1/Rac1 as well as
IQGAP1; (iv) down-regulation of Rac1/IQGAP1 blocked hyperoxia-induced Src and
cortactin tyrosine phosphorylation, p47phox translocation,
and ROS production; and (v) down-regulation of PLD2 either with siRNA or
dominant-negative PLD2 attenuated activation of Rac1, IQGAP1, Src, cortactin,
and p47phox redistribution to cell periphery.
Our results show a requirement for both PLD1 and PLD2 in the assembly and activation of NADPH oxidase and ROS generation. Although it is not clear how both PLD1 and PLD2 regulate hyperoxia-induced NADPH oxidase activation and ROS generation, it has been proposed that increased PIP2 generated by PLD1 leads to PLD2 activation (49). Further, PA generated through PLD1/PLD2 activation can be metabolically converted to either lysophosphatidic acid or DAG (21-23); therefore, we have used 1- and tertiary-butanol (47) in some of the key experiments to demonstrate the participation of PLD pathway in NADPH oxidase activation and ROS generation. These experiments were based on the literature that showed PLD also catalyzes transphosphatidylation of PA to primary, but not secondary or tertiary, short-chain alcohols such as propanol or butanol, and the formation of phosphatidyl alcohol mitigates PA- or DAG-dependent second messenger functions (22, 47). Thus, incubation of cells with 1-, but not tertiary-, butanol resulted in increased PBt formation by hyperoxia and attenuated hyperoxia-induced Rac1, IQGAP1, cortactin, and p47phox translocation to cell periphery, confirming a role for PLD pathway in the assembly and activation of endothelial NADPH oxidase and ROS generation (supplemental Figs. S3-S6). However, our results do not exclude the role of DAG-dependent activation of PKC isoforms in the assembly and activation of vascular NADPH oxidase and ROS generation (12, 50). In human lung ECs, overexpression of dominant-negative PKCα and PKCδ partly blocked hyperoxia-induced ROS generation (12) suggesting involvement of DAG-dependent activation of PKC in NADPH oxidase activation. Similarly, earlier studies have shown that, in activated leukocytes, p47phox is phosphorylated by PKC α, β II, δ, and ζ (51, 52), and upon stimulation with angiotensin II, p47phox is phosphorylated at serine and tyrosine residues in vascular smooth muscle cells (53).
Accumulating evidence supports an absolute requirement for Rac in the activation of phagocytic and non-phagocytic NADPH oxidase in vivo and in vitro (37, 54); however, mechanisms of Rac1 or Rac2 activation and downstream targets of Rac that regulate NADPH oxidase assembly and ROS generation have not been well defined. In the present study, we demonstrate for the first time a critical role for PLD in Rac1-dependent ROS production by hyperoxia in HPAECs. Hyperoxia-induced Rac1 activation was mediated by Tiam1 (data not shown), one of several Rac GEFs, and down-regulation of PLD2 with siRNA or overexpression of a catalytically inactive mutant of PLD2 attenuated Tiam1 as well as Rac1 activation and its translocation to cell periphery (Fig. 3, A and B). The role of PLD in Rac activation seems to depend on the stimulus and cell type studied. Our current results on PLD-dependent activation of Rac1 by hyperoxia are in agreement with sphingosine-1-phosphate-mediated Rac1 activation by PLD2 in human lung EC motility (55) and regulation of integrin-mediated spreading and migration by GTP-Rac1 localization through the elevating PLD activity in OVCAR-3 cells (56). Although the mechanisms of regulation of Rac1 by PLD have not been well defined, the elegant studies of Chae et al. (56) have shown that PA generated from PLD activation in OVCAR-3 cells acts as a membrane anchor of Rac1 with the C-terminal polybasic motif of Rac1 being responsible for the direct interaction with PA. Further, it seems that PA may also induce dissociation of the Rho-guanine nucleotide dissociation inhibitor from Rac1 for PA-mediated Rac1 localization (56, 57). Alternatively, PA based on its anionic properties (58, 59) may support NADPH oxidase assembly as evidenced in vitro experiments (57). These results on PLD-dependent activation of Rac1 are in contrast to earlier reports on small GTP-protein-dependent activation of PLD by growth factors and antigens in human promyelocytic HL60 (60) and RBL-2H3 (61).
In defining effectors of Rac1 that regulate NADPH oxidase assembly, we have identified IQGAP1 as a novel downstream target of PLD-Rac1 signal transduction in human lung ECs. IQGAP1 is an IQ domain-containing protein with a region containing sequence that has homology to RasGAP and has several interaction sites of well established calponin homology, WW, IQ motif, GRD, and RGCT domains (42). IQGAP1 interacts with several proteins, including signaling molecules like Cdc42, Rac1, calmodulin, β-catenin, E-cadherin, actin filaments, and microtubule plus end tracking proteins (CLIP170), and adenomatous polyposis coli suggesting its role in cell polarity, adhesion, and migration (13, 42-46, 62-65). A recent study indicates that IQGAP1 may link Nox2 to actin at the leading edge, thereby enhancing ROS production and contributing to cell motility in ECs (13). Here, we demonstrate that hyperoxia induces IQGAP1 translocation to cell periphery, IQGAP1 siRNA attenuated hyperoxia-induced ROS generation, and down-regulation of Tiam1/Rac1 attenuated IQGAP1 translocation to cell periphery in HPAECs. We also show that hyperoxia enhances association between IQGAP1 and Rac1, PLD2, cortactin, and p47phox and that siRNA for Rac1 and PLD2 mitigated IQGAP1 activation. The functional significance of interaction of IQGAP1 with p47phox is demonstrated by the observation that down-regulation of Rac1 and IQGAP1 expression with siRNA inhibits tyrosine phosphorylation and translocation of p47phox to cell periphery. The observed effects of Rac1 and IQGAP1 siRNA were not due to off-target responses to siRNA (supplemental Fig. S7). IQGAP1, an effector of Rho-GTPases, is a key regulator of cellular processes such as cell migration, wound closure, and proliferation (66-68). IQGAP1 has been shown to mediate repair of bronchial epithelial cells through activating Tcf signals independent of Rac1 and Cdc42 (69). In the current study, IQGAP1 is the effector of Rac1, because IQGAP1 siRNA did not affect hyperoxia-induced activation of Rac1 and blocking Rac1 had no effect on PLD activation by hyperoxia (data not shown) suggesting a PLD → Rac1 → IQGAP1 cascade in p47phox translocation and ROS generation in HPAECs.
Actin cytoskeleton and other cytoskeletal proteins play an essential role in the assembly and activation of NADPH oxidase (11, 17, 70, 71). We have recently demonstrated that the interaction between cortactin and p47phox, which facilitates NADPH oxidase activation and ROS (17), is dependent on Src-mediated tyrosine phosphorylation of cortactin and association of Src with cortactin (16). In the present study, we have identified that PLD/PA signaling is part of the regulatory mechanism of hyperoxia-mediated assembly of p47phox with Rac1/IQGAP1/Src/cortactin and ROS generation. Addition of exogenous PA stimulated phosphorylation of p47phox in intact neutrophils and cell-free preparations via a novel PA-activated protein kinase (18, 20, 27). In a subsequent study, several neutrophil agonists such as phorbol myristate acetate, opsonized zymosan, and formyl-Met-Leu-Phe stimulated p22phox phosphorylation and NADPH oxidase activity in neutrophils (72) suggesting a role of PA derived from PLD in the phosphorylation of the components and activation of the phagocytic oxidase. However, the signaling cascade of the PLD/PA pathway in relation to assembly and activation of NADPH oxidase are still not well defined. In the present study, we have defined some of the downstream targets of PLD, including Rac1, IQGAP1, and Src, in the assembly of cortactin with p47phox and ROS generation. Our results also indicate that Rac1 is the immediate effector of PLD as down-regulation of Rac1 attenuates PLD-dependent IQGAP1, Src, cortactin, and p47phox translocation as well as ROS generation. It is unclear if the PA-dependent activation of Rac1 and subsequent signaling cascades involves either a direct or indirect interaction between Rac1 with IQGAP1, cortactin, and p47phox.
In summary, our results demonstrate a novel and important mechanism linking PLD-generated PA in the assembly and activation of endothelial NADPH oxidase and ROS generation in response to hyperoxia. A role for ROS in pulmonary leak and injury is a clinically relevant condition in bronchopulmonary dysplasia and ventilator-induced lung injury. Future studies targeting PLD or some of the downstream targets of PLD such as Rac1 or IQGAP1 may provide new therapeutic avenues in minimizing excess ROS generation and lung injury.
Supplementary Material
Acknowledgments
We thank Dr. Michael E. Kleinberg from the University of Maryland, School of Medicine at Baltimore, Maryland for providing p47phox-GFP plasmid. The services of the University of Iowa Gene Transfer Vector Core, supported in part by the National Institutes of Health, and Roy J. Carver Foundation, for viral amplification and generation of purified wild type hPLD1 and mPLD2, and dominant-negative Rac1 adenoviral constructs.
This work was supported, in whole or in part, by National Institutes of Health Grant PO1 HL 58064 (to V. N. and J. G. N. G.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S7.
Footnotes
The abbreviations used are:
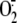 , superoxide; DAG,
diacylglycerol; DCFDA, 6-carboxy-2′,7′-dichlorofluorescein
diacetate; EC, endothelial cell; ERK, extracellular-signal-regulated kinase;
GEF, guanine nucleotide exchange factor; HPAEC, human pulmonary artery
endothelial cell; HO, hyperoxia; MAPK, mitogen-activated protein kinase; Mn,
mutant; NF-κB, nuclear factor κB; phox, phagocytic
oxidase; PA, phosphatidic acid; PBS, phosphate-buffered saline; PC,
phosphatidylcholine; PLA2, phospholipase A2; PKC,
protein kinase C; PLC, phospholipase C; PLD, phospholipase D; PI,
phosphatidylinositol; PI3K, phosphatidylinositol 3-kinase; ROS, reactive
oxygen species; TBST, Tris-buffered saline with 0.1% Tween 20; TNF-α,
tumor necrosis factor-α; Wt, wild type; PBt, phosphatidylbutanol.
, superoxide; DAG,
diacylglycerol; DCFDA, 6-carboxy-2′,7′-dichlorofluorescein
diacetate; EC, endothelial cell; ERK, extracellular-signal-regulated kinase;
GEF, guanine nucleotide exchange factor; HPAEC, human pulmonary artery
endothelial cell; HO, hyperoxia; MAPK, mitogen-activated protein kinase; Mn,
mutant; NF-κB, nuclear factor κB; phox, phagocytic
oxidase; PA, phosphatidic acid; PBS, phosphate-buffered saline; PC,
phosphatidylcholine; PLA2, phospholipase A2; PKC,
protein kinase C; PLC, phospholipase C; PLD, phospholipase D; PI,
phosphatidylinositol; PI3K, phosphatidylinositol 3-kinase; ROS, reactive
oxygen species; TBST, Tris-buffered saline with 0.1% Tween 20; TNF-α,
tumor necrosis factor-α; Wt, wild type; PBt, phosphatidylbutanol.
References
- 1.Cave, A. C., Brewer, A. C., Narayanapanicker, A., Ray, R., Grieve, D. J., Walker, S., and Shah, A. M. (2006) Antioxid. Redox. Signal 8 691-728 [DOI] [PubMed] [Google Scholar]
- 2.Griendling, K. K., Sorescu, D., and Ushio-Fukai, M. (2000) Circ. Res. 86 494-501 [DOI] [PubMed] [Google Scholar]
- 3.Ushio-Fukai, M. (2006) Cardiovasc. Res. 71 226-235 [DOI] [PubMed] [Google Scholar]
- 4.Allen, L. A., DeLeo, F. R., Gallois, A., Toyoshima, S., Suzuki, K., and Nauseef, W. M. (1999) Blood 93 3521-3530 [PubMed] [Google Scholar]
- 5.Clark, R. A. (1999) J. Infect. Dis. 179 Suppl. 2, S309-317 [DOI] [PubMed] [Google Scholar]
- 6.Henderson, L. M., and Chappel, J. B. (1996) Biochim. Biophys. Acta 1273 87-107 [DOI] [PubMed] [Google Scholar]
- 7.Ago, T., Kitazono, T., Ooboshi, H., Iyama, T., Han, Y. H., Takada, J., Wakisaka, M., Ibayashi, S., Utsumi, H., and Iida, M. (2004) Circulation 109 227-233 [DOI] [PubMed] [Google Scholar]
- 8.Bokoch, G. M., and Zhao, T. (2006) Antioxid. Redox. Signal 8 1533-1548 [DOI] [PubMed] [Google Scholar]
- 9.Sohn, H. Y., Keller, M., Gloe, T., Morawietz, H., Rueckschloss, U., and Pohl, U. (2000) J. Biol. Chem. 275 18745-18750 [DOI] [PubMed] [Google Scholar]
- 10.Wolfson, M., McPhail, L. C., Nasrallah, V. N., and Snyderman, R. (1985) J. Immunol. 135 2057-2062 [PubMed] [Google Scholar]
- 11.Tamura, M., Kai, T., Tsunawaki, S., Lambeth, J. D., and Kameda, K. (2000) Biochem. Biophys. Res. Commun. 276 1186-1190 [DOI] [PubMed] [Google Scholar]
- 12.Pendyala, S., Gorshkova, I. A., Usatyuk, P., He, D., Pennathur, A., Lambeth, D., Thannickal, V. J., and Natarajan, V. (2009) Antioxid. Redox Signal. 11 747-764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikeda, S., Yamaoka-Tojo, M., Hilenski, L., Patrushev, N. A., Anwar, G. M., Quinn, M. T., and Ushio-Fukai, M. (2005) Arterioscler. Thromb. Vasc. Biol. 25 2295-2300 [DOI] [PubMed] [Google Scholar]
- 14.Parinandi, N. L., Kleinberg, M. A., Usatyuk, P. V., Cummings, R. J., Pennathur, A., Cardounel, A. J., Zweier, J. L., Garcia, J. G., and Natarajan, V. (2003) Am. J. Physiol. Lung Cell Mol. Physiol. 284 L26-38 [DOI] [PubMed] [Google Scholar]
- 15.Usatyuk, P. V., Vepa, S., Watkins, T., He, D., Parinandi, N. L., and Natarajan, V. (2003) Antioxid. Redox. Signal 5 723-730 [DOI] [PubMed] [Google Scholar]
- 16.Chowdhury, A. K., Watkins, T., Parinandi, N. L., Saatian, B., Kleinberg, M. E., Usatyuk, P. V., and Natarajan, V. (2005) J. Biol. Chem. 280 20700-20711 [DOI] [PubMed] [Google Scholar]
- 17.Usatyuk, P. V., Romer, L. H., He, D., Parinandi, N. L., Kleinberg, M. E., Zhan, S., Jacobson, J. R., Dudek, S. M., Pendyala, S., Garcia, J. G., and Natarajan, V. (2007) J. Biol. Chem. 282 23284-23295 [DOI] [PubMed] [Google Scholar]
- 18.McPhail, L. C., Waite, K. A., Regier, D. S., Nixon, J. B., Qualliotine-Mann, D., Zhang, W. X., Wallin, R., and Sergeant, S. (1999) Biochim. Biophys. Acta 1439 277-290 [DOI] [PubMed] [Google Scholar]
- 19.Palicz, A., Foubert, T. R., Jesaitis, A. J., Marodi, L., and McPhail, L. C. (2001) J. Biol. Chem. 276 3090-3097 [DOI] [PubMed] [Google Scholar]
- 20.Regier, D. S., Waite, K. A., Wallin, R., and McPhail, L. C. (1999) J. Biol. Chem. 274 36601-36608 [DOI] [PubMed] [Google Scholar]
- 21.Brown, H. A., Henage, L. G., Preininger, A. M., Xiang, Y., and Exton, J. H. (2007) Methods Enzymol. 434 49-87 [DOI] [PubMed] [Google Scholar]
- 22.Cummings, R., Parinandi, N., Wang, L., Usatyuk, P., and Natarajan, V. (2002) Mol. Cell Biochem. 234-235 99-109 [PubMed] [Google Scholar]
- 23.Exton, J. H. (2002) Rev. Physiol. Biochem. Pharmacol. 144 1-94 [DOI] [PubMed] [Google Scholar]
- 24.Hammond, S. M., Jenco, J. M., Nakashima, S., Cadwallader, K., Gu, Q., Cook, S., Nozawa, Y., Prestwich, G. D., Frohman, M. A., and Morris, A. J. (1997) J. Biol. Chem. 272 3860-3868 [DOI] [PubMed] [Google Scholar]
- 25.Pettitt, T. R., McDermott, M., Saqib, K. M., Shimwell, N., and Wakelam, M. J. (2001) Biochem. J. 360 707-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agwu, D. E., McPhail, L. C., Sozzani, S., Bass, D. A., and McCall, C. E. (1991) J. Clin. Invest. 88 531-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waite, K. A., Wallin, R., Qualliotine-Mann, D., and McPhail, L. C. (1997) J. Biol. Chem. 272 15569-15578 [DOI] [PubMed] [Google Scholar]
- 28.Zhao, Y., Ehara, H., Akao, Y., Shamoto, M., Nakagawa, Y., Banno, Y., Deguchi, T., Ohishi, N., Yagi, K., and Nozawa, Y. (2000) Biochem. Biophys. Res. Commun. 278 140-143 [DOI] [PubMed] [Google Scholar]
- 29.Parinandi, N. L., Scribner, W. M., Vepa, S., Shi, S., and Natarajan, V. (1999) Antioxid. Redox. Signal 1 193-210 [DOI] [PubMed] [Google Scholar]
- 30.Parinandi, N. L., Roy, S., Shi, S., Cummings, R. J., Morris, A. J., Garcia, J. G., and Natarajan, V. (2001) Arch. Biochem. Biophys. 396 231-243 [DOI] [PubMed] [Google Scholar]
- 31.Natarajan, V., Scribner, W. M., and Vepa, S. (1996) Chem. Phys. Lipids 80 103-116 [DOI] [PubMed] [Google Scholar]
- 32.Pendyala, S., Gorshkova, I. A., Usatyuk, P., He, D., Pennathur, A., Lambeth, J. D., Thannickal, V. J., and Natarajan, V. (2008) Antioxid. Redox. Signal
- 33.Brueckl, C., Kaestle, S., Kerem, A., Habazettl, H., Krombach, F., Kuppe, H., and Kuebler, W. M. (2006) Am. J. Respir. Cell Mol. Biol. 34 453-463 [DOI] [PubMed] [Google Scholar]
- 34.Roy, S., Parinandi, N., Zeigelstein, R., Hu, Q., Pei, Y., Travers, J. B., and Natarajan, V. (2003) Antioxid. Redox. Signal 5 217-228 [DOI] [PubMed] [Google Scholar]
- 35.Jenkins, G. M., and Frohman, M. A. (2005) Cell Mol. Life Sci. 62 2305-2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang, L., Cummings, R., Usatyuk, P., Morris, A., Irani, K., and Natarajan, V. (2002) Biochem. J. 367 751-760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abo, A., Webb, M. R., Grogan, A., and Segal, A. W. (1994) Biochem. J. 298 585-591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ando, S., Kaibuchi, K., Sasaki, T., Hiraoka, K., Nishiyama, T., Mizuno, T., Asada, M., Nunoi, H., Matsuda, I., and Matsuura, Y. (1992) J. Biol. Chem. 267 25709-25713 [PubMed] [Google Scholar]
- 39.Gorzalczany, Y., Sigal, N., Itan, M., Lotan, O., and Pick, E. (2000) J. Biol. Chem. 275 40073-40081 [DOI] [PubMed] [Google Scholar]
- 40.Hordijk, P. L. (2006) Circ. Res. 98 453-462 [DOI] [PubMed] [Google Scholar]
- 41.Quinn, M. T., Evans, T., Loetterle, L. R., Jesaitis, A. J., and Bokoch, G. M. (1993) J. Biol. Chem. 268 20983-20987 [PubMed] [Google Scholar]
- 42.Brown, M. D., and Sacks, D. B. (2006) Trends Cell Biol. 16 242-249 [DOI] [PubMed] [Google Scholar]
- 43.Fukata, M., Nakagawa, M., Itoh, N., Kawajiri, A., Yamaga, M., Kuroda, S., and Kaibuchi, K. (2001) Mol. Cell Biol. 21 2165-2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuroda, S., Fukata, M., Kobayashi, K., Nakafuku, M., Nomura, N., Iwamatsu, A., and Kaibuchi, K. (1996) J. Biol. Chem. 271 23363-23367 [DOI] [PubMed] [Google Scholar]
- 45.Owen, D., Campbell, L. J., Littlefield, K., Evetts, K. A., Li, Z., Sacks, D. B., Lowe, P. N., and Mott, H. R. (2008) J. Biol. Chem. 283 1692-1704 [DOI] [PubMed] [Google Scholar]
- 46.Yamaoka-Tojo, M., Ushio-Fukai, M., Hilenski, L., Dikalov, S. I., Chen, Y. E., Tojo, T., Fukai, T., Fujimoto, M., Patrushev, N. A., Wang, N., Kontos, C. D., Bloom, G. S., and Alexander, R. W. (2004) Circ. Res. 95 276-283 [DOI] [PubMed] [Google Scholar]
- 47.Morris, A. J., Frohman, M. A., and Engebrecht, J. (1997) Anal. Biochem. 252 1-9 [DOI] [PubMed] [Google Scholar]
- 48.Al-Mehdi, A. B., Zhao, G., Dodia, C., Tozawa, K., Costa, K., Muzykantov, V., Ross, C., Blecha, F., Dinauer, M., and Fisher, A. B. (1998) Circ. Res. 83 730-737 [DOI] [PubMed] [Google Scholar]
- 49.Foster, D. (2007) Cancer Res. 67 1-4 [DOI] [PubMed] [Google Scholar]
- 50.Frey, R. S., Ushio-Fukai, M., and Malik, A. B. (2009) Antioxid. Redox Signal. 11 791-810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fontayne, A., Dang, P. M., Gougerot-Pocidalo, M. A., and El-Benna, J. (2002) Biochemistry 41 7743-7750 [DOI] [PubMed] [Google Scholar]
- 52.Bengis-Garber, C., and Gruener, N. (1995) Cell Signal 7 721-732 [DOI] [PubMed] [Google Scholar]
- 53.Touyz, R. M., Yao, G., and Schiffrin, E. L. (2003) Arterioscler. Thromb. Vasc. Biol. 23 981-987 [DOI] [PubMed] [Google Scholar]
- 54.Heyworth, P. G., Bohl, B. P., Bokoch, G. M., and Curnutte, J. T. (1994) J. Biol. Chem. 269 30749-30752 [PubMed] [Google Scholar]
- 55.Gorshkova, I., He, D., Berdyshev, E., Usatuyk, P., Burns, M., Kalari, S., Zhao, Y., Pendyala, S., Garcia, J. G., Pyne, N. J., Brindley, D. N., and Natarajan, V. (2008) J. Biol. Chem. 283 11794-11806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chae, Y. C., Kim, J. H., Kim, K. L., Kim, H. W., Lee, H. Y., Heo, W. D., Meyer, T., Suh, P. G., and Ryu, S. H. (2008) Mol. Biol. Cell 19 3111-3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ugolev, Y., Berdichevsky, Y., Weinbaum, C., and Pick, E. (2008) J. Biol. Chem. 283 22257-22271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faraudo, J., and Travesset, A. (2007) Biophys. J. 92 2806-2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kooijman, E. E., Tieleman, D. P., Testerink, C., Munnik, T., Rijkers, D. T., Burger, K. N., and de Kruijff, B. (2007) J. Biol. Chem. 282 11356-11364 [DOI] [PubMed] [Google Scholar]
- 60.Ohguchi, K., Nakashima, S., Tan, Z., Banno, Y., Dohi, S., and Nozawa, Y. (1997) J. Biol. Chem. 272 1990-1996 [DOI] [PubMed] [Google Scholar]
- 61.Powner, D. J., Hodgkin, M. N., and Wakelam, M. J. (2002) Mol. Biol. Cell 13 1252-1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamaoka-Tojo, M., Tojo, T., Kim, H. W., Hilenski, L., Patrushev, N. A., Zhang, L., Fukai, T., and Ushio-Fukai, M. (2006) Arterioscler. Thromb. Vasc. Biol. 26 1991-1997 [DOI] [PubMed] [Google Scholar]
- 63.Briggs, M. W., and Sacks, D. B. (2003) EMBO Rep. 4 571-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukata, M., Watanabe, T., Noritake, J., Nakagawa, M., Yamaga, M., Kuroda, S., Matsuura, Y., Iwamatsu, A., Perez, F., and Kaibuchi, K. (2002) Cell 109 873-885 [DOI] [PubMed] [Google Scholar]
- 65.Watanabe, T., Wang, S., Noritake, J., Sato, K., Fukata, M., Takefuji, M., Nakagawa, M., Izumi, N., Akiyama, T., and Kaibuchi, K. (2004) Dev. Cell 7 871-883 [DOI] [PubMed] [Google Scholar]
- 66.Mataraza, J. M., Li, Z., Jeong, H. W., Brown, M. D., and Sacks, D. B. (2007) Cell Signal 19 1857-1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jadeski, L., Mataraza, J. M., Jeong, H. W., Li, Z., and Sacks, D. B. (2008) J. Biol. Chem. 283 1008-1017 [DOI] [PubMed] [Google Scholar]
- 68.Wang, Y., Wang, M., Wang, F., Zhu, M., Ma, Y., Wang, X., and Wu, R. (2008) Int. J. Mol. Med. 22 79-87 [PubMed] [Google Scholar]
- 69.Wang, Y., Wang, A., Wang, F., Wang, M., Zhu, M., Ma, Y., and Wu, R. (2008) Exp. Mol. Pathol. 85 122-128 [DOI] [PubMed] [Google Scholar]
- 70.Touyz, R. M., Yao, G., Quinn, M. T., Pagano, P. J., and Schiffrin, E. L. (2005) Arterioscler. Thromb. Vasc. Biol. 25 512-518 [DOI] [PubMed] [Google Scholar]
- 71.Zhan, Y., He, D., Newburger, P. E., and Zhou, G. W. (2004) J. Cell Biochem. 92 795-809 [DOI] [PubMed] [Google Scholar]
- 72.Regier, D. S., Greene, D. G., Sergeant, S., Jesaitis, A. J., and McPhail, L. C. (2000) J. Biol. Chem. 275 28406-28412 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.