

Eukaryotic proliferating cell nuclear antigen (PCNA), an essential accessory factor in DNA replication and repair, is a ring-shaped homotrimer. A novel nontrimeric structure of E113G-mutant PCNA protein is reported, which shows that this protein forms alternate subunit interactions. It is concluded that the charged side chain of Glu113 promotes normal trimer formation by destabilizing these alternate subunit interactions.
Keywords: DNA replication, DNA repair, protein–protein interactions, protein–DNA interactions, translesion synthesis, proliferating cell nuclear antigen
Abstract
Eukaryotic proliferating cell nuclear antigen (PCNA) is an essential replication accessory factor that interacts with a variety of proteins involved in DNA replication and repair. Each monomer of PCNA has an N-terminal domain A and a C-terminal domain B. In the structure of the wild-type PCNA protein, domain A of one monomer interacts with domain B of a neighboring monomer to form a ring-shaped trimer. Glu113 is a conserved residue at the subunit interface in domain A. Two distinct X-ray crystal structures have been determined of a mutant form of PCNA with a substitution at this position (E113G) that has previously been studied because of its effect on translesion synthesis. The first structure was the expected ring-shaped trimer. The second structure was an unanticipated nontrimeric form of the protein. In this nontrimeric form, domain A of one PCNA monomer interacts with domain A of a neighboring monomer, while domain B of this monomer interacts with domain B of a different neighboring monomer. The B–B interface is stabilized by an antiparallel β-sheet and appears to be structurally similar to the A–B interface observed in the trimeric form of PCNA. The A–A interface, in contrast, is primarily stabilized by hydrophobic interactions. Because the E113G substitution is located on this hydrophobic surface, the A–A interface should be less favorable in the case of the wild-type protein. This suggests that the side chain of Glu113 promotes trimer formation by destabilizing these possible alternate subunit interactions.
1. Introduction
Proliferating cell nuclear antigen (PCNA) is an essential eukaryotic replication accessory factor that interacts with and promotes DNA binding by a variety of proteins involved in DNA replication and repair (reviewed in Naryzhny, 2008 ▶; Moldovan et al., 2007 ▶; Maga & Hübscher, 2003 ▶; Tsurimoto, 1999 ▶). Normally, PCNA exists as a stable ring-shaped homotrimer with a central cavity through which double-stranded DNA passes (Krishna et al., 1994 ▶). The PCNA ring is loaded and unloaded from the DNA in an ATP-dependent manner by replication factor C (RFC; reviewed in Mossi & Hübscher, 1998 ▶; Majka & Burgers, 2004 ▶; Indiani & O’Donnell, 2006 ▶). Once on the DNA, the PCNA trimer functions as a sliding clamp to enhance the processivity of DNA polymerases. In addition to serving as a processivity factor for DNA replication, PCNA also interacts with proteins functioning in a wide range of other processes including Okazaki fragment joining, base-excision repair, nucleotide-excision repair, mismatch repair, translesion DNA synthesis, cell-cycle control and chromatin remodeling (reviewed in Naryzhny, 2008 ▶; Moldovan et al., 2007 ▶; Maga & Hübscher, 2003 ▶; Tsurimoto, 1999 ▶).
The trimeric form of PCNA possesses pseudo-sixfold symmetry because each monomer is comprised of two independent domains with similar folds (Krishna et al., 1994 ▶). The N-terminal domain (domain A) is linked to the C-terminal domain (domain B) through a long flexible linker called the interdomain connector loop (IDCL). The IDCL is the binding site for many of PCNA’s interacting partners, which contain a conserved PCNA-interacting protein motif (PIP motif; reviewed in Hingorani & O’Donnell, 2000 ▶; Maga & Hübscher, 2003 ▶). When three PCNA monomers associate to form the trimeric ring-shaped structure, they arrange in a head-to-tail manner in which domain A of one monomer interacts with domain B of the neighboring monomer. This subunit interaction is stabilized via the backbone hydrogen bonds of an antiparallel β-sheet comprised of one β-strand from domain A of one monomer and a second β-strand from domain B of the other monomer (Krishna et al., 1994 ▶).
Recently, it has been shown that yeast cells expressing a mutant form of PCNA (a Glu113-to-Gly substitution) are unable to carry out translesion DNA synthesis (Northam et al., 2006 ▶). These cells have a slightly increased sensitivity to DNA-damaging agents, but otherwise have no noticeable growth defects. During our studies of the impact of this PCNA mutant protein on translesion DNA synthesis (discussed in Freudenthal et al., 2008 ▶), we performed X-ray crystallographic analyses to determine the structure of the E113G PCNA mutant protein. To our surprise, we obtained two distinct types of protein crystals and we determined the X-ray crystal structures of this mutant protein from both types. One structure was of the trimeric form of PCNA. The other structure was of a nontrimeric form of PCNA. The focus of the present report is the structure of this nontrimeric form of PCNA. The monomers in the nontrimeric form interact in two ways. The first interaction is a tail-to-tail contact in which domain B of one monomer interacts with domain B of its neighbor. The B–B interface of this nontrimeric form of PCNA is surprisingly similar structurally to the A–B interface of the trimeric form. The second interaction is a head-to-head contact in which domain A of one monomer interacts with domain A of a different neighboring monomer. Analysis of this mutant protein structure indicated that the A–A interface would be significantly less favorable in the presence of the wild-type Glu113 side chain. This implies that this conserved charged amino-acid residue plays an important role in promoting trimer formation by destabilizing these possible alternate subunit interactions.
2. Materials and methods
2.1. Protein expression and purification
Overexpression of wild-type and mutant PCNA proteins from the yeast Saccharomyces cerevisiae were carried out in Escherichia coli Rosetta-2 (DE3) cells harboring pET-11a vectors, into which were cloned the wild-type or mutant PCNA gene. The PCNA proteins were tagged with an N-terminal FLAG sequence for easy purification. Cells were grown in Overnight Express Instant TB Medium (Novagen) at 310 K for 12 h. Lysis was carried out in 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM PMSF and 1 mg ml−1 lysozyme with a Complete Mini Protease-Inhibitor Cocktail tablet (Roche). Cell debris was removed by ultracentrifugation and the resulting crude extract was loaded onto an Anti-FLAG M2 Affinity Gel (Sigma) column (15 ml bed volume) and purified as per the manufacturer’s instructions. The eluted protein was then further purified using a Superdex G-75 column equilibrated with 20 mM Tris–HCl pH 7.5, 1 mM DTT and 250 mM NaCl. Purified PCNA was stored at 193 K.
2.2. Crystallization of the E113G-mutant PCNA protein
Crystallization was performed manually using the hanging-drop method with 4 µl drops. An initial screen utilizing conditions similar to those which produced wild-type PCNA crystals (Krishna et al., 1994 ▶) was used to identify ideal crystallization conditions. Crystals of the trimeric form of the E113G-mutant PCNA protein that diffracted optimally were generated within 16 h by combining an equal volume of protein solution (20 mg ml−1) with reservoir solution containing 2.03 M ammonium sulfate and 0.1 M sodium citrate pH 5.8 at 291 K. Crystals of the nontrimeric form of the E113G-mutant PCNA protein that diffracted optimally were generated within 14–20 d by combining an equal volume of protein solution (20 mg ml−1) with reservoir solution containing 1.6 M ammonium sulfate and 0.1 M sodium citrate pH 5.8 at 291 K.
2.3. Data collection and structural determination
PCNA protein crystals were flash-frozen at 100 K after being presoaked in mother liquor containing 10%(v/v) glycerol. Data were collected from these crystals at 100 K on the 4.2.2 synchrotron beamline at the Advanced Light Source at the Ernest Orlando Lawrence Berkeley National Laboratory. Data were collected with a 150 mm crystal-to-detector distance. d*TREK was used to analyze and scale the data (Pflugrath, 1999 ▶). The cubic crystals of the trimeric form of the mutant PCNA protein diffracted to a resolution of 3.8 Å and the space group was determined to be P213, which is the same space group into which the wild-type PCNA protein crystallized (Krishna et al., 1994 ▶). The orthorhombic crystals of the nontrimeric form of the mutant PCNA protein diffracted to 2.5 Å resolution and the space group was determined to be C2221.
Molecular replacement was carried out using the structure of wild-type PCNA (PDB code 1plq) and Phaser (Read, 2001 ▶). Prior to refinement, simulated annealing to remove any structural bias was performed using PHENIX (Adams et al., 2002 ▶). Refinement and model building were carried out using PHENIX (Adams et al., 2002 ▶), REFMAC5 (Collaborative Computational Project, Number 4, 2004 ▶) and Coot (Emsley & Cowtan, 2004 ▶).
2.4. Size-exclusion chromatography
Wild-type and mutant PCNA proteins were diluted to a final volume of 200 µl with 1×TBS (150 mM NaCl, 50 mM Tris–HCl pH 7.5) and 3 µl acetone (used as a volume marker) and were loaded onto a 24 ml Superose 6 HR10/30 column (GE Biosciences) equilibrated at 277 K with 1× TBS. Samples were eluted at 0.5 ml min−1 and monitored by UV absorbance using an ÄKTA FPLC system (GE Biosciences). The elution volume of each protein was calculated using the UNICORN evaluation software (Amersham Biosciences). The Stokes radius of each protein was determined using the Porath correlation with standard proteins. The following standards were used to calibrate the column: thyroglobulin (670 kDa, 85 Å), ferritin (440 kDa, 61 Å), catalase (232 kDa, 52.2 Å), aldolase (158 kDa, 48.1 Å), bovine serum albumin (67 kDa, 35.5 Å), ovalbumin (43 kDa, 30.5 Å) and RNaseA (16.4 kDa, 14 Å).
3. Results
3.1. Overview of the structures of two forms of the E113G-mutant PCNA protein
We obtained two distinct types of crystals of the E113G-mutant PCNA protein. Crystals of the first type, which formed overnight, were cubic and diffracted to a resolution of 3.8 Å (Table 1 ▶). These crystals contained one PCNA monomer per asymmetric unit and had the same space group and unit-cell parameters as the crystals used to determine the structure of wild-type PCNA (Krishna et al., 1994 ▶). Similar to what was observed for the wild-type PCNA protein, each monomer contained an N-terminal domain A (residues 1–118) and a C-terminal domain B (residues 135–240) linked by a long flexible interdomain connector loop (IDCL; residues 119–134). The biologically relevant trimeric structure was formed by generating neighboring monomers along the threefold axis of the cubic symmetry as was performed for the wild-type protein. The three monomers form head-to-tail contacts, with domain A of each monomer interacting with domain B of its neighbor (Fig. 1 ▶ a). The only significant structural difference observed between the wild-type protein and the E113G-mutant protein is within loop J (residues 105–110), an extended loop in domain A near the subunit interface. As previously described in detail elsewhere, the conformation of loop J is likely to be responsible for the inability of this mutant protein to carry out translesion DNA synthesis (Freudenthal et al., 2008 ▶). In the present report, we focus primarily on the structure of the E113G-mutant protein determined with the second type of protein crystals.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Trimeric form | Nontrimeric form | |
|---|---|---|
| Data-collection statistics | ||
| Resolution () | 29.613.80 (3.943.80) | 28.912.50 (2.592.50) |
| Wavelength () | 0.97 | 0.97 |
| Space group | P213 | C2221 |
| Unit-cell parameters () | a = 122.09, b = 122.09, c = 122.09 | a = 74.59, b = 147.51, c = 81.44 |
| Completeness (%) | 100 (100) | 98.1 (96.8) |
| Redundancy | 10.56 (9.84) | 4.75 (3.79) |
| I/(I) | 17.9 (6.8) | 11.4 (3.2) |
| R merge † (%) | 7.2 (29.4) | 9.0 (36.5) |
| Refinement statistics | ||
| Resolution range () | 29.63.80 | 28.92.50 |
| R ‡ (%) | 0.27 | 0.23 |
| R free § (%) | 0.1 | 0.27 |
| R.m.s.d. bonds () | 0.011 | 0.017 |
| R.m.s.d. angles () | 1.5 | 1.7 |
| No. of water molecules | 0 | 0 |
| No. of protein atoms | 1981 [254 amino acids] | 1967 [253 amino acids] |
| Ramachandran analysis | ||
| Most favored (%) | 82 | 93 |
| Allowed (%) | 18 | 7 |
| PDB code | 3gpm | 3gpn |
R
merge = 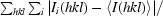
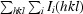 , where I
i(hkl) is the ith measurement of reflection hkl and I(hkl) is a weighted mean of all measurements of hkl.
, where I
i(hkl) is the ith measurement of reflection hkl and I(hkl) is a weighted mean of all measurements of hkl.
R = 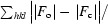
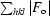 , where F
o and F
c are the observed and calculated structure factors, respectively.
, where F
o and F
c are the observed and calculated structure factors, respectively.
R free is defined in Brnger (1992 ▶).
Figure 1.

Structures of two forms of the E113G-mutant PCNA protein. (a) The trimeric form of the mutant protein with monomers shown in orange, purple and light blue. Domains A and B, the interdomain connecting loop (IDCL), loop J and the E113G substitution are indicated. (b) The nontrimeric form of the mutant protein with monomers shown in green, red, blue and yellow. Domains A and B are indicated.
Crystals of the second type, which formed over a period of two weeks, were orthorhombic and diffracted to a resolution of 2.5 Å (Table 1 ▶). These crystals also contained one monomer per asymmetric unit and generation of the symmetry-related neighbors showed that they do not pack to form the usual head-to-tail ring-shaped trimer. Instead, the monomers are arranged in a nontrimeric structure in which domain A of one monomer interacts with domain A of a neighboring monomer in a head-to-head contact and in which domain B of the original monomer interacts with domain B of a different neighboring monomer in a tail-to-tail contact (Fig. 1 ▶ b). To our knowledge, this is the first structure to be determined of a nontrimeric form of eukaryotic PCNA.
Comparing the structures of the E113G-mutant PCNA monomers in the trimeric and nontrimeric forms showed that there is flexibility between the domains within individual monomers as well as between neighboring monomers. For example, in the trimeric form of PCNA the angle between domain A and domain B within the same monomer is 125° (Fig. 2 ▶ a). In the nontrimeric form of PCNA the angle between domains within the same monomer is reduced slightly to 114° (Fig. 2 ▶ b). The change in angle between the two domains is likely to be possible because of the inherent flexibility of the interdomain connector loop. The angles between the domains of neighboring monomers are even more variable. In the trimeric form of PCNA the angle between domain A and domain B of the neighboring monomers (i.e. the A–B interface) is 115° (Fig. 2 ▶ a). In the nontrimeric form of PCNA the angle between the two A domains on neighboring monomers (i.e. the A–A interface) is 180° (Fig. 2 ▶ b). The angle between the two B domains on neighboring monomers (i.e. the B–B interface) is 100°. As discussed below, the A–B interface of the trimeric form of PCNA and the B–B interface of the nontrimeric form of PCNA are structurally similar. At a global level, the principal difference between these two interfaces is that the angle between the interacting domains is smaller in the nontrimeric B–B interface (100°) than in the trimeric A–B interface (115°).
Figure 2.

Subunit interfaces of the trimeric and nontrimeric forms of the E113G-mutant PCNA protein. (a) The A–B interface of the trimeric form of the mutant protein is shown with a schematic indicating the values of the angles within each monomer and the angles between monomers. (b) The B–B interface (top) and the A–A interface (bottom) of the nontrimeric form of the mutant protein is shown with a schematic indicating the values of the angles within each monomer and the angles between monomers.
3.2. Subunit interactions of the nontrimeric form of the E113G-mutant PCNA protein
In the trimeric form of PCNA, the A–B subunit interface consists of two antiparallel β-strands: β-I 1 (residues 109–117) in domain A of one monomer and β-D 2 (residues 175–183) in domain B of the other monomer (Fig. 3b ▶). The interactions between the two monomers are stabilized by seven hydrogen bonds between the backbone carbonyl and amide groups of these two β-strands and this interface buries a total of 1310 Å2 of solvent-accessible surface area. In the nontrimeric form of PCNA, the B–B interface is surprisingly similar in structure to the trimeric A–B interface. The B–B interface also consists of two antiparallel β-strands: the β-D 2 strands from the two interacting B domains (Fig. 3a ▶). In this case, the interactions between the monomers are stabilized by eight hydrogen bonds between the backbones of these two β-strands and a total of 1580 Å2 of solvent-accessible surface area is buried. It should be noted that the E113G substitution does not directly influence the formation of the B–B interface because this amino-acid substitution is in domain A on the opposite end of the monomers, 50 Å away from the B–B interface. Thus, it seems likely that the B–B interface observed with this mutant PCNA protein would be equally favorable in the wild-type PCNA protein.
Figure 3.

The B–B interface of the nontrimeric form of the E113G-mutant PCNA protein. (a) The B–B interface of the nontrimeric form of the mutant protein is stabilized by the β-D 2 strands (residue 175–183) from domain B of each monomer forming an antiparallel β-sheet. The positions of the eight backbone hydrogen bonds are shown. (b) The A–B interface of the trimeric form of the mutant protein is stabilized by the β-D 2 strand (residues 175–183) from domain B of one monomer forming an antiparallel β-sheet with the β-I 1 strand (residues 109–117) from domain A of the other monomer. The positions of the seven backbone hydrogen bonds are shown.
The A–A interface of the nontrimeric form of PCNA, in contrast, is dramatically different from the A–B interface of the trimeric form. The region of each monomer near the A–A interface is comprised of a β-sheet containing five β-strands (Fig. 4 ▶ a): β-A 1 (residues 2–6), β-E 1 (residues 59–61), β-G 1 (residues 87–92), β-H 1 (residues 98–104) and β-I 1 (residues 109–117). The side of this β-sheet facing the neighboring monomer is hydrophobic and the hydrophobic contacts between Ile91 (in β-G 1) and Ile100 (in β-H 1) stabilize the subunit interaction at the A–A interface (Fig. 4 ▶ b). This interface buries 1650 Å2 of solvent-accessible surface area. The E113G substitution plays an important role in favoring the formation of the A–A interface. As shown in Fig. 4 ▶(c), if the wild-type Glu113 (in β-I 1) residues were present their negatively charged side chains would project toward and be likely to interfere with the hydrophobic contacts made by the four Ile91 and Ile100 residues. Therefore, the A–A interface observed in this mutant PCNA protein is in all likelihood less stable in the case of the wild-type protein.
Figure 4.

The A–A interface of the nontrimeric form of the E113G mutant PCNA protein. (a) The A–A interface of the nontrimeric form of the mutant protein is stabilized by hydrophobic interactions between the surface of each monomer comprised of five β-strands: β-A 1, β-E 1, β-G 1, β-H 1 and β-I 1. These surfaces are highlighted by the dashed square. (b) Close-up view of the A–A interface of the E113G mutant protein. The side chains of the hydrophobic Ile91 and Ile100 residues are shown in stick format (left) and in space-filling format (right). (c) Model of the A–A interface of the wild-type protein. This panel is identical to (b), except that the side chain of Glu113 has been modeled and shown in yellow.
3.3. Comparison of the domains of the trimeric and nontrimeric forms of the E113G-mutant PCNA protein
To determine whether the novel A–A and B–B interfaces of the nontrimeric form of the mutant PCNA protein affected the structure of the individual domains of each monomer, we determined the root-mean-square deviation between the Cα atoms of the monomers of the trimeric form of the wild-type protein and of the nontrimeric form of the mutant protein. Surprisingly, there are very few differences between the structures of the protein backbones of the individual domains of PCNA in the trimeric and nontrimeric forms (Fig. 5 ▶). In the case of domain B, which made structurally similar contacts in both the trimeric form of PCNA (the A–B interface) and the nontrimeric form of PCNA (the B–B interface), the backbone structures of the domain are very similar, with an r.m.s.d. of 0.3 Å over 105 Cα atoms. In the case of domain A, which made dramatically different contacts in the trimer form (the A–B interface) and the nontrimeric form (the A–A interface), the backbone structures are slightly less similar, with an r.m.s.d. of 0.9 Å over 118 Cα atoms. Overall, these results indicate that the structures of the individual domains within the PCNA monomers are not significantly impacted by the oligomeric form of PCNA.
Figure 5.

Superimposition of the protein backbone of a monomer of the trimeric form of the wild-type PCNA protein and a monomer of the nontrimeric form of the E113G-mutant PCNA protein. Domains A and B of the monomers are separated by a dashed line and the r.m.s.d. values were independently determined for each domain.
3.4. Stability of the trimeric form of the E113G-mutant PCNA protein
Because we observed a nontrimeric form of the E113G-mutant PCNA protein, we carried out size-exclusion chromatography at various concentrations of the wild-type and mutant PCNA proteins to examine the stability of the trimeric form of this mutant protein in solution. Fig. 6 ▶ shows the Stokes radius plotted as a function of PCNA monomer concentration. The wild-type PCNA protein was in the trimeric form at all concentrations used, with a Stokes radius equal to 45 Å, which corresponds closely to its actual radius (46 Å). A comparison with molecular-weight standards provided a predicted molecular weight of 109 kDa, which is in reasonable agreement with the actual molecular weight of the PCNA trimer (87 kDa) given the unusually flat shape of the PCNA ring. In the case of the E113G-mutant PCNA protein, the trimeric form predominated at high monomer concentration (>5 µM). At lower monomer concentrations (<0.5 µM), the mutant protein appeared to exist predominantly as a dimer. The Stokes radius of the dimer was 33 Å; this corresponds to a predicted molecular weight of 54 kDa, which is in close agreement with the actual molecular weight of the PCNA dimer (58 kDa). Thus, the trimeric form of the E113G-mutant PCNA protein is significantly less stable than the trimeric form of the wild-type protein.
Figure 6.

Stability of the trimeric forms of the wild-type and E113G-mutant PCNA proteins. Size-exclusion chromatography was carried out and the Stokes radius was determined for various concentrations of the wild-type PCNA (black circles) and mutant PCNA (white triangles). Estimated molecular weights derived from protein standards are provided.
4. Discussion
In the structure of the trimeric form of PCNA only one type of subunit interface is observed. This is the A–B interface, which is stabilized by the formation of an antiparallel β-sheet between two β-strands, one from domain A of one monomer and one from domain B of a neighboring monomer (Krishna et al., 1994 ▶). Here, we report the structure of a nontrimeric form of the E113G-mutant PCNA protein that reveals two alternate subunit interfaces. The first is the B–B interface, in which an antiparallel β-sheet is formed between two β-strands: one from domain B of one monomer and one from domain B of a neighboring monomer. Because the E113G substitutions are at the other end of the PCNA monomers from this interface, it is likely that the B–B interface observed in this mutant protein is at least as favorable in the wild-type protein. The second is the A–A interface, which is stabilized primarily through hydrophobic contacts between domain A of one monomer and domain A of a neighboring monomer. Because in the wild-type protein the side chain of Glu113 projects toward and is likely to interfere with these hydrophobic interactions (see Fig. 4 ▶ c), the A–A interface observed here is in all likelihood significantly less favorable in the wild-type protein.
There are compelling reasons why the A–A and B–B interfaces observed in the structure of the nontrimeric form of PCNA are not merely the result of crystal packing but are instead actual contacts that are likely to occur in solution under some conditions. Firstly, the B–B interface of the nontrimeric form and the A–B interface of the trimeric form are structurally similar. In fact, the B–B interface may be slightly more stable than the A–B interface. The B–B interface is stabilized by eight backbone hydrogen bonds, while the normal A–B interface is stabilized by seven. In addition, more surface area (1580 Å2) is buried at the B–B interface than at the A–B interface (1310 Å2). This is important because large buried surface areas are characteristics of actual subunit interfaces as opposed to crystal contacts (Dasgupta et al., 1997 ▶). Similarly, the A–A interface buries 1650 Å2, which also implies that it is not a result of crystal packing but instead can occur in solution, at least in the case of this mutant protein. We should point out that in addition to the A–A and B–B contacts there is another contact between two monomers in the crystal of the nontrimeric form of PCNA and this contact is along the IDCL of each monomer. We believe that this contact is indeed the result of crystal packing because it has a lower buried surface area (1060 Å2) and is very similar to the crystal contact observed in the trimeric structure of wild-type PCNA.
The stable trimeric form of PCNA is generally believed to assemble in two steps. Firstly, two PCNA monomers come together to form the head-to-tail dimer, with domain A of one monomer contacting domain B of the other monomer (see the structure in Fig. 2 ▶ a). Next, a third monomer comes together with the dimer, with domain A and domain B of the monomer contacting the available domain B and domain A of the dimer, respectively. Our finding of possible alternative subunit interactions in PCNA complicates this scenario because our results suggest that three distinct types of PCNA dimers can be formed. In addition to the standard head-to-tail dimer described above, dimers can be formed when PCNA monomers come together in a head-to-head manner, with domain A of one monomer contacting domain A of another (see the bottom structure in Fig. 2 ▶ b), or a tail-to-tail manner, with domain B of one monomer contacting domain B of another (see the top structure in Fig. 2 ▶ b). In the case of the E113G-mutant PCNA protein, all three types of dimers are likely to co-exist at low and intermediate protein concentrations (<1 µM). In the case of the wild-type PCNA protein, however, the presence of the charged Glu113 side chain should destabilize any head-to-head A–A interfaces and greatly favor the formation of the standard PCNA trimer even at lower protein concentrations.
While to our knowledge this is the first structure that has been determined of a nontrimeric form of eukaryotic PCNA, an X-ray crystal structure has been determined of a nontrimeric form of the unrelated prokaryotic PCNA from the archeaon Pyrococcus furiosus. Although lacking homology in amino-acid sequence to eukaryotic PCNA, prokaryotic PCNA is also normally a trimeric ring-shaped protein that is generally similar in overall structure to eukaryotic PCNA (Matsumiya et al., 2001 ▶). The A–B interface of the prokaryotic PCNA trimer, however, differs from that of the eukaryotic PCNA trimer in that this interface is stabilized by several electrostatic interactions between charged side chains of residues from different monomers. X-ray crystal structures have been determined for two mutant forms of this prokaryotic PCNA with substitutions of these charged residues in domain B at the subunit interface (Matsumiya et al., 2003 ▶). The oligomeric forms of both of these mutant proteins are dimers, which are held together by a mutant B–B interface that is roughly analogous to that we describe here for eukaryotic PCNA. Incidentally, no A–A interfaces have been observed for prokaryotic PCNA. The primary conclusion of the study of the nontrimeric form of the prokaryotic protein was that the charged residues in question promote trimer formation by directly stabilizing the trimeric structure. This is quite different from our observations regarding the role of Glu113 in eukaryotic PCNA. Here, we conclude that the eukaryotic PCNA trimer formation is favored by Glu113 owing to destabilization of the A–A interface.
Finally, given that the tail-to-tail B–B dimer could exist at low protein concentration in the case of the wild-type PCNA protein, it is tempting to speculate about a possible biological role of this species. Clearly, the trimeric form of PCNA is the most important oligomeric form of this protein as it participates in DNA replication and many DNA-repair processes. However, at estimated cellular concentrations of PCNA a significant population of stable wild-type PCNA dimers has been observed by several in vitro experimental techniques (Zhang et al., 1995 ▶). Given that PCNA plays a role in so many other biological processes, including cell-cycle control and survival, chromatin assembly and remodeling and regulation of transcription (reviewed in Naryzhny, 2008 ▶; Moldovan et al., 2007 ▶; Maga & Hübscher, 2003 ▶; Tsurimoto, 1999 ▶), and given that the oligomeric state of the PCNA molecules participating in these processes has not been determined, it is possible that PCNA functions in some biological contexts as a stable dimer. The alternate subunit interactions reported here suggest that a stable B–B dimer may in fact be a biologically important molecular species. The formation of a stable B–B dimer would allow novel protein–protein interactions between the large hydrophobic surface of domain A and potential protein partners.
Supplementary Material
PDB reference: E113G-mutant PCNA, trimeric form 3gpm, r3gpmsf
PDB reference: nontrimeric form, 3gpn, r3gpnsf
Acknowledgments
The project described here was supported by Award No. R01GM081433 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. We thank Lynne Dieckman, Christine Kondratick, John Pryor, Marc Wold and Manju Hingorani for valuable discussions.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dasgupta, S., Iyer, G. H., Bryant, S. H., Lawrence, C. E. & Bell, J. A. (1997). Proteins, 28, 494–514. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Freudenthal, B. D., Ramaswamy, S., Hingorani, M. M. & Washington, M. T. (2008). Biochemistry, 47, 13354–13361. [DOI] [PMC free article] [PubMed]
- Hingorani, M. M. & O’Donnell, M. (2000). Curr. Biol. 10, R25–R29. [DOI] [PubMed]
- Indiani, C. & O’Donnell, M. (2006). Nature Rev. Mol. Cell Biol. 7, 751–761. [DOI] [PubMed]
- Krishna, T. S., Kong, X. P., Gary, S., Burgers, P. M. & Kuriyan, J. (1994). Cell, 79, 1233–1243. [DOI] [PubMed]
- Maga, G. & Hübscher, U. (2003). J. Cell Sci. 116, 3051–3060. [DOI] [PubMed]
- Majka, J. & Burgers, P. M. (2004). Prog. Nucleic Acid Res. Mol. Biol. 78, 227–260. [DOI] [PubMed]
- Matsumiya, S., Ishino, S., Ishino, Y. & Morikawa, K. (2003). Protein Sci. 12, 823–831. [DOI] [PMC free article] [PubMed]
- Matsumiya, S., Ishino, Y. & Morikawa, K. (2001). Protein Sci. 10, 17–23. [DOI] [PMC free article] [PubMed]
- Moldovan, G.-L., Pfander, B. & Jentsch, S. (2007). Cell, 129, 665–679. [DOI] [PubMed]
- Mossi, R. & Hübscher, U. (1998). Eur. J. Biochem. 254, 209–216. [PubMed]
- Naryzhny, S. N. (2008). Cell. Mol. Life Sci. 65, 3789–3808. [DOI] [PMC free article] [PubMed]
- Northam, M. R., Garg, P., Baitin, D. M., Burgers, P. M. J. & Shcherbakova, P. V. (2006). EMBO J. 25, 4316–4325. [DOI] [PMC free article] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Tsurimoto, T. (1999). Front. Biosci. 4, d849–d858. [DOI] [PubMed]
- Zhang, P., Zhang, S.-J., Zhang, Z., Woessner, J. F. & Lee, M. Y. W. T. (1995). Biochemistry, 34, 10703–10712. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: E113G-mutant PCNA, trimeric form 3gpm, r3gpmsf
PDB reference: nontrimeric form, 3gpn, r3gpnsf