

Abstract
Metabolomics provides a readout of the state of metabolism in cells or tissue and their responses to external perturbations. For this reason, the approach has great potential in clinical diagnostics. For more than two decades,, we have been using stable isotope tracer approaches to probe cellular metabolism in greater detail. The ability to enrich common compounds with rare isotopes such as carbon (13C) and nitrogen (15N) is the only practical means by which metabolic pathways can be traced, which entails following the fate of individual atoms from the source molecule to products via metabolic transformation. Changes in regulation of pathways are therefore captured by this approach, which leads to deeper understanding of the fundamental biochemistry of cells. Using lessons learned from pathways tracing in cells and organs, we have been applying this methodology to human cancer patients in a clinical setting. Here we review the methodologies and approaches to stable isotope tracing in cells, animal models and in humans subjects.
Keywords: stable isotopes, isotopomer pathway analysis, mass spectrometry, NMR
Introduction
Metabolomics is a rapidly advancing field that complements genomics and proteomics. It is concerned with the identification and quantification of the metabolome and its response to perturbations. As differentiated tissues have their own characteristic metabolic profiles, which may be impacted differently depending on the origin of the disease, the metabolome of tissues provides a direct readout of the organ or cellular responses to external perturbation. Thus, normal cells in tissue respond to environmental factors including nutrients and xenobiotics according to the prior physiological state of the organism (history) and specific genetic variations. Further, specific aspects of the metabolome may vary in response to disease progression, depending on whether it is of genetic origin (e.g. cancers) or via infection by for example bacteria or viruses. Thus specific target organs contribute different variations to the basal metabolome of biofluids such as blood, urine, saliva, CSF, sputum etc. Many metabolic biomarker studies have been initiated to examine the variability of the metabolome of such common and readily obtained biofluids and how they differ for specific disease states (Lindon et al. 1999; Lindon et al. 2004; Griffin 2006; Weljie et al. 2006) (Fan et al. 2004; Ippolito et al. 2005; Beger et al. 2006; Griffin et al. 2007; Kind et al. 2007; Zimmermann et al. 2007). These approaches rely on detecting changes in concentrations of metabolites, within a background of hundreds or thousands of species.
As the metabolic rates in individuals reflect absorption, metabolism and excretion of xenobiotic compounds such as pharmaceutical drugs, the metabolomic signatures reflect the functional biochemistry of the individual, specific changes in the metabolome can be used to identify the likely response of an individual to specific drugs, i.e. distinguish between responders and non-responders prior to the start of therapeutic interventions. The inherent variability, and lack of mechanistic information, makes statistical approaches essential. Certainly, such statistical approaches can be extremely valuable for establishing reliable diagnostic tools or to determine whether a particular individual is likely to respond to a drug or not (Kaddurah-Daouk et al. 2008).
The lack of specificity, (i.e. the origin of the altered metabolic profile) makes mechanistic interpretation of the observations effectively impossible, yet such information is essential for understanding the progress of a disease, and for designing new, more specific therapeutics. In order to obtain mechanistic information about the metabolome in the appropriate tissue, it is necessary to turn to metabolic tracer techniques, in cells animal models, and ultimately human patients, including analysis of the metabolome of actual tissue.
At the J.G. Brown Cancer Center and the recently established Center for Regulatory Environmental Analytical Metabolomics (‘CREAM’) we have been developing stable isotope tracer methods in conjunction with NMR and a variety of mass spectrometry methods. We have been applying these methods to human cancers both in vitro and in vivo in order to understand the metabolic differences between tumor cells at different stages of transformation compared with the cell type of origin (Fan et al. 2004; Fan et al. 2005; Fan et al. 2006; Telang et al. 2006; Fan et al. 2007; Telang 2007; Clem et al. 2008; Fan et al. 2008; Lane et al. 2008). To do this requires a multi stage approach that enables translation of the information obtained from model systems to clinically relevant procedures.
The metabolism of differentiated cells reflects their biochemical specialization; different cell types display a characteristic metabolic phenotype. Cancer cells show an altered metabolic phenotype compared with the untransformed cell. Perhaps the best-known example is the increased aerobic glycolysis in cancer cells both in tissue and cell culture, known as the Warburg effect (Warburg 1956; Altenberg et al. 2004; Robey et al. 2005). Superchargers of glycolysis in cancer as therapeutic targets are described in the chapters by Yalcin et al. and Dailey et al. (this volume). The enhanced glucose consumption of cancer cells is exploited in FDG PET imaging (Dhital et al. 2000; Gatenby et al. 2007; Cermik et al. 2008).
The hallmark of cancer cells is uncontrolled proliferation. In order to divide, a cancer cell must double it contents, including the macromolecules that collectively account for most of the biomass. As macromolecular biosynthesis is thermodynamically unfavorable, this requires metabolic energy over and above that needed for normal cellular maintenance and repair processes. Furthermore, although many molecular precursors such as the essential amino acids can only be obtained from the diet, others must be synthesized de novo. Hence a proliferating cell must up-regulate uptake/utilization of building blocks such as glucose, energy metabolism, biosynthesis, and specific anabolic pathways. Only the first is addressed by the Warburg effect, while the rest comprise a rich metabolic signature that holds the promise of novel biomarkers of cancer progression, for diagnosis, and also in response to particular therapies.
Monocultured cancer and normal cells represent the highest level of experimental control in the laboratory, in which essentially all variables can be altered and controlled at will, such as specific nutrient supply, oxygenation, nature of the culture conditions, gene expression levels, and so forth, The information that can be derived from systematic studies is very detailed, and can be used to map out the variation in pathways in response to a wide range of environmental perturbation, that encompass any conditions that might be present within a tumor. Thus, the potential behavior of a given cell type can be established. However, as the biochemistry of a cell is affected by its local environment (cf. tumor microenvironment) it is not obvious a priori what the actual response of a cell type would be in situ. To translate the detailed response potential into a more realistic situation, the next level of complexity must be addressed, such as a tumor xenograft in a suitable host organism (usually an immune compromised mouse strain). Here it is important to obtained paired samples from the same subject. For example, a tumor xenograft can be excised at a given time point or size, along with nearby tissue, and in the case of animals, other organs altogether. Such paired samples decrease the intrinsic variability as they have been exposed to the same diet, as well as being in an identical genetic background. This paired-sample approach can be extremely important at the next level of investigations, namely human subjects. In fact, we have found in a study with lung cancer patients that the comparison of paired samples the statistical power can be very high with small numbers of subjects when stable isotope tracing is used (Fan, Lane, Bousamra, Higashi and Miller, unpublished).
A Technologies for stable isotope metabolomics
Clearly, the appropriate biological model must be coupled with stable isotope investigations of metabolomes. The first step is to quantify metabolites, for which there are several methods in use. A long-standing technique is to use purified enzymes to convert the substrates to delectable products quantitatively. This by definition is a targeted approach, and is generally limited to a small number of metabolites. More generally, a variety of chromatographic separations can be used in conjunction with a suitable detector either on line or off line. This is commonly done by HPLC with absorbance detection of a class of compounds, or using mass spectrometry for detection, which has a much wider range of applicability. The highest resolution is probably achieved by GC-MS, which requires an additional derivatization step, but is often capable of resolving a large number of compounds in a complex mixture such as a biofluid or cell extract.
However, for the detection of isotopomers, the common metabolite methods such as enzymes assays and analysis by absorption, electrochemistry or other tools are unsuitable because they are not sensitive to the presence of heavy isotopes. The only two analytical platforms that are capable of stable isotopomer analysis and also provide the highest information content are mass spectrometry and NMR (Fan et al. 1986) (Fan et al. 2006; Fan et al. 2008; Lane et al. 2008). The former in most implementations requires a separation by chromatography prior to the mass determination, commonly GC or LC. However, the advent of the FT-ICR MS is making it feasible to measure individual metabolites by direct infusion, making use of the ultra high mass resolution of the ICR (Brown et al. 2005).
NMR and FT-ICR MS are true simultaneous devices, in that the mixtures of metabolites in a fluid or an extract are simultaneously detected, and the resolution arises from the dispersion of the chemical shifts by frequency in one or more dimensions. In NMR isotopes are distinguished by magnetic properties. Thus 13C is NMR active whereas the more abundant isotope 12C is inactive. Thus a metabolite that contains a 13C atom is readily distinguished from one that does not contain 13C. Both forms of the molecule can be simultaneously detected and quantified, making it possible to determine the particular atom within a molecule that has received 13C from a source such as 13C-glucose. In MS, the differentiation is via the difference in mass (approx. 1 amu), so that 13C or 15N labeled metabolites are heavier than the unlabeled compound. The number of labeled atoms in a molecule can therefore be counted straightforwardly.
In our FT-ICR-MS configuration a mixture of metabolites from a crude extract or biofluid is infused directly via a nanoelectrospray into a linear ion trap that is in tandem with the FT-ICR mass spectrometer. The mixture requires no derivatization or chromatographic separation. The individual ions are temporarily stored in the ion trap, where they can be selectively retained for injection into the FT-ICR MS for accurate mass determination, and/or fragmented to give product ions. For relatively small metabolites, the exact mass (to < 1 ppm mass error) is often sufficient to identify the compounds (and isomers). Using the mass-matching software PREMISE (Higashi et al. 2008) the class of compounds, and often the identity of metabolites is identified by comparing the experiment mass with those in a list of thousands of calculated theoretical masses based on chemical formulae.
Phospholipids, are an outstanding example of key metabolites that have been difficult to analyze in detail by any means. However, the FT-ICR-MS can discriminate readily between phosphatidyl moieties of cholines, serines, ethanolamines, inositols, sphingomyelins, etc., including differences in the acyl chain moieties; the FT-ICR-MS can analyze for hundreds of intact phospholipids simultaneously. Isomers, e.g.. which chain is at the 1 or 2 position on a glycerol headgroup cannot be distinguished by mass alone. However, mild fragmentation and MS2 or MS3 can often distinguish between such isomers.
Furthermore, each of these hundreds of phospholipids, in turn, can have many dozens of 13C isotopomeric forms introduced from the experiment, resulting in (typically) about 2,000 phospholipid species observed by the FT-ICR-MS in a single analysis. The number of 13Cs in the molecule – the “mass isotopomers” – is readily determined simultaneously with the non-labeled forms. The utility of this high information throughput is, for example, quantifying the metabolic transformation of 13C- labeled glucose entering either the fatty acyl chains or the glycerol moieties of the various phopholipids. As the relative intensity of the MS peaks is very reliable (our current estimate is that the precision is better than 0.2%). Accurate mass isotopomer distributions can be precisely quantified, and with appropriate modeling and knowledge of metabolic pathways, a detailed picture of the flux into lipid biosynthesis can be obtained, and how this responds to external perturbations such as diet, hypoxia, drug treatment etc.
Isotopomer Analysis and Pathway Tracing
Although metabolite concentrations in biofluids can be diagnostic of particular disease states, as well as providing information about metabolism and excretion of drugs, the mechanistic information this approach supplies is quite limited, not least because the tissue of origin cannot be uniquely assigned. A simple (hypothetical) example shows the additional information that can be obtained using isotope tracers. Pyruvate is a so-called hub metabolite, that can be made from several precursors, and is also the precursor of many other metabolites. As shown in Figure 1, in addition to being the product of glycolysis, pyruvate can also be generated by amino acid oxidation, such as glutaminolysis (Petch et al. 1994; Portais et al. 1996; Mazurek et al. 2000; DeBerardinis et al. 2007), which is also an important anaplerotic reaction. Changes in pyruvate concentration could therefore be a consequence of changes in any or all of these pathways, the biochemical significance of which depends on the cell and the conditions. The concentration of pyruvate is generally low, and therefore difficult to measure. Furthermore, it is likely to be strongly regulated (homeostasis). and this its concentration may not vary significantly.
Figure 1. Simple metabolic scheme.

Carbon flow from [U-13C]-glucose through glycolysis, Krebs cycle, PPP, pyruvate carboxylation, malate/Asp shuttle, and synthesis of GSH and pyrimidine nucleotides. Solid oval represents the plasma membrane and the dashed oval represents the mitochondrial space. Double-headed arrows: exchange or reversible processes.
Glc = glucose, G6P = glucose-6-phosphate, Rib = nucleotide ribose, DHAP, GAP = dihydroxyacetone phosphate and glyceraldehyde-3-phosphate, PEP = phosphoenolpyruvate, Pyr=pyruvate, Lac = lactate, Cit=citrate, 2=OG = 2-oxoglutarate, Mal=malate, OAA = oxalacetate, U = uracil base.
Critical enzymes are shown in cyan. HK= hexokinase (entry to glycolysis), G6PDH = glucose-6-phosphate dehydrogenase (entry to the oxidative branch of the pentose phosphate pathway), TK, TA transketolase and transaldolase (non –oxidative pentose phosphate pathway), PK = pyruvate kinase, PDH = pyruvate dehydrogenase, PC = pyruvate carboxylase, AAT = aspartate amino transferase, MDH= malate dehydrogenase, ME = malic enzyme. Glutaminolysis is the pathway from Gln to pyruvate via ME, leading to unlabeled malate, Asp, Ala and lactate.
On the other hand, lactate, a pyruvate reporter (cf. Figure 1) is easy to measure, and is often secreted at a very high rate from cancer cells, accounting for a substantial fraction of the glucose consumed by the cells (DeBerardinis et al. 2007) and see below. However, if only the total concentration of lactate is measured it is not possible to determine how much derived from glucose (i.e. by glycolysis) or by amino acid oxidation. However, if the glucose supplied to the cells is labeled with 13C, then not only can the consumption of the 13C glucose be measured, but also the fraction of the lactate produced that contains 13C using NMR or MS as described above, as shown in Figure 2. Figure 2A shows a 1D NMR spectrum of the medium 24 h after addition of uniformly labeled 13C glucose to A549 cells. Two forms of lactate are clearly observable, namely those that contain 12C at the methyl position, and those that contain 13C. Indeed, the 13C satellites demonstrate that all three carbons are 13C (Lloyd et al. 2004; Lane et al. 2007), as expected for lactate deriving exclusively from uniformly labeled glucose (Lane et al. 2008). The unlabeled lactate molecules must have originated from some other (unlabeled) source. Additional experiments with other labeled source molecules can be used in a similar way. For example, 13C-labeled glutamine could produce 13C lactate if the glutaminolysis pathway including malic enzyme is active (DeBerardinis et al. 2007) (cf. Figure 1).
Figure 2. Time course of glucose consumption and lactate secretion from A549 cells.


Human lung adenocarcinoma cells A549 were grown at 37 °C in RPMI 1640 medium containing [U-13C]-glucose in place of natural 12C glucose in 5% CO2 and 10% dialyzed FCS. Medium was sampled at different times, extracted with cold trichloracetic acid (Fan et al. 2008; Lane et al. 2008) and lyophilized. The polar metabolites remaining were redissolved in D2O and analyzed by NMR at 600 MHz.
A. 1D spectrum showing the presence of 13C lactate.
B. Time course of 12C (○) and 13C (●) lactate production and consumption of 13C glucose (■), Gln (□), Val (◆) and Thr (✧). Average of 4 parallel experiments.
C. The fraction of lactate that is U-13C, and the fraction of glucose consumed converted to lactate was calculated as previously described (Lane et al. 2008). Average of 4 parallel experiments.
In even a simple experiment of this kind, the information content is relatively high when isotopomer tracing is used.. Not only can the rate of glucose and glutamine consumption as well as essential amino acid such as Val and Thr, but also the concentrations of both labeled and unlabeled lactate (Figure 2B). In these A549 cells, the rate of consumption of glucose was nearly ten fold higher than of glutamine, and 90 fold higher than valine. In contrast the consumption of the Val and Thr could be accounted for by the needs of protein biosynthesis, rather than for energy production.
It is possible both to determine both the enrichment of individual atoms in a product from a given labeled source, and fraction of the source that is consumed, that ends in the product. For example, cancer cells may convert more than 90% of the glucose consumed into secreted lactate and alanine and the enrichment in the lactate may exceed 90% (DeBerardinis et al. 2007). The enrichment in lactate in these A549 reached a plateau value of > 97% (Fig. 2C) that reflects the relative rate of production from different sources. This then provides an estimate of the relative importance of glycolysis versus other pathways (e.g. glutaminolysis) to lactate production. Furthermore, the fraction of glucose consumed that was converted to lactate approached 30%, which is much lower than observed in a glioblastoma cell line (DeBerardinis et al. 2007), and more similar to other cancer cells of epithelial origin we have examined (Fan et al. 2008). Although the glucose consumption was high, the glutamine consumption was eightfold lower, though considerably higher than that accounted for by unlabeled lactate secretion. A key interpretation of this metabolic pattern, is that the glutamine was being used for other purposes, such as anaplerosis and biosynthesis (DeBerardinis et al. 2007; Fan et al. 2008).
B. Intracellular components
To obtain direct information on intracellular fates of source carbon, the isotopomer approach must be used, for which there are two main options available- in vivo NMR (also known as MRS) and NMR plus MS analysis of cell extracts. For MRS approaches, the resolution and sensitivity is generally lower than for extracts but has the obvious advantage of measurements in the living organism in situ (Macdonald et al. 2002). This is an extremely promising but very advanced topic beyond the scope of this present paper.
With extracts, a much larger range of metabolites can be identified and quantified, thus providing more information, at the expense of losing direct measurement of compartmentation. However, even in extracts, stable isotope tracers can detect the influence of compartmentation in vivo,. This is because the distribution in intracellular metabolites depends on the specific pathways, which include compartmentalized reactions such as those in the cytosol versus the in the mitochondria, resulting in separate “pools” of differently labeled metabolites at different times. Provided that there are non-mixing pools of metabolites that are utilized in different pathways, these can be detected by isotopomer analysis. Using the previous discussion of pyruvate and lactate as an example, with isotopomer analysis now it is possible to distinguish separate pools of pyruvate that are involved in lactate production via glycolysis, or pyruvate carboxylation (an anaplerotic reaction) in brain (Zwingmann et al. 2001; Zwingmann et al. 2003)and in isolated islet cells (Lu et al. 2002; Fransson et al. 2006).
In fact, with stable isotope tracing, many of the central anabolic and energy producing pathways can be directly monitored as shown in Figure 1. For delineating a wide variety of pathway activities, [U-13C]-glucose is a useful and inexpensive source that rapidly enters many of the key reporter metabolites of central metabolism, such as Ala and Lactate that report on glycolysis, ribose moieties in the free nucleotides pool (pentose phosphate pathway), glutamate, citrate, malate and aspartate (Krebs’ cycle), pyrimidine rings (nucleotide biosynthesis), fatty acid and glycerol moieties of phospholipids and reduced glutathione (Fan et al. 2004; Fan et al. 2006; Fan et al. 2008; Lane et al. 2008).
In order to resolve and identify a large number of metabolites in cells, we make use of two-dimensional NMR and several MS methods (Fan et al. 1986; Fan 1996; Fan et al. 2008; Lane et al. 2008). A particularly useful NMR experiment is the so-called TOCSY, with which it is possible to determine 13C enrichments at individual carbon atoms in numerous metabolites simultaneously (Lane et al. 2007; Fan et al. 2008; Lane et al. 2008). Figure 3 shows a portion of a TOCSY spectrum of an extract of A549 cells grown in the presence of [U-13C]-glucose. As expected, essential metabolites such as Thr are not labeled with 13C, whereas newly synthesized intracellular metabolites like Glu, Asp, Ala and Lac are extensively labeled. The specific patterns of peaks provide specific quantitative information about which atoms in the metabolites have incorporated 13C originating from glucose. Thus, both Ala and Lac show a pattern characteristic of a mixture of unlabeled metabolites and where all three atoms derived intact from glucose. There was no evidence of scrambling which would be manifested as additional peaks in the form of a cross (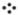 ) rather than the observed square (
) rather than the observed square ( ).
).
Figure 3. 2-D 1H TOCSY identified unlabeled and 13C-labeled metabolites in extracts.

TCA extract of A549 cells grown in the presence of 5 mM [U-13C]-glucose for 24 h, as described for Figure 2. Boxes show the 13C satellite peaks of selected metabolites (Lac, Ala, Glu and Asp).
Prima facie evidence for a functioning Krebs cycle is also obtained from the labeling pattern. The Glu and Asp peaks, showed the presence of metabolic scrambling which occurs at the succinate/fumarate step in the Krebs’ cycle (both of these amino aids are transamination products of Krebs’ cycle intermediates). Furthermore for the Krebs’ cycle and the glycolysis to continue, the aspartate malate shuttle must also be active, which further requires that the respiratory chain is functional (i.e. to reoxidize both glycolysis and Krebs’ cycle generated NADH). Similar considerations apply to the synthesis of ribose and the pyrimidine rings (Fan et al. 2004; Fan et al. 2005; Fan et al. 2006).
In order to differentiate between oxidative (NADPH producing) and non-oxidative activity of the PPP requires experiments using different glucose isotopomers, such as 13C1,2 (Lee et al. 1998) (Haberg et al. 1998; Miccheli et al. 2006) or 13C1,6 (Yang et al. 2005; DeBerardinis et al. 2007), whereas determining the roles of alternative substrate for oxidation or biosynthesis requires 13C-labeled glutamine for example (e.g., to evaluate glutaminolysis (DeBerardinis et al. 2007)). Glutamine is also a central component of nitrogen metabolism, so 15N-labeled Gln can be used to monitor glutaminase activity (up-regulated in many cancers (Perez-Gomez et al. 2005)), and the incorporation of the side chain nitrogen into purine rings for example.
The specific isotopomer patterns of metabolically labeled reporters also make possible the distinction between entry of carbon into the Krebs cycle via PDH (acetyl CoA, or 2 carbons) versus the anaplerotic reaction catalyzed by PC (pyruvate, or three carbons). These inputs give rise to characteristic labeling patterns of Glu, Asp and malate, and thus provide a means to estimate the relative Krebs cycle rate versus anaplerosis. Anaplerosis is essential if Krebs’ cycle intermediates are used for biosynthesis. For example, proliferating cells must make nucleotides to synthesize both ribosomes and DNA. As OAA is the direct precursor of uracil, removing OAA from the Krebs’ cycle requires that the carbon is replaced. Pyruvate carboxylase and glutaminolysis are two major pathways for achieving this.
As the forgoing examples illustrate, the extremely information-rich isotopomer distribution patterns – both among metabolites as well as the pattern within a metabolite clearly define whether specific pathways are active in a given cell type and can provide information about relative fluxes. Such information is critical also for understanding mechanism of regulation. For example, suppose that the isotopomer analysis had shown an elevated flux to nucleotide biosynthesis, and elevated anaplerosis via PC activation. The reaction rate could be elevated for a variety of reasons, including substrate availability, increased enzyme concentration due to increased transcription/translation or decreased degradation, or increased intrinsic activity via post translation modification or allosteric activation. Glutaminase activity is modulated by the glutaminase interacting protein (GIP) (Banerjee et al. 2008), and PC is modulated by AcCoA (Jitrapakdee et al. 2008). Thus an increase in enzyme concentration or better enzyme activity in an extract would be consistent with the observed rate increase deduced directly through isotopomer metabolomics. However, the absence of a corresponding change in protein or mRNA level would indicate an alternative mechanism that bears directly on the enzyme activity, due to specific regulators of the enzyme, allosteric modulation, substrate availability due to compartmentation, etc. We note here that protein levels do not always correlate well with any change in mRNA level (Jansen et al. 2002; Kadota et al. 2003; Pascal et al. 2008). However, in an extract the influence of non-covalent modulators of enzyme activity may well be different from that in vivo; clearly the in vivo derived flux information using the isotopomer approach is functionally the most important measurements to make.
Metabolomics in whole organisms
In animal models or a clinical setting the systemic consumption of glucose and production of lactate is straightforward to determine using NMR. This is essential for deciding on suitable time points for tissue extraction, as with surgical patients only one tissue sample is usually possible, especially when paired normal and cancerous samples are required. We have made a series of experiments in which human patients (70–80 kg), rats (ca. 250 g) and nude mice (20 g) were injected with [U-13C]-glucose, and blood was collected immediately after injection and at timed intervals thereafter The blood was rapidly chilled, and the plasma separated within minutes, before being flash frozen. The frozen plasma was then extracted with cold TCA (Gradwell et al. 1998; Lane et al. 2008), and the metabolites analyzed by 1D NMR (cf. Fig 2). The glucose and lactate molecules (both 12C and 13C) were quantified, from which the supplied glucose uptake, and the production of lactate could be measured. As expected, the peak concentration of plasma lactate occurred at times depending on the mass of the organism, and in a manner consistent with the well-established 3/4 power scaling law relating metabolic rate to body mass (Downs et al. 2008; Glazier 2008; White et al. 2008). This suggests that the optimal sampling time can be estimated from the weight of the organism.
In parallel, we obtained tissue samples from the human subjects (Lung), mice (lung, liver, kidney) and rats (skeletal muscle, liver, adipose tissue), and measured 13C enrichments in various positions of intracellular metabolites including lactate, Ala, Glu and Asp. Substantial enrichment was observed for these metabolites at the time at which the labeled lactate in the plasma was maximal, indicating that the plasma lactate enrichment is a good surrogate for tissue metabolism. Thus for tumor bearing nude mice, the optimal sampling time of tissue was determined to be 15–20 minutes, whereas for rats it was 45–60 min and for human lung cancer patients it was about 3 h. Furthermore, because NMR provides positional isotopomer distributions, analysis of the labeled lactate and alanine provides an estimate of the degree of metabolic scrambling owing to the Cori and alanine cycles, in which lactate produced from tissues is secreted into the blood and reconverted into glucose in the liver (Jin et al. 2004). If a uniformly labeled lactate moiety is condensed with an unlabeled lactate moiety, the resulting glucose is partly scrambled owing to the utilization of Pyr carboxylase and PEPCK activity in gluconeogenesis. This glucose is returned to the plasma can then be metabolized and produces lactate that is not labeled at all three positions. In our experience, because the sampling times were short, the degree of scrambling was quite small (less than 5% in the worst case, and not significantly different from natural abundance istotopomer patterns in the best cases). Furthermore, the only significantly labeled metabolites observed in the blood plasma were uniformly labeled glucose, uniformly labeled Lac and Ala; other amino acids in the plasma showed insignificant degrees of labeling. Therefore, the metabolites isolated from tissues quite accurately reflect the transformation of glucose into various metabolites in situ, with a small contribution to uptake of labeled metabolites released from other tissues and taken up by e.g. the target tumor.
In the absence of prior laboratory and animal based model systems, it would be difficult to determine what to look for in the human tissue, which is the ultimate goal of all cancer studies, either for early diagnosis, prognosis or in drug development. As already stated, most measurements such as mRNA expression level or protein content per se does not equate to enzyme activity that is frequently modulated by covalent or non-covalent interactions (cf. AcCoA for pyruvate carboxylase), and other such unknown in vivo conditions. Thus, only actual metabolic flux via tracers can demonstrate activities directly, and recent analytical advances in isotopomer approaches enable this capability. Our long term goal has been to establish a coherent methodology, obtain relevant biochemical information about different types of cancer cells under defined conditions, and then use stable isotope tracing in a clinical setting. We have in fact obtained good results with this general approach on resectable NSCLC.
C. Application to clinical samples
Unlike the medium bathing cells in culture, biofluids such as blood and urine are systemic, so that detected biomarkers can have their origin in any tissue. Thus plasma biomarkers have limited value for understanding mechanism, as not even the source of changes in biofluid composition is clear especially for systemic fluids like plasma though other fluids may retain some specificity (e.g. CSF, nipple aspirates). This is perhaps where the real power of the isotopomer approach manifests itself, as the specific isotopic distribution in source and products may be indicative of tissue-specific activity. For example, [U-13C]-glucose can become partially scrambled by the Cori and alanine cycles, as explained above, resulting in isotope distributions that may differ in tissue metabolites from those in the plasma.. Furthermore the overall isotope enrichment of a tissue metabolite may differ from that in the serum, indicating the likelihood of tissue specific metabolism. We have used this isotope tracing approach in both rats and mice, and more extensively in human NSCLC patients.
Sequential sampling of blood after administering [U-13C]-glucose i.v. showed the rate of uptake of glucose in fasted patients, and the appearance of 13C lactate. Figure 4A shows 1D NMR spectra of normal and lung cancer tissue from a cancer patient 3 h after consuming U-13C glucose. The tissue the lung cancer patient shows not only an elevated level of lactate compared with the normal lung tissue, but also a higher enrichment in 13C. The fine structure of the 13C satellite peak pattern is characteristic of fully labeled lactate as discussed above.
Figure 4. NMR spectra of plasma and tissue of NSCLC cancer patient.


A patient was infused with 10 g [U-13C]-glucose 3 h prior to resection of the lung. The tumor and non-tumorous lung tissue was flash frozen and then extracted with cold TCA as described (Gradwell et al. 1998; Lane et al. 2008).
A. 1D tissue: normal and cancer
B. TOCSY spectrum of cancer
Similar isotopomer patterns were obtained in the NSCLC extracts as with A549 cells in culture (Fig. 4B and Fig. 3), implying a similar underlying biochemistry. For example, both Ala and Lac are significantly labeled with 13C deriving from glucose, with very little detectable scrambling. This supports the scheme that glucose enters the lung tissue directly from the blood, and is metabolized in situ. Furthermore, the glutamate is also labeled with 13C indicating that the Krebs cycle is active in the tumor cells. In general, the labeling level in lung tumors was substantially higher than in the normal tissues, despite the poorer vascularization of the tumors. Additional HSQC and GC-MS experiments showed specific 13C enrichments of aspartate that are consistent with elevated pyruvate carboxylase activity, suggesting that this cam be an important anaplerotic reaction in some tumors (cf. (Fan et al. 2007; Fan et al. 2008).
Another example illustrating the information richness of the isotopomer approach is for lipids. As cancer cells grow, they must make new membranes, which requires up regulation of lipid biosynthesis. Lipid biosynthesis and biochemistry is in general greatly up-regulated in many cancers (Swinnen et al. 2006), and critical enzymes such as fatty acid synthase and acetyl CoA carboxylase are potential anticancer targets (Piyathilake et al. 2000; Orita et al. 2007). Furthermore, increased oxidative stress in lung tumors is associated with lipid peroxidation, which alters membrane and signaling properties. Some lung cancer cells synthesize large quantities of lipids, and the distribution of phospholipids (PL) and triglyceride (TG) varies according to cell type (Garattini 2007; Trombetta et al. 2007; Guo et al. 2008). Thus lipid analysis can be highly informative about the proliferative state of cells.
We have been developing ultra-high-resolution, extreme mass accuracy FT-ICR-MS methods for direct infusion analysis of unfractionated lipids extracted from cultured cells and tissue. This technique is both very fast (about 10 min per sample) and can yield virtually completely resolved phospholipid speciation. Most importantly, the FT-ICR-MS is currently the only method that can identify and quantify mass isotopomers most phospholipid species simultaneously. The assignment of thousands of peaks generated in each analysis becomes the bottleneck, and we have partially solved this biochemoinformatic problem using proprietary software. Figure 5 A and B shows typical FT-ICR–MS spectra of phospholipids extracted from a human lung tumor and non-cancerous tissue from the same subject. As illustrated in Figure 5, each 10 minute sample analysis can typically yield hundreds to thousands of results on a lipid-specific basis; currently, limitations in biochemoinformatic data processing is our most serious bottleneck to this otherwise extremely high information throughput (HIT) technique.
Figure 5. FT-ICRMS spectra of lung tumor lipids.

Part A shows FT-ICR-MS spectra of phospholipid extracts, from the lungs of a single human subject. Note that overall, the spectra appear similar to each other, which is expected since most of the phospholipids serve functional roles as membrane structure. Part B shows the expansion of the region indicated in Part A, illustrating the three general categories of results for each of the hundreds to thousands of peaks obtained from each 10 minute sample run. Thus, this is a high-information throughput (HIT) technique. As illustrated, the most abundant lipids tend to be similar in profile, while many of the less abundant lipids decrease or increase in lung cancer relative to the non-cancerous lung tissue from the same patient. In this particular example, there are >1,000% differences in the indicated lipids, which are only two out of dozens of differences at this magnitude that are present.
D. Prospects and Conclusions
Non-isotopomer approaches to metabonomic analysis of biofluids have been shown to be useful for diagnosis. However, to understand the origins and progression of diseases requires more direct information, i.e. tissue, and the dynamic information obtainable only through the isotopomer approach. We have obtained extensive isotopomer data on lung and breast cancer cells, as well as representative non-transformed cells from these tissues, in conjunction with analysis of the relevant biofluid (medium, plasma, urine etc.). The analysis of the human NSCLC was dependent on the availability of matched pairs of cancer and normal lung tissue (each patient is also the control), which provides information on basic lung biochemistry.
The goals are to use these approaches both in the area of early detection, but also given the detailed mechanistic information, as part of the molecular targeting and drug discovery process. For targeting specific metabolic lesion associated with a particular disease state such as a cancer drug development requires a deep understanding of the actual biochemistry of the relevant cell type, in its appropriate environment, i.e. in situ. For example, the accelerated glycolysis that is a hallmark of most tumors is a general anticancer target. There are possible enzymes as targets to inhibit, presumably to down-regulate to a more normal level. For example, 2-deoxyglucose and similar compounds have been proposed to inhibit glycolysis at the post hexokinase step (Kurtoglu et al. 2007; Kurtoglu et al. 2007; Boutrid et al. 2008), or the so-called supercharger of glycolysis, PFK-2 (Telang et al. 2006; Clem et al. 2008) described in detail the article in this issue by Yalcin et al. and other metabolic targets such as aspartate aminotransferase (Thornburg et al. 2008).
Metabolic profiling, using stable isotope tracers, can also be used to verify that the drug under development is functioning in the way expected, and can also reveal off-target effects in the same samples. If the metabolic targets are understood, then the influence of the inhibitor can be predicted, and compared to the actual biochemical response of the cells or tissue. Any additional unexpected effects are evidence of off-target interactions, which may be identified by the perturbed metabolic signature. This rich information source is invaluable for the development phase of a drug, either by additional in silico development (see article by Dailey et al., this issue) or chemical optimization.
Metabolomics approaches may also be useful in the realm of personalized medicine. The response to a particular drug is well known to vary enormously among individuals (Clarke 1937); the IC50 dose can vary by more than an order of magnitude within a population (Roden et al. 2006; Li et al. 2008). This leads to the stratification into responders and non-responders. As metabolism is the result of the interaction of gene expression with complex environmental cues and history, it can be anticipated that the functional biochemistry discovered through stable isotope-based metabolomics could provide valuable sets of biochemical markers of individual response to a particular drug. Clinically this would be used either to control the dose, or to use an alternative drug.
As alluded to above, one of the problem areas with all ‘omics approaches to biofluids is specificity and proximity. Furthermore, most of the clinically relevant studies have been limited to late stage disease. Yet a major goal of such research is to establish reliable methods for early detection of asymptomatic malignancies. This is especially relevant lung cancer, as early presentation of the disease is uncommon (ca. 15% of cases are found in early stages prior to spread to lymph nodes). The present 5-year survival rate for non-small cell lung cancer (NSCLC) often exceeds 80% when treatment is given at stage IA (Phillips et al. 1999; Greenberg et al. 2007) versus 15–17% when treated at later stages (Hayat et al. 2007).
In contrast, the 5-year survival of stage 3 SCLC or NSCLC is dismal. In particular, SCLC is generally not suited to surgical intervention because by the time it is diagnosed, it has usually spread to the lymph nodes and beyond (Edelman et al. 2004). Lung biopsy is rather invasive; a non-invasive technique is urgently needed, but non-invasive modalities rely on biofluids. Proximal biofluids relevant to the lung include bronchial lavage fluid (Schiller et al. 2001; Ohwada et al. 2003), and exhaled air (Andreoli et al. 2003; Rahman et al. 2003; Gessner et al. 2004). These methods are still under development and evaluation, as there are numerous technical problems associated with sampling and analysis, not least contamination from recent dietary intake and environmental air. Here, the isotopomer approach to studies would greatly reduce or eliminate many of these current problems. The analytical methods we have been developing in our laboratories are being applied to a variety of cancer problems. Although clinically the isotope tracing approach has been used predominantly for lung cancers, the technology can be adapted to any malignancy. We are currently using this approach to examine chemotherapeutics effects in breast cancers, and for analyses of metastatic melanoma.
Acknowledgments
This work has been supported by NIH Grant RR018733 from the National Center for Research Resources, the Kentucky Challenge for Excellence, 1R01CA118434-01 and the KY Lung Cancer Research Program.
Abbreviations
- CAC
Citric Acid Cycle
- FT-ICR-MS
fourier transform ion cyclotron resonance mass spectrometry
- HMEC
human mammary epithelial cells
- MS
mass spectrometry
- PEPCK
phosphoenolpyruvate carboxykinase
- PET
Positron Emission Tomography
- PPP
Pentose Phosphate Pathway
- PC
pyruvate carboxylase
- PDH
pyruvate dehydrogenase
Footnotes
Conflict of Interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014–1020. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Andreoli R, Manini P, Corradi M, Mutti A, Niessen WMA. Determination of patterns of biologically relevant aldehydes in exhaled breath condensate of healthy subjects by liquid chromatography/atmospheric chemical ionization tandem mass spectrometry. Rapid Communications in Mass Spectrometry. 2003;17:637–645. doi: 10.1002/rcm.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M, Huang C, Marquez J, Mohanty S. Probing the structure and function of human glutaminase-interacting protein: A possible target for drug design. Biochemistry. 2008;47:9208–9219. doi: 10.1021/bi800287v. [DOI] [PubMed] [Google Scholar]
- Beger RD, Schnackenberg LK, Holland RD, Li DH, Dragan Y. Metabonomic models of human pancreatic cancer using 1D proton NMR spectra of lipids in plasma. Metabolomics. 2006;2:125–134. [Google Scholar]
- Boutrid H, Jockovich ME, Murray TG, Pina Y, Feuer WJ, Lampidis TJ, Cebulla CM. Targeting hypoxia, a novel treatment for advanced retinoblastoma. Investigative Ophthalmology & Visual Science. 2008;49:2799–2805. doi: 10.1167/iovs.08-1751. [DOI] [PubMed] [Google Scholar]
- Brown SC, Kruppa G, Dasseux JL. Metabolomics applications of FT-ICR mass spectrometry. Mass Spectrometry Reviews. 2005;24:223–231. doi: 10.1002/mas.20011. [DOI] [PubMed] [Google Scholar]
- Cermik TF, Mavi A, Basu S, Alavi A. Impact of FDG PET on the preoperative staging of newly diagnosed breast cancer. European Journal of Nuclear Medicine and Molecular Imaging. 2008;35:475–483. doi: 10.1007/s00259-007-0580-5. [DOI] [PubMed] [Google Scholar]
- Clarke AJ. Individual Variation in Repsonse to Drugs. Brit Med J. 1937:307–310. doi: 10.1136/bmj.2.3997.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem BF, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane AN, Trent JOJAC. Small Molecule Inhibition of 6-Phosphofructo-2-Kinase Activity Suppresses Glycolytic Flux and Tumor Growth. Molecular Cancer Therapeutics. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhital K, Saunders CA, Seed PT, O’Doherty MJ, Dussek J. (18)F]Fluorodeoxyglucose positron emission tomography and its prognostic value in lung cancer. European journal of cardio-thoracic surgery. 2000;18:425–428. doi: 10.1016/s1010-7940(00)00535-2. [DOI] [PubMed] [Google Scholar]
- Downs CJ, Hayes JP, Tracy CR. Scaling metabolic rate with body mass and inverse body temperature: a test of the Arrhenius fractal supply model. Functional Ecology. 2008;22:239–244. [Google Scholar]
- Edelman RJ, Gandara DR. Lung Cancer. In: Casciato DA, editor. Manual of Clinical Oncology. Philadelphia: Lippincott Williams & Wilkins; 2004. [Google Scholar]
- Fan T, Bandura L, Higashi R, Lane A. Metabolomics-edited transcriptomics analysis of Se anticancer action in human lung cancer cells. Metabolomics. 2005;1:1–15. [Google Scholar]
- Fan T-M, Lane A, Farag M, Arumugam V, Higashi R, Bousamra M, Miller D. Human Lung Cancers Have Altered Anaplerotic (“Replenishing”) Pathways Discerned By 13C-Isotopomer-Based Metabolomics. 16th Triennial Conference for the International Society of Magnetic Resonance Kenting; Taiwan. 2007. [Google Scholar]
- Fan TW, Lane AN. Structure-based profiling of Metabolites and Isotopomers by NMR. Progress in NMR Spectroscopy. 2008;52:69–117. [Google Scholar]
- Fan TWM. Metabolite profiling by one- and two-dimensional NMR analysis of complex mixtures. Progress in Nuclear Magnetic Resonance Spectroscopy. 1996;28:161–219. [Google Scholar]
- Fan TWM, Kucia M, Jankowski K, Higashi RM, Rataczjak MZ, Rataczjak J, Lane AN. Proliferating Rhabdomyosarcoma cells shows an energy producing anabolic metabolic phenotype compared with Primary Myocytes. Molecular Cancer. 2008;7:79. doi: 10.1186/1476-4598-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TWM, Higashi RM, Lane AN. Integrating metabolomics and transcriptomics for probing Se anticancer mechanisms. Drug Metabolism Reviews. 2006;38:707–732. doi: 10.1080/03602530600959599. [DOI] [PubMed] [Google Scholar]
- Fan TWM, Higashi RM, Lane AN, Jardetzky O. Combined Use of H-1-Nmr and Gc-Ms for Metabolite Monitoring and Invivo H-1-Nmr Assignments. Biochimica Et Biophysica Acta. 1986;882:154–167. doi: 10.1016/0304-4165(86)90150-9. [DOI] [PubMed] [Google Scholar]
- Fan TWM, Lane AN, Higashi RM. The promise of metabolomics in cancer molecular therapeutics. Current Opinion in Molecular Therapeutics. 2004;6:584–592. [PubMed] [Google Scholar]
- Fransson U, Rosengren AH, Schuit FC, Renstrom E, Mulder H. Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP: ADP ratio in rat pancreatic islets. Diabetologia. 2006;49:1578–1586. doi: 10.1007/s00125-006-0263-y. [DOI] [PubMed] [Google Scholar]
- Garattini S. Long-chain n-3 fatty acids in lipid rafts: implications for anti-inflammatory effects. Journal of Cardiovascular Medicine. 2007;8:S30–S33. doi: 10.2459/01.JCM.0000289277.10675.e8. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Glycolysis in cancer: A potential target for therapy. International Journal of Biochemistry & Cell Biology. 2007;39:1358–1366. doi: 10.1016/j.biocel.2007.03.021. [DOI] [PubMed] [Google Scholar]
- Gessner C, Kuhn H, Toepfer K, Hammerschmidt S, Schauer J, Wirtz H. Detection of p53 gene mutations in exhaled breath condensate of non-small cell lung cancer patients. Lung Cancer. 2004;43:215–222. doi: 10.1016/j.lungcan.2003.08.034. [DOI] [PubMed] [Google Scholar]
- Glazier DS. Effects of metabolic level on the body size scaling of metabolic rate in birds and mammals. Proceedings of the Royal Society B-Biological Sciences. 2008;275:1405–1410. doi: 10.1098/rspb.2008.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwell MJ, Fan TWM, Lane AN. Analysis of phosphorylated metabolites in crayfish extracts by two-dimensional H-1-P-31 NMR heteronuclear total correlation spectroscopy (heteroTOCSY) Analytical Biochemistry. 1998;263:139–149. doi: 10.1006/abio.1998.2789. [DOI] [PubMed] [Google Scholar]
- Greenberg AK, Lee MS. Biomarkers for lung cancer: clinical uses. Curr Opin Pulm Med. 2007;13:249–255. doi: 10.1097/MCP.0b013e32819f8f06. [DOI] [PubMed] [Google Scholar]
- Griffin JL. Understanding mouse models of disease through metabolomics. Current Opinion in Chemical Biology. 2006;10:309–315. doi: 10.1016/j.cbpa.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Griffin JL, Kauppinen RA. Tumour metabolomics in animal models of human cancer. Journal of Proteome Research. 2007;6:498–505. doi: 10.1021/pr060464h. [DOI] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberg A, Qu H, Bakken IJ, Sande LM, White LR, Haraldseth O, Unsgard G, Aasly J, Sonnewald U. In vitro and ex vivo C-13-NMR spectroscopy studies of pyruvate recycling in brain. Developmental Neuroscience. 1998;20:389–398. doi: 10.1159/000017335. [DOI] [PubMed] [Google Scholar]
- Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the surveillance, epidemiology, and end results (SEER) program. Oncologist. 2007;12:20–37. doi: 10.1634/theoncologist.12-1-20. [DOI] [PubMed] [Google Scholar]
- Higashi R, Lane A, Yang T, Xie Z, Fan T. Isotopomer Lipid Metabolomics in Cancer Cells by 2D-NMR and FTICR-MS. Reveals Detailed Lipid Metabolism Metabolomics Society 4th Annual Meeting; Boston MA. 2008. [Google Scholar]
- Ippolito JE, Xu J, Jain SJ, Moulder K, Mennerick S, Crowley JR, Townsend RR, Gordon JI. An integrated functional genomics and metabolomics approach for defining poor prognosis in human neuroendocrine cancers. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9901–9906. doi: 10.1073/pnas.0500756102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RC, Nap JP, Mlynarova L. Errors in genomics and proteomics. Nature Biotechnology. 2002;20:19–19. doi: 10.1038/nbt0102-19b. [DOI] [PubMed] [Google Scholar]
- Jin ES, Jones JG, Merritt M, Burgess SC, Malloy CR, Sherry AD. Glucose production, gluconeogenesis, and hepatic tricarboxylic acid cycle fluxes measured by nuclear magnetic resonance analysis of a single glucose derivative. Analytical Biochemistry. 2004;327:149–155. doi: 10.1016/j.ab.2003.12.036. [DOI] [PubMed] [Google Scholar]
- Jitrapakdee S, St Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV. Structure, mechanism and regulation of pyruvate carboxylase. Biochemical Journal. 2008;413:369–387. doi: 10.1042/BJ20080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R, Kristal BS, Weinshilboum RM. Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol. 2008;48:23.21–32.31. doi: 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- Kadota K, Tominaga D, Asai R, Takahashi K. Correlation Analysis of mRNA and Protein Abundances in Human Tissues. Genome Letters. 2003;2:39–148. [Google Scholar]
- Kind T, Tolstikov V, Fiehn O, Weiss RH. A comprehensive urinary metabolomic approach for identifying kidney cancer. Analytical Biochemistry. 2007;363:185–195. doi: 10.1016/j.ab.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Kurtoglu M, Gao N, Maher JC, Lehrman MA, Wangpaichitr M, Savaraj N, Lane AN, Lampidis TJ. Under normoxia 2-deoxy-D-Glucose, but not 2-fluoro-deoxy-D-glucose, kills select tumor cells by interfering with N-linked glycosylation. Molecular Cancer Therapeutics. 2007;6:3049–3058. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- Kurtoglu M, Maher JC, Lampidis TJ. Differential toxic mechanisms of 2-deoxy-D-glucose versus 2-fluorodeoxy-D-glucose in hypoxic and normoxic tumor cells. Antioxidants & Redox Signaling. 2007;9:1383–1390. doi: 10.1089/ars.2007.1714. [DOI] [PubMed] [Google Scholar]
- Lane AN, Fan TW. Quantification and identification of isotopomer distributions of metabolites in crude cell extracts using 1H TOCSY. Metabolomics. 2007;3:79–86. [Google Scholar]
- Lane AN, Fan TW, Higashi RM. Isotopomer-based metabolomic analysis by NMR and mass spectrometry. In: John HWD, Correia J, editors. Biophysical Tools for Biologists. 84. Vol. 1. San Diego: Academic Press; 2008. pp. 541–588. [DOI] [PubMed] [Google Scholar]
- Lane AN, Fan TW-M, Higashi RM. Stable isotope assisted metabolomics in cancer research. IUBMB Life. 2008;60:124–129. doi: 10.1002/iub.17. [DOI] [PubMed] [Google Scholar]
- Lee WNP, Boros LG, Puigjaner J, Bassilian S, Lim S, Cascante M. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am J Physiol Endocrinol Metab. 1998;274:E843–851. doi: 10.1152/ajpendo.1998.274.5.E843. [DOI] [PubMed] [Google Scholar]
- Li L, Fridley B, Kalari K, Jenkins G, Batzler A, Safgren S, Hildebrandt M, Ames M, Schaid D, Wang LW. Gemcitabine and cytosine arabinoside cytotoxicity: Association with lymphoblastoid cell expression. Cancer Research. 2008;68:7050–7058. doi: 10.1158/0008-5472.CAN-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon JC, Holmes E, Nicholson JK. Metabonomics: Systems biology in pharmaceutical research and development. Current Opinion in Molecular Therapeutics. 2004;6:265–272. [PubMed] [Google Scholar]
- Lindon JC, Nicholson JK, Everett JR. Annual Reports on Nmr Spectroscopy. Vol. 38. San Diego: Academic Press Inc; 1999. NMR spectroscopy of biofluids; pp. 1–88. [Google Scholar]
- Lloyd SG, Zeng HD, Wang PP, Chatham JC. Lactate isotopomer analysis by H-1 NMR spectroscopy: Consideration of long-range nuclear spin-spin interactions. Magnetic Resonance in Medicine. 2004;51:1279–1282. doi: 10.1002/mrm.20075. [DOI] [PubMed] [Google Scholar]
- Lu DH, Mulder H, Zhao PY, Burgess SC, Jensen MV, Kamzolova S, Newgard CB, Sherry AD. C-13 NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS) Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2708–2713. doi: 10.1073/pnas.052005699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald JM, Xu A, Kubota H, LeCluyse E, Hamilton G, Liu H, Rong Y, Moss N, Lodestro C, Luntz T, Wolfe SP, Reid LM. Metabolic Tissue Engineering. San Diego: Academic Press; 2002. Liver Cell Culture and Lineage Biology; pp. 151–201. [Google Scholar]
- Mazurek S, Grimm H, Oehmke M, Weisse G, Teigelkamp S, Eigenbrodt E. Tumor M2-PK and glutaminolytic enzymes in the metabolic shift of tumor cells. Anticancer Research. 2000;20:5151–5154. [PubMed] [Google Scholar]
- Miccheli A, Tomassini A, Puccetti C, Valerio M, Peluso G, Tuccillo F, Calvani M, Manetti C, Conti F. Metabolic profiling by C-13-NMR spectroscopy: [1,2-C-13(2)] glucose reveals a heterogeneous metabolism in human leukemia T cells. Biochimie. 2006;88:437–448. doi: 10.1016/j.biochi.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Ohwada A, Yoshioka Y, Iwabuchi K, Nagaoka I, Dambara T, Fukuchi Y. VEGF regulates the proliferation of acid-exposed alveolar lining epithelial cells. Thorax. 2003;58:328–332. doi: 10.1136/thorax.58.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orita H, Coulter J, Lemmon C, Tully E, Vadlamudi A, Medghalchi SM, Kuhajda FP, Gabrielson E. Selective inhibition of fatty acid synthase for lung cancer treatment. Clinical Cancer Research. 2007;13:7139–7145. doi: 10.1158/1078-0432.CCR-07-1186. [DOI] [PubMed] [Google Scholar]
- Pascal LE, True LD, Campbell DS, Deutsch EW, Risk M, Coleman IM, Eichner LJ, Nelson PS, Liu AY. Correlation of mRNA and protein levels: Cell type-specific gene expression of cluster designation antigens in the prostate. Bmc Genomics. 2008;9 doi: 10.1186/1471-2164-9-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Gomez C, Campos-Sandoval JA, Alonso FJ, Segura JA, Manzanares E, Ruiz-Sanchez P, Gonzalez ME, Marquez J, Mates JM. Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochemical Journal. 2005;386:535–542. doi: 10.1042/BJ20040996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petch D, Butler M. Profile of Energy-Metabolism in a Murine Hybridoma - Glucose and Glutamine Utilization. Journal of Cellular Physiology. 1994;161:71–76. doi: 10.1002/jcp.1041610110. [DOI] [PubMed] [Google Scholar]
- Phillips M, Gleeson K, Hughes JMB, Greenberg J, Cataneo RN, Baker L, McVay WP. Volatile organic compounds in breath as markers of lung cancer: a cross-sectional study. Lancet. 1999;353:1930–1933. doi: 10.1016/S0140-6736(98)07552-7. [DOI] [PubMed] [Google Scholar]
- Piyathilake CJ, Frost AR, Manne U, Bell WC, Weiss H, Heimburger DC, Grizzle WE. The expression of fatty acid synthase (FASE) is an early event in the development and progression of squamous cell carcinoma of the lung. Human Pathology. 2000;31:1068–1073. doi: 10.1053/hupa.2000.9842. [DOI] [PubMed] [Google Scholar]
- Portais JC, Voisin P, Merle M, Canioni P. Glucose and glutamine metabolism in C6 glioma cells studied by carbon 13 NMR. Biochimie. 1996;78:155–164. doi: 10.1016/0300-9084(96)89500-9. [DOI] [PubMed] [Google Scholar]
- Rahman I, Kelly F. Biomarkers in breath condensate: A promising new non-invasive technique in free radical research. Free Radical Research. 2003;37:1253–1266. doi: 10.1080/10715760310001623331. [DOI] [PubMed] [Google Scholar]
- Robey IF, Lien AD, Welsh SJ, Baggett BK, Gillies RJ. Hypoxia-inducible factor-1 alpha and the glycolytic phenotype in tumors. Neoplasia. 2005;7:324–330. doi: 10.1593/neo.04430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Altman RB, Benowitz NL, Flockhart DA, Giacomini KM, Johnson JA, Krauss RM, McLeod HL, Ratain MJ, Relling MV, Ring HZ, Shuldiner AR, Weinshilboum RM, Weiss ST. Pharmacogenomics: Challenges and opportunities. Annals of Internal Medicine. 2006;145:749–757. doi: 10.7326/0003-4819-145-10-200611210-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Hammerschmidt S, Wirtz H, Arnhold J, Arnold K. Lipid analysis of bronchoalveolar lavage fluid (BAL) by MALDI-TOF mass spectrometry and 31P NMR spectroscopy. Chemistry & Physics of Lipids. 2001;112:67–79. doi: 10.1016/s0009-3084(01)00163-3. [DOI] [PubMed] [Google Scholar]
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Current Opinion in Clinical Nutrition and Metabolic Care. 2006;9:358–365. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- Telang S, Lane AN, Nelson KK, Arumugam S, Chesney JA. The oncoprotein H-RasV12 increases mitochondrial metabolism. Molec Cancer. 2007;6:77. doi: 10.1186/1476-4598-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telang S, Yalcin A, Clem AL, Bucala R, Lane AN, Eaton JW, Chesney J. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene. 2006;25:7225–7234. doi: 10.1038/sj.onc.1209709. [DOI] [PubMed] [Google Scholar]
- Thornburg JM, Nelson KK, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Targeting Aspartate Aminotransferase in Breast Cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta A, Maggiora M, Martinasso G, Cotogni P, Canuto RA, Muzio G. Arachidonic and docosahexaenoic acids reduce the growth of A549 human lung-tumor cells increasing lipid peroxidation and PPARs. Chemico-Biological Interactions. 2007;165:239–250. doi: 10.1016/j.cbi.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weljie AM, Newton J, Mercier P, Carlson E, Slupsky CM. Targeted profiling: Quantitative analysis of H-1 NMR metabolomics data. Analytical Chemistry. 2006;78:4430–4442. doi: 10.1021/ac060209g. [DOI] [PubMed] [Google Scholar]
- White CR, Terblanche JS, Kabat AP, Blackburn TM, Chown SL, Butler PJ. Allometric scaling of maximum metabolic rate: the influence of temperature. Functional Ecology. 2008;22:616–623. [Google Scholar]
- Yang TH, Heinzle E, Wittmann C. Theoretical aspects of C-13 metabolic flux analysis with sole quantification of carbon dioxide labeling. Computational Biology and Chemistry. 2005;29:121–133. doi: 10.1016/j.compbiolchem.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Zimmermann D, Hartmann M, Moyer MP, Nolte J, Baumbach JI. Determination of volatile products of human colon cell line metabolism by GC/MS analysis. Metabolomics. 2007;3:13–17. [Google Scholar]
- Zwingmann C, Leibfritz D. Regulation of glial metabolism studied by C-13-NMR. Nmr in Biomedicine. 2003;16:370–399. doi: 10.1002/nbm.850. [DOI] [PubMed] [Google Scholar]
- Zwingmann C, Richter-Landsberg C, Leibfritz D. C-13 isotopomer analysis of glucose and alanine metabolism reveals cytosolic pyruvate compartmentation as part of energy metabolism in astrocytes. Glia. 2001;34:200–212. doi: 10.1002/glia.1054. [DOI] [PubMed] [Google Scholar]