

Abstract
We have uncovered a significant allosteric response of the D2 dopamine receptor to physiologically relevant concentrations of sodium (140 mM), characterized by a sodium-enhanced binding affinity for a D4-selective class of agonists and antagonists. This enhancement is significantly more pronounced in a D2-V2.61(91)F mutant and cannot be mimicked by an equivalent concentration of the sodium replacement cation N-methyl-d-glucamine. This phenomenon was explored computationally at the molecular level by analyzing the effect of sodium binding on the dynamic properties of D2 receptor model constructs. Normal mode analysis identified one mode (M19), which is involved in the open/closed motions of the binding cleft as being particularly sensitive to the sodium effect. To examine the consequences for D2 receptor ligand recognition, one of the ligands, L-745,870 [3-{[4-(4-chlorophenyl) piperazin-1-yl]-methyl}-1H-pyrrolo[2,3-b]pyridine or CPPMA, chlorophenylpiperazinyl methylazaindole], was docked into conformers along the M19 trajectory. Structurally and pharmacologically well established ligand-receptor interactions, including the ionic interaction with D3.32(114) and interactions between the ligand aryl moieties and V2.61(91)F, were achieved only in “open” phase conformers. The docking of (-)-raclopride [3,5-dichloro-N-(1-ethylpyrrolidin-2-ylmethyl)-2-hydroxy-6-methoxybenzamide] suggests that the same binding cleft changes in response to sodium-binding perturbation account as well for the enhancements in binding affinity for substituted benzamides in the wild-type D2 receptor. Our findings demonstrate how key interactions can be modulated by occupancy at an allosteric site and are consistent with a mechanism in which sodium binding enhances the affinity of selected ligands through dynamic changes that increase accessibility of substituted benzamides and 1,4-DAP ligands to the orthosteric site and accessibility of 1,4-DAPs to V2.61(91)F.
Sodium ions have been shown to modulate dopamine receptors, and allosteric modulation by sodium ions has been shown to drive the conformational equilibrium of heterotrimeric G protein-coupled receptors (GPCRs) toward an agonist low-affinity state (for review, see Schetz, 2005). In dopamine receptors, like in other heterotrimeric GPCRs, the highly conserved and negatively charged aspartic acid at position 2.50 (the generic numbering system is defined in Ballesteros and Weinstein, 1995) has been identified as a sodium interaction site. For example, charge-neutralizing mutations in the D2 or the D4 receptor [e.g., D2.50(80)N or D2.50(80)A] make them sodium-insensitive, whereas a charge-sparing mutation [e.g., D2.50(80)E] retains much of the sodium sensitivity (Neve et al., 1991; Schetz and Sibley, 2001). D2 receptor mutations at other positions [e.g., S3.39(121)A and S7.46(391)A] also diminish sensitivity, presumably by reducing the H-bonding capacity at the sodium binding site (Neve et al., 2001). The latter studies, in the structural context of the high-resolution crystalline structure of bovine rhodopsin (Palczewski et al., 2000), led to a revised model of the sodium binding site (Neve et al., 2001), in which sodium is at the center of a square-pyramidal hydrogen-bonding network whose vertices are formed by D2.50(80), S3.39(121), N7.45(390), and S7.46(391); sodium binding is thought to neutralize the negative charge at D2.50(80). Allosteric modulation of dopamine receptors by sodium has been shown previously to reduce the affinity of endogenous agonists and zinc, increase the affinity or binding capacity (Bmax) of substituted benzamide antagonists, and alter the rate of chemical modification (Neve, 1991; Schetz et al., 1999, 2001; Vivo et al., 2006). However, despite the prevalence of studies across multiple GPCR families indicating allosteric modulation by sodium, an exhaustive search of the literature failed to identify a mechanism for the sodium-induced effects.
In the process of determining the source of large discrepancies in binding affinity reported for 1,4-disubstituted aromatic piperidines/piperazines (1,4-DAPs) for the D2-V2.61(91)F mutant (Simpson et al., 1999; Floresca et al., 2005), we discovered a change, elicited by sodium binding, in the dynamic properties of the receptor that correlate with a dramatic increase in sodium sensitivity for both agonists and antagonists belonging to a similar structural class. This finding offers an opportunity to understand the allosteric mechanism of the effect produced by sodium binding. To this end, we carried out a normal mode analysis (NMA) of the dynamic properties of various D2 receptor constructs using three-dimensional molecular models of the receptor. Normal modes are calculated from the molecular structure model and provide information about the component harmonic motions or vibrations of the molecule that characterize its dynamic fluctuation as it occupies a stable conformational state (e.g., inactive state, etc.). The modes constitute a set of orthogonal vectors ranked by energy (or the corresponding frequency), which indicates the direction in which each particle (the component atoms, or residues, or Cα) is moving at that particular level of energy (frequency). Thus, the superposition of all the normal mode vectors describes the entire intrinsic motion of the molecule based on its shape and molecular connectivity, but often one or a few low-frequency (low energy) modes contribute most significantly to this thermal “breathing” motion of the molecule. It was demonstrated for many proteins that the directions of the lowest frequency modes also tend to indicate the path of molecular movements associated with functionally relevant conformational changes (Cui and Bahar, 2006). Here, the comparison of the normal modes between sodium-bound and sodium-free structures of the receptor models allowed us to identify a specific sodium-responsive normal mode motion that indicates distinct dynamic changes in the environment of position 2.61(91) and accounts for the hypersensitivity in the mutant D2-V2.61(91)F. Thus, when backbone movements associated with this mode are explored as an additional degree of freedom in the ligand docking process, the “open” conformations produced within the trajectory of this normal mode are found to promote ligand binding poses that are consistent with experimentally verified interactions. These findings connect the dynamic properties characterized by the NMA of the wild-type and highly sensitive mutant receptor with experimentally observed sodium-dependent allosteric effects and suggest a mechanism by which the presence of sodium alters ligand affinity.
Materials and Methods
Reagents. Cell culture media were purchased from HyClone Laboratories (Logan, UT). For radioligand studies, NET-856 (70–80 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA), and wash buffer reagents were purchased from United States Biological (Swampscott, MA). The source of (-)-quinpirole and forskolin was Sigma-Aldrich (St. Louis, MO). Other drugs were purchased from Tocris Bioscience (Ellisville, MO).
Site-Directed Mutagenesis. Mutagenesis was accomplished using a QuikChange kit (Stratagene, La Jolla, CA). The integrity of mutations and the lack of unwanted mutations were confirmed by full-length sequencing at the University of Maine DNA sequencing facility (Orono, ME). Mutant receptors are named employing the system created by Ballesteros and Weinstein (1995) and other nomenclature conventions. In brief, for each residue in a transmembrane segment, the first digit denotes the transmembrane segment (TMS), followed by a period and a relative position index within the transmembrane segment. The most conserved amino acid in a TMS is assigned the position index 50, and the other amino acids within this TMS are numbered relative to the conserved amino acid. The number in parentheses is the residue number in the sequence of the rat D2 dopamine receptor short isoform. Our naming system for the mutants begins with a letter to designate the species (e.g., “r” for rat or “h” for human) followed by the receptor subtype abbreviation, e.g., rD2 for rat D2 dopamine receptor. Next, the single-letter abbreviation for the amino acid is listed followed by its position and then amino acid substitution. For example, rD2-V2.61(91)F denotes a rat D2 receptor with valine at position 2.61(91) being substituted for phenylalanine (Fig. 1).
Fig. 1.

Depiction of the D2-V2.61(91)F mutant dopamine receptor as a
monomer in a section of lipid bilayer. This figure represents the unfolded
D2L receptor showing the amino terminus (-NH2) on the
extracellular side and the carboxyl terminus (-CO2H) on the
intracellular side. Open circles (○) are used to indicate wild-type amino
acids, whereas closed circles (• and
 ) are used to represent
specific amino acids. As shown in the sequence, a valine-to-phenylalanine
mutation at amino acid residue 91 (•) results in the
D2-V2.61(91)F mutant dopamine receptor. The purpose of the
V2.61(91)F mutation is to modify the binding pocket of the D2
receptor with the corresponding residue of the D4 receptor and make
it more accommodating to D4-selective 1,4-DAPs
(Simpson et al., 1999;
Schetz et al., 2000;
Kortagere et al., 2004;
Floresca et al., 2005).
Although most ligands bind an orthosteric binding site accessible from the
extracellular face of the receptor
(Floresca and Schetz, 2004),
the sodium ion binds the receptor through an intracellular allosteric binding
site formed by the interactions of transmembrane segments 2, 3, and 7
(Neve et al., 2001). Also
shown in the diagram and the sequence is the relative position of the
conserved negatively charged D2.50(80) that is critical for the interaction of
sodium ions with the dopamine receptor (Neve et al.,
1991,
2001).
) are used to represent
specific amino acids. As shown in the sequence, a valine-to-phenylalanine
mutation at amino acid residue 91 (•) results in the
D2-V2.61(91)F mutant dopamine receptor. The purpose of the
V2.61(91)F mutation is to modify the binding pocket of the D2
receptor with the corresponding residue of the D4 receptor and make
it more accommodating to D4-selective 1,4-DAPs
(Simpson et al., 1999;
Schetz et al., 2000;
Kortagere et al., 2004;
Floresca et al., 2005).
Although most ligands bind an orthosteric binding site accessible from the
extracellular face of the receptor
(Floresca and Schetz, 2004),
the sodium ion binds the receptor through an intracellular allosteric binding
site formed by the interactions of transmembrane segments 2, 3, and 7
(Neve et al., 2001). Also
shown in the diagram and the sequence is the relative position of the
conserved negatively charged D2.50(80) that is critical for the interaction of
sodium ions with the dopamine receptor (Neve et al.,
1991,
2001).
Transfections. DNA constructs subcloned into the pcDNA3.1 vector were transfected into human embryonic kidney (HEK) 293 cells by CaPO4 precipitation. In brief, 20 μg of DNA was mixed with 60 μl of 2 M CaCl2, and the mixture was added to an appropriate volume of sterile water to make 500 μl of solution. This DNA-CaCl2 solution was then added drop-wise to 500 μl of 2× HEPES-buffered saline while bubbling the HEPES-buffered saline with a 1-ml serological pipet. The resultant final transfection mixture was allowed to sit for 30 min before drop-wise addition to 150-cm2 culture dishes seeded with an appropriate number of cells. For stable transfections, HEK293 cells were seeded at a density of 200,000 cells/150-cm2 culture dish. For transient transfections, COS7 cells were seeded at a density of 1.5 million cells/150-cm2 culture dish. Both cell lines were allowed to grow overnight in 20 ml of sterile growth media containing DMEM supplemented with 10% bovine calf serum, 100 μM sodium pyruvate, and 1% penicillin/streptomycin (5000 units). This resulted in approximately 30% confluent COS-7 cells and less than 5% confluent HEK293 cells. The transfected cells were incubated overnight with the final transfection mixture, after which the media were replaced. Two to 4 h before transfection, the media were replaced with 20 ml of new sterile growth media. Again, the next day, the media were replaced. For the HEK293 cells, the media were supplemented with 2 mg/ml G-418 to allow for clonal selection. Stable clones of HEK293 cells containing the mutant receptor were generated after several weeks of G-418-selective pressure and expanded for further use in radioligand binding and functional assays.
Membrane Preparation. Cell membranes were prepared by first detaching healthy cells with lifting buffer (Dulbecco's phosphate-buffered saline without Ca2+ and Mg2+; 5 mM EDTA) and then pelleting the detached cells by centrifugation in a sterile conical tube for 10 min at 700g. After centrifugation, the supernatant was decanted from the pellet. The pellet was then resuspended in 10 ml of lysis buffer (5 mM Tris, 5 mM MgCl2, pH 7.4 at 4°C) and allowed to lyse on ice for 5 to 10 min before transfer to an ice-cold Dounce homogenizer. Eight full strokes of the Dounce homogenizer were used to disrupt the whole cells by glass-on-glass homogenization. The resulting homogenate was poured into a centrifugation tube, balanced by the addition of cold binding buffer (50 mM Tris, pH 7.4 at 4°C), and then centrifuged at 28,000g for 45 min. The supernatant was decanted from the resulting membrane pellet. This membrane pellet was then resuspended in cold binding buffer and recentrifuged. The final membrane pellet was resuspended in an appropriate amount of cold binding buffer for the experiment, rehomogenized by four strokes in an ice-cold Dounce homogenizer, and then stored on ice for same day use.
Radioligand Binding Studies. Both receptor saturation experiments and radiolabeled competition assays were used to characterize the receptors in this study. In brief, the binding and wash buffers consist of 50 mM Tris, pH 7.4, at 25°C, with 1 N KOH used for the fine pH adjustment. For sodium shift assays, the binding and wash buffers were supplemented with 140 mM NaCl. The membrane density of the receptors and their affinity for the radioligand [3H]methylspiperone ([3H]MSP) was assessed by saturation isotherm analysis. For this type of assay, the cell membranes were allowed to equilibrate with increasing nanomolar concentrations of [3H]MSP in the presence or absence of 5 μM(+)-butaclamol, a dopamine receptor antagonist used to define the nonspecific interactions of [3H]MSP. After 90 min of equilibration at room temperature, the samples were rapidly filtrated and washed with ice-cold binding buffer (50 mM Tris, pH 7.4, at 0°C) through GF/C filters pretreated for 10 min with 0.3% polyethylenimine. The filters were allowed to dry before cutting them into vials. Vials were then filled with 3.5 ml of scintillation fluid and mixed before quantifying the amount of radioactivity in a scintillation counter. Radiolabeled competition assays were performed in a similar fashion to saturation assays, except that a fixed concentration of 0.5 nM [3H]MSP was utilized in conjunction with increasing concentrations of nonradiolabeled competitive ligand. Membrane protein concentration was determined by bicinchoninic acid assay (Pierce Chemical, Rockford, IL) according to the manufacturer's instructions.
cAMP Functional Assays. Intracellular cAMP concentrations were determined using a PerkinElmer Fusion plate analyzer and a cAMP Alphascreen detection kit (PerkinElmer Life and Analytical Sciences). The assay was performed essentially according to the manufacturer's specifications, except for adaptations we devised to measure cAMP levels in attached cells. In brief, HEK293 cells stably expressing mutant receptor were resuspended in sterile growth media (DMEM with 10% bovine calf serum, 1% penicillin/streptomycin, 100 μM sodium pyruvate) and then plated at a density of 50,000 cells/well in 96-well microtiter plates coated with poly-l-lysine (Sigma-Aldrich). The next morning, stimulation buffer (DMEM, 20 mM HEPES, 100 μM sodium metabisulfite, 30 μM Ro 20-1724, pH 7.4 at 25°C), cell lysis buffer (0.3% Tween 20, 20 mM HEPES, 1 μg/μl bovine serum albumin, pH 7.4 at 25°C), and bead buffer (20 mM HEPES, 30 μM Ro 20-1724, 1 μg/μl bovine serum albumin, 1× Hanks' balanced salt solution, pH 7.4 at 25°C) were freshly prepared and pH adjusted to 7.4 with 1 N cell culture tested sodium hydroxide (Sigma-Aldrich). To examine the Gi protein-mediated inhibition of adenylyl cyclase, the levels of cAMP were first raised with 6 μM forskolin, a direct stimulator of adenylyl cyclase. Drug dilutions were prepared in stimulation buffer, and 200 μl of dilution was added per well in an empty 96-well microtiter plate and allowed to equilibrate in the 37°C incubator for 30 min. Culture medium was removed from the cells and the temperature- and carbon dioxide-equilibrated drug dilutions were rapidly added to the cells using a multichannel pipet. Cells were then incubated the presence of the drug dilutions at 37°C for 20 min and then centrifuged at 1500g for 5 min. Drug dilutions were carefully removed by pipetting, and 100 μl of cell lysis buffer was added to each well. The cells were lysed by shaking at 600 rpm on a microtiter plate shaker for 1.5 h. A portion of the resulting lysate (30 μl) was transferred to an opaque 96-well Costar plate (catalog no. 07-200-309; Corning Inc., Corning, NY) and challenged with 0.5 units of acceptor and donor beads (9.35 and 12.5 μg/ml, respectively) containing 5 units of biotinylated cAMP (3.76 nM). Before reading, this reaction was allowed to equilibrate for 1 h with shaking at 600 rpm, protected from light with aluminum foil.
Calculations and Data Analysis. Data points for each experiment were sampled in triplicate, and each experiment repeated three times, except where noted. The geometric mean and S.D. are reported for each experiment; however, the errors in the graphs are S.E.M. values. The equilibrium dissociation constant (KD) of [3H]MSP was determined from saturation isotherm analysis. The inhibition constants (Ki) for all radioligand competition assays were calculated with the Cheng-Prusoff equation: Ki = IC50/(1 + [radioligand]/KD), where KD is the equilibrium dissociation constant of the radioligand. A K0.5 value is reported in cases where the Hill slope is significantly different from unity. All data were analyzed using Prism version 4.0 (GraphPad Software Inc., San Diego, CA). For the inhibition assays, data from three or more assays were combined and then interpreted by extrapolating all dose-response curves to zero to generate IC50 values. These were subsequently converted to Ki values before analysis by one-way ANOVA with a Dunnett's post hoc analysis. For saturation isotherm binding assays, specific binding curves were obtained by subtracting nonspecific binding [defined as binding in the presence of 5 μM (+)-butaclamol] from the total binding at each concentration of radioligand. Values for KD and Bmax were determined from the specific binding curve. Sodium shift binding assays were analyzed by comparing the receptor data with and without sodium in a paired two-tailed Student's t test. cAMP functional assays were assessed by first quantifying the amount of cAMP generated per milligram of protein in each sample and then normalizing this value as a percentage of the cAMP generated by unopposed 6 μM forskolin. Efficacy was determined by subtracting the best fit values for the bottom of the curve (lowest horizontal asymptote) from the top of the curve (highest horizontal asymptote). Functional assays are graphed as sigmoidal semilog dose-response curves. Statistical analyses of the curve fitting procedure included the run test, F test, and Pearson's correlation coefficient. Potency and efficacy values generated from three or more replicate curves were analyzed by one-way ANOVA with a Dunnett's post hoc analysis. Significance was established at the 95% confidence level (p ≤ 0.05).
Construction of Receptor Homology Models. A wild-type D2 receptor model was constructed using Modeler 9v1 (Sali and Blundell, 1993) simultaneously using as templates the (1GZM) structure of bovine rhodopsin (Li et al., 2004) and the recently determined structure of the β2 adrenergic receptor (Cherezov et al., 2007) (2RH1). Initially, 1000 D2 receptor structures were generated and ranked by Modeler's objective function. The models were then structurally aligned and clustered using the GROMACS version 3.3 package (van der Spoel et al., 2005) gcluster utility (cutoff = 0.20 Å). The most representative (central) structure from the best scoring cluster was selected for further analysis and mutated to V2.61F(91) before energy minimization runs.
Building Sodium-Bound D2 Receptor Models. Sodium-bound models were constructed with the sodium cation placed at the putative sodium binding pocket near D2.50(80) proposed by Neve et al. (2001). One negative control was constructed by positioning the sodium far from the TM region, at the intracellular carboxylate terminus (C415) where it would not affect the dynamic properties through direct interaction with the TM region. A second negative control used in this study was the sodium-free “null” system. All the structures were subjected to energy minimization runs in vacuo with the low-memory Broyden-Fletcher-Goldfarb-Shanno (quasi-Newtonian algorithm for energy minimization) method, in three stages, each carried out to convergence (Fmax < 10 kJ/mol/nm) using GROMACS version 3.3 (van der Spoel et al., 2005), with the molecular systems parameterized according to the optimized potentials for liquid simulations all-atom force field (Jorgensen et al., 1996). Electrostatic interactions were treated by the particle-mesh Ewald method (Essman et al., 1995). In the first stage, all heavy protein atoms were restrained with half-harmonic force restraints (k = 1000 kJ/mol/nm), with only the sodium ion unrestrained. In the second minimization stage, sodium and all side chains of residues within 6 Å of the sodium were unrestrained. Finally, sodium and all atoms of residues within 6 Å of the sodium position were unrestrained during minimization, with residues outside of this region restrained.
Normal Mode Analysis. To examine the effect of sodium binding on the dynamic properties of the receptor molecule, we used NMA, which determines a spectrum of independent harmonic (vibrational) motions available to a particular stable molecular conformation within a harmonic approximation (for further description, see Introduction). Our analysis focused on the lower frequency modes that comprise more facile motions (thus, higher amplitude) along directions that coincide with the more shallow curvatures along the potential well (Tama and Sanejouand, 2001) and have been shown to indicate function-related dynamics of proteins (Cui and Bahar, 2006). Each minimized structure was submitted to the NOMAD-Ref Web server for NMA (Lindahl et al., 2006). Elastic network models (Tirion, 1996) were built from Cα positions, with the sodium ion represented as an additional Cα at its optimized binding position. The first 106 normal modes (M1–106) were calculated for each elastic network model using default parameters, with the exception of the distance-weighting parameter in which a nondefault value of 3.0 Å was applied as recommended for Cα-only models.
To examine divergence in dynamical behavior between sodium-bound and
control (null) structures, dot products were computed for each sodium-bound
normal mode vector against all computed modes from the null system. A window
of the null spectrum was then chosen by centering it at the most analogous
mode 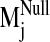 (highest
dot product with
(highest
dot product with
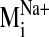 ). The dot
product squares were then summed over the selected null spectrum window to
provide a Pij value
(
). The dot
product squares were then summed over the selected null spectrum window to
provide a Pij value
(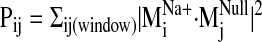 ).
A range of vectors from the control structures must be included because what
might appear as a unique sodium-bound mode can be recapitulated by a set of
such vectors, when combined. This overcomes a potential pitfall in comparative
NMA resulting from a comparison of only pairs of corresponding modes between
structures (Ming and Wall,
2005). Pij was then plotted as a function of
).
A range of vectors from the control structures must be included because what
might appear as a unique sodium-bound mode can be recapitulated by a set of
such vectors, when combined. This overcomes a potential pitfall in comparative
NMA resulting from a comparison of only pairs of corresponding modes between
structures (Ming and Wall,
2005). Pij was then plotted as a function of
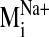 using various
window sizes to identify the difference in normal mode(s) between sodium-bound
structures and control.
using various
window sizes to identify the difference in normal mode(s) between sodium-bound
structures and control.
Ligand Docking into D2 Receptor Conformers from the
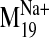 Trajectory. The
trajectory of the sodium-sensitive
Trajectory. The
trajectory of the sodium-sensitive
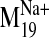 was selected to explore
effects of sodium-related motions on ligand docking because: 1) its
Pij value demonstrates significant divergence from the
conformational space of the other modes; 2) it was the lowest frequency mode
among those showing divergence and represents a softer and, thus, higher
amplitude collective domain motion; and 3) visual inspection revealed that as
part of its characteristic motion, an open phase appears to widen the binding
pocket, increasing accessibility (from the extracellular milieu) and its
volume in the V2.61(91)F mutant. Therefore, we constructed a series of
D2 receptor models representing points in the trajectory of this
particular mode by rebuilding all-atom structures on the Cα
frames from the M19 trajectory (output from NOMAD-Ref). Backbone
and side chain atoms were built onto the fixed Cα template
with Modeler 9v1, followed by minimization, a short 15-ps MD run, and
minimization with Cα atoms fixed for each procedure. The
structure was then minimized to convergence with positional restraints on the
Cα carbons in GROMACS, as described under Materials and
Methods.
was selected to explore
effects of sodium-related motions on ligand docking because: 1) its
Pij value demonstrates significant divergence from the
conformational space of the other modes; 2) it was the lowest frequency mode
among those showing divergence and represents a softer and, thus, higher
amplitude collective domain motion; and 3) visual inspection revealed that as
part of its characteristic motion, an open phase appears to widen the binding
pocket, increasing accessibility (from the extracellular milieu) and its
volume in the V2.61(91)F mutant. Therefore, we constructed a series of
D2 receptor models representing points in the trajectory of this
particular mode by rebuilding all-atom structures on the Cα
frames from the M19 trajectory (output from NOMAD-Ref). Backbone
and side chain atoms were built onto the fixed Cα template
with Modeler 9v1, followed by minimization, a short 15-ps MD run, and
minimization with Cα atoms fixed for each procedure. The
structure was then minimized to convergence with positional restraints on the
Cα carbons in GROMACS, as described under Materials and
Methods.
Ligands were constructed in Discovery Studio (Accelrys, San Diego, CA), and their geometries were optimized with ab initio quantum mechanical calculations using the HF6–31G** basis set in Gaussian03 (Frisch et al., 2004). The partial charges were set according to the AutoDockTools 1.4.5 automated Gasteiger partial charge assignment. Ligands were then docked 50 times into each M19-based D2 receptor frame using the Lamarckian genetic algorithm search routine in AutoDock 4.0 (Morris et al., 1998), with default parameters and a maximal number of energy evaluations of 4.0 × 106. Selective side chain flexibility was allowed within the docking routine; we explored the rotation of dihedrals in the side chains of F2.61(91), F3.28(110), V3.29(111), D3.32(114), W6.48(358), F6.51(361), and H6.55(375), chosen because of their apparently critical position in the ligand binding site. After docking into each receptor frame, poses were clustered using an RMSD tolerance of 3.0 Å, and clusters were ranked by mean energies.
Results
When transiently expressed in COS-7 cells, the D2-V2.61(91)F mutant receptor had a level of expression and an affinity for the moderately D2-selective 1,4-DAP [3H]MSP that was similar to the wild-type D2 receptor (1.1- and 4.1-fold, respectively) (Table 1; Fig. 2). It also had an affinity comparable with that of the wild-type receptor for the agonist (-)-quinpirole, which lacks a 1,4-DAP structural motif (1.2-fold, Table 1). The 1,4-DAP structural motif consists of two aromatic rings linked to positions 1 and 4 of a central six-membered piperidine or piperazine ring. L-745,870, for example, has two distinct aryl substituents that extend from positions 1 and 4 of the central piperazine ring directly and via a methylene spacer (Table 1). One of these ring nitrogens is likely protonated at physiological pH and interacts with the acidic pocket residue D3.32(114). For all ligands tested within this structure class, the D2-V2.61(91)F mutant had affinities very similar to those measured for the wild-type D2 receptor, including L-745,870 and six other D4-selective 1,4-DAPs (1.1–3.3-fold changes, Table 1). All of these 1,4-DAPs have been tested in previous binding studies designed to investigate molecular determinants of ligand selectivity for D4 receptors versus D2 receptor subtypes (Schetz et al., 2000; Kortagere et al., 2004), but only one of them (L-745,870 or CPPMA) had been tested on the D2-V2.61(91)F mutant, and it was reported to have a large improvement in binding affinity (Simpson et al., 1999).
TABLE 1.
Affinity of D4-selective ligands for the wild-type D2 and D2-V2.61(91)F mutant receptors expressed in COS-7 cells Affinities for the wild-type D4 receptor are shown for comparison. Affinity values (Ki or KD, nanomolar) are expressed as geometric averages of the mean of three experiments ± S.D. The fold changes in affinity values relative to the wild-type D2 (D2-WT) are shown in parentheses with the direction of the change indicated by arrows: ↑, increase in Ki value corresponding to a decreased affinity; and ↓, decrease in Ki value corresponding to an increased affinity.
| Ligand | Structure | D2-WT | D2-V2.61(91)F | D4-WT |
|---|---|---|---|---|
| L-745,870 (CPPMA) |  |
656 ± 227 (1) | 482 ± 251 (↓ 1.4) | 0.32 ± 0.14 (↓ 2050) |
| L-750,667c |  |
1400 ± 950 (1) | 1100 ± 510 (↓ 1.3) | 0.11 ± 0.02a (↓ 13,400) |
| RBI-257 | 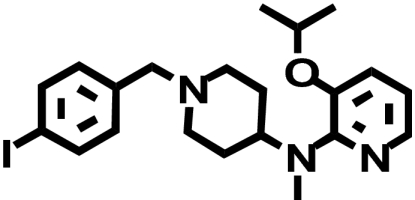 |
85 ± 12 (1) | 78 ± 3.3 (↑ 1.1) | 0.27 ± 0.10b (↓ 315) |
| FAUC213c |  |
1300 ± 640 (1) | 1200 ± 730 (↓ 1.1) | 1.1 ± 0.22b (↓ 1030) |
| Ro61-6270 | 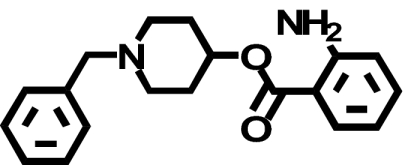 |
655 ± 274 (1) | 1121 ± 70* (↑ 1.7) | 0.89 ± 0.12b (↓ 736) |
| NGD 94-1 | 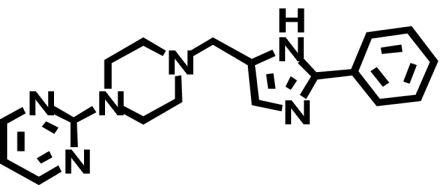 |
817 ± 284 (1) | 3358 ± 266* (↑ 3.3) | 0.3 ± 0.04b (↓ 2720) |
| PD168,077 | 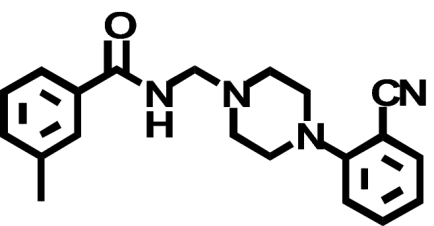 |
1380 ± 64 (1) | 3601 ± 474*(↑ 2.6) | 1.5 ± 0.41b (↓ 540) |
| (—)-Quinpirole | 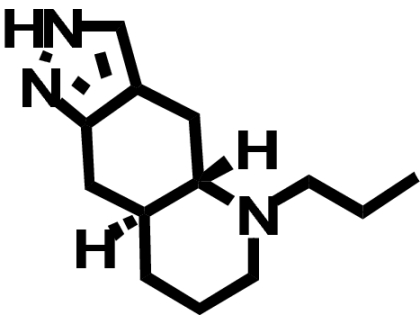 |
812 ± 617 (1) | 673 ± 466 (↓ 1.2) | N.D. |
| [3H]Methylspiperone | 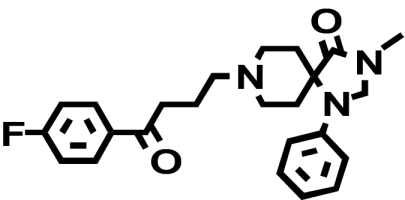 |
0.016 ± 0.0032 (1) | 0.066 ± 0.030* (↑ 4.1) | 0.29 ± 0.030a (↑ 18) |
Fig. 2.

L-745,870, L-750,667, and NGD 94-1 display sodium-sensitive binding to the D2-V2.61(91)F mutation. Shown are [3H]-methylspiperone competition binding experiments with L-745,870, L-750,667, or NGD 94-1 competing for wild-type D2 or D2-V2.61(91)F receptors stably expressed in HEK293 cells. The following graphs are composites of three or more parallel runs statistically examined by one-way ANOVA with Dunnett's post hoc analysis (*, p < 0.05). A, L-745,870 at the wild-type D2 receptor in the presence (•) and absence (○) of sodium. Sodium does not significantly increase the affinity for L-745,870 at the wild-type D2 receptor. B, L-745,870 at the D2-V2.61(91)F receptor in the presence (▪) and absence (□) of sodium. Sodium significantly increases (p ≤ 0.05, 37-fold increase) the affinity of L-745,870 for the D2-V2.61(91)F mutant receptor. C, L-750,667 at the wild-type D2 receptor in the presence (•) and absence (○) of sodium. In the absence of sodium, binding to the wild-type D2 receptor is negligible. In the presence of sodium, very weak binding is observed. D, L-750,667 at the D2-V2.61(91)F receptor in the presence (▪) and absence (□) of sodium. Only in the presence of sodium does L-750,667 have significant binding (p < 0.05, 35-fold increase). E, NGD 94-1 at the wild-type D2 receptor in the presence (•) and absence (○) of sodium. In the absence of sodium, the wild-type D2 receptor has negligible binding. In the presence of sodium, very weak binding is observed. F, NGD 94-1 at the D2-V2.61(91)F receptor in the presence (▪) and absence (□) of sodium. Only in the presence of sodium does NGD 94-1 have detectable binding (p < 0.05, greater than a 16-fold increase). All affinity values are given in Table 2.
In an effort to determine the source of the discrepancy between the reported 97-fold increase in affinity for L-745,870 for an N- and C-terminally epitope-tagged human D2-V2.61(91)F mutant (Simpson et al., 1999) and the lack of change for the identical mutation in the rat receptor for the same ligand (Table 1) and several other ligands belonging to the same structural class (Table 1; Floresca et al., 2005), we systematically eliminated differences between the two experimental systems. Initially, we expressed our rat D2-V2.61(91)F mutant in the same HEK293 cell line used in the previous report. Changing the cell background had only a moderate effect on the relative affinity measured for L-745,870 at the rD2-V2.61(91)F mutant versus the wild-type rD2 receptor that was difficult to accurately quantify because of low affinity and limited drug solubility under the conditions tested (∼5.5-fold increase based on estimates from extrapolated values, Fig. 2, A and B; Table 2).
TABLE 2.
The affinities of L-745,870, L-750,667, and NGD 94-1 for the D2-V2.61(91)F mutant receptor are significantly enhanced in the presence of 140 mM NaCl Receptors were stably expressed in the HEK293 cell line. Binding affinities (Ki) were calculated from the Cheng-Prusoff equation, Ki = IC50/(1+[radioligand]/KD, and expressed as geometric mean values (nanomolar) ± S.E.M. (n = 3). The D2-V2.61(91)F was run in three paired experiments, whereas wild-type D2 was run only in two paired experiments.
|
D2-WT*
|
D2-V2.61(91)F
|
|||
|---|---|---|---|---|
| No NaCl | 140 mM NaCl | No NaCl | 140 mM NaCl | |
| L-745,870 | 5232 ± 8419 (1) | 778 ± 89 (↓7) | 943 ± 328* (1) | 27.0 ± 4.34 (↓35) |
| L-750,667 | >10,000 (1) | 1331 ± 927 (>↓7) | 1271 ± 924* (1) | 34 ± 24 (↓37) |
| NGD 94-1 | >10,000 (1) | 3827 ± 3299 (>↓3) | >10,000* (1) | 616 ± 128* (>↓16) |
These are approximate values based upon extrapolation. The fold changes in affinity values relative to the receptor in the absence of sodium are shown in parentheses with the direction of the change indicated by arrows: ↓, decrease in Ki value corresponding to an increased affinity
The stable expression of these receptors in the HEK293 cell line also allowed us to study D2-like dopamine receptor cAMP functional responses. Although useful for the initial binding studies, transient expression in a COS-7 cell line lacked a suitable functional response in our assay system (data not shown). The HEK293 cell line lacks endogenous receptors for dopamine but can mediate a cAMP functional response for transfected dopamine receptors (data not shown). In preparation for functional assays, the cell surface density of dopamine receptors expressed in several stable HEK293 clones was determined with [3H]MSP saturation isotherm analysis, after which selected clones were matched by receptor density to avoid discrepancies because of spare receptors. HEK293 cell lines expressing 8.6 ± 3.4 pmol/mg protein of the wild-type D2 receptor and 11.6 ± 0.23 pmol/mg protein of the D2-V2.61(91)F receptor were selected for use in all subsequent experiments. The corresponding [3H]MSP affinity values (KD) were 74 ± 9.7 pM for the wild-type D2 receptor and 95 ± 18 pM for the D2-V2.61(91)F receptor. The absolute affinities of [3H]MSP for the D2-V2.61(91)F mutant receptor expressed in COS-7 and HEK293 cells were similar (1.4-fold different), although small differences were found for the wild-type receptor (4.6-fold different). For this same reason, the relative differences between wild-type and mutant D2 receptors within the same cell line are more pronounced in COS-7 than in HEK293 cells (4.1- versus 1.3-fold, respectively).
Our next step was to assess whether differences in the binding buffer could account for the large difference observed between our initial COS-7 binding data (Table 1) and published HEK293 binding data from Simpson et al. (1999). The striking finding was that the addition of 140 mM sodium chloride to the binding (and wash) buffer, to mimic the buffer conditions in the previous report (Simpson et al., 1999), resulted in a large (35-fold) increase in L-745,870 affinity for the rD2-V2.61(91)F mutant receptor (Fig. 2B; Table 2), although the wild-type D2 receptor displayed only a very limited sodium sensitivity for this ligand (∼7-fold, Fig. 2A; Table 2). These affinity values in the presence of sodium are consistent with those for hD2 wild-type and hD2-V2.61(91)F published in an earlier report (920 ± 200 and 9.5 ± 4.0 nM, respectively, Simpson et al., 1999). A similarly strong pattern of sodium-sensitive binding is evident for L-750,667, the iodinated derivative of L-745,870 (Fig. 2, C and D; Table 2); the affinity of L-750,667 for the rD2-V2.61(91)F mutant was increased 37-fold in the presence of 140 mM sodium, even though there is only a moderate sodium sensitivity for this ligand at the wild-type receptor (>∼7-fold). Another D4-selective 1,4-DAP, NGD 94-1, chosen because its structure is less similar to L-745,870 and L-750,667 (Table 1), still displayed comparably enhanced affinity for the D2-V2.61(91)F mutant in the presence of sodium (Fig. 2, E and F; Table 2) but only a very small sodium-dependent increase in affinity for the wild-type receptor (Fig. 2E; Table 1). Similar patterns of sodium sensitivity were also observed for several other D4-selective 1,4-DAPs and the sodium-sensitive substituted benzamide antagonist, (-)-raclopride (Fig. 3), and these patterns were not mimicked by the same concentration of N-methyl-d-glucamine, a sodium replacement ion (Fig. 3). Note that differences in (in the absence of sodium) affinities appear in Tables 1 and 2, but these are not comparable. The affinities for expressed wild-type and mutant D2 receptors in Table 1 are from membranes isolated from COS7 cells, whereas those in Table 2 are from membranes isolated from HEK293 cells.
Fig. 3.

Single-point competition measurements for displacement of [3H]MSP by various 1,4-DAPs. [3H]MSP-bound D2 receptors were incubated with various 1,4-DAPs in the presence of different buffer conditions. A, the presence of 140 mM sodium enhances the affinity of D2 receptor for 1,4-DAP ligands. B, the effect of sodium is greatly enhanced in the D2 V2.61(91)F mutant. As a control for ionic strength and nonspecific charge effects, N-methyl-d-glucamine was tested and displayed no significant changes from binding buffer alone. * means significantly different (P < 0.05) from both binding buffer and NMDG. ++ denotes significantly different (P < 0.05) from only NMDG. +++ denotes significantly different from only binding buffer.
In whole-cell attached functional assays, the full agonists dopamine and (-)-quinpirole both strongly reversed forskolin-stimulated increases in cAMP for both wild-type and D2-V2.61(91)F mutant receptors (Fig. 4A). Spiperone was able to fully reverse the (-)-quinpirole-stimulated inhibition of cAMP (Fig. 4B), but neither (-)-quinpirole nor L-750,667 were able to reduce forskolin-stimulated cAMP accumulation in untransfected HEK293 cells (data not shown).
Fig. 4.

L-750,667 has agonist activity at only the wild-type D2 receptor. A, agonists dopamine, (-)-quinpirole, and NGD 94-1 inhibit forskolin-stimulated cAMP response for wild-type D2 and D2-V2.61(91)F mutant receptors stably expressed in HEK293 cells, indicating that both receptors are functional. L-745,870 is an antagonist at both receptors, but L-750,667 displays (partial) agonist activity at only the D2 wild-type receptor. B, blockade of a low-dose (-)-quinpirole functional response by L-750,667 indicates that, like L-745,870 and spiperone, it is an antagonist at the D2-V2.61(91)F receptor. *, significantly different from forskolin alone for the wild-type D2 receptor; +, significantly different from forskolin alone for the D2-V2.61(91)F mutant receptor at p ≤ 0.05. For each experiment, the values are an average of triplicate determinations. The data are expressed as the geometric means from four separate experiments (n = 4) for all groups in 4A, except for dopamine, L-750,667, and NGD 94-1, where n = 3. For all groups in B, n = 3. B, all groups are statistically different from (-)-quinpirole alone at p ≤ 0.05.
It is notable that we found that L-750,667 has partial agonist properties at the wild-type D2 receptor (Fig. 4A) but acts as an antagonist at the D2-V2.61(91)F mutant receptor (Fig. 4B). However, the chlorinated derivative, L-745,870, has no agonist properties at either receptor, whereas NGD 94-1 has agonist properties at both receptors (Fig. 4A). Concentrations of L-745,870 five times higher than that shown in Fig. 4 did not change its functional profile (data not shown). Despite these drugs having enhanced affinity in the presence of a high concentration of sodium (Fig. 2), no attempt was made to measure such sodium effects in the functional assays.
Dynamic Properties of the Receptor Constructs. NMA performed on
molecular model constructs of the D2 receptor was used to examine
the dynamic response to sodium ion binding. In an effort to characterize
changes in the receptor's harmonic motions imparted by the perturbation of
sodium ion, we compared the spectrum of normal modes calculated for: 1) the
sodium-bound structure [near D2.50(80)], 2) a negative control with sodium ion
bound outside the protein core (at the carboxylate terminus), and 3) a system
without any sodium, hereafter referred to as the null system. To ascertain
which intrinsic low-frequency (and relatively high-amplitude) motions are
sensitive to the perturbation from sodium binding within the receptor, we
calculated the sum of dot product squares,
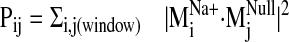 (see Fig. 5) for each
sodium-bound normal mode
(
(see Fig. 5) for each
sodium-bound normal mode
(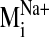 ) over ranges
(windows) of modes in the null spectrum
) over ranges
(windows) of modes in the null spectrum
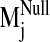 (for details,
see Materials and Methods). A plot of Pij as a function of
sodium-bound normal mode vectors
(
(for details,
see Materials and Methods). A plot of Pij as a function of
sodium-bound normal mode vectors
(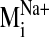 ) indicates that
sodium ion binding influences the characteristic movements of only a few
low-frequency modes. Pij values < 0.9 are indicative of motions
different from those described by the normal modes of the null system. Such
values were observed for modes 19, 26, 29, and 31 in the sodium-bound
structure, hereafter referred to as
) indicates that
sodium ion binding influences the characteristic movements of only a few
low-frequency modes. Pij values < 0.9 are indicative of motions
different from those described by the normal modes of the null system. Such
values were observed for modes 19, 26, 29, and 31 in the sodium-bound
structure, hereafter referred to as
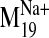 ,
,
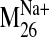 , and so forth. We
explored different window sizes (window = 7, 15, 21, and 106) in the null
spectrum to see whether divergent
, and so forth. We
explored different window sizes (window = 7, 15, 21, and 106) in the null
spectrum to see whether divergent
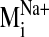 could be
recapitulated (Pij ∼ 1.0) by comparison against larger windows
of the null spectrum.
could be
recapitulated (Pij ∼ 1.0) by comparison against larger windows
of the null spectrum. 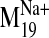 ,
however, consistently provided Pij values significantly lower than
the other
,
however, consistently provided Pij values significantly lower than
the other 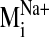 modes.
Pij calculations for the negative control spectrum
(
modes.
Pij calculations for the negative control spectrum
(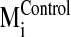 ) against
the null spectrum (not shown) do not yield uniquely low Pij values
comparable with
) against
the null spectrum (not shown) do not yield uniquely low Pij values
comparable with 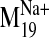 , which
also suggests that the
, which
also suggests that the 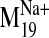 divergence is site specific in regards to sodium perturbation.
divergence is site specific in regards to sodium perturbation.
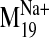 had no clearly
equivalent mode in the entire spectrum calculated for the null system. The
modes
had no clearly
equivalent mode in the entire spectrum calculated for the null system. The
modes 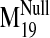 and
and
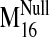 were most analogous,
with
were most analogous,
with
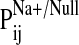 values of 0.25 and 0.36, respectively. Together, these findings suggest that
the different motion described by
values of 0.25 and 0.36, respectively. Together, these findings suggest that
the different motion described by
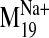 is because of the effect
of sodium binding on the dynamic properties of the receptor molecule.
is because of the effect
of sodium binding on the dynamic properties of the receptor molecule.
Fig. 5.

Divergence in sodium-bound normal modes
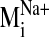 from the null
spectrum,
from the null
spectrum,  ,
calculated from the normal mode analysis of the control (no sodium in binding
site) system. Dot product squares were computed for each sodium-bound normal
mode vector against modes within a window of the null spectrum centered at the
most analogous mode
,
calculated from the normal mode analysis of the control (no sodium in binding
site) system. Dot product squares were computed for each sodium-bound normal
mode vector against modes within a window of the null spectrum centered at the
most analogous mode
 (highest dot
product with
(highest dot
product with  ).
Each dot product square is then summed over the entire null spectrum window to
provide a Pij value
(
).
Each dot product square is then summed over the entire null spectrum window to
provide a Pij value
(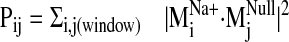 ).
Plotted here is Pij for each normal mode
).
Plotted here is Pij for each normal mode
 , calculated over
a window size of seven null modes. Among the lowest frequency nontrivial
normal modes (7–36, ranked in increasing frequency),
, calculated over
a window size of seven null modes. Among the lowest frequency nontrivial
normal modes (7–36, ranked in increasing frequency),
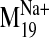 is the lowest frequency
and most divergent (lowest Pij) mode from the null system as it is
not reproduced by a set of normal modes from the null system. A similar trend
in Pij values is obtained regardless of the window size used in the
Pij calculation.
is the lowest frequency
and most divergent (lowest Pij) mode from the null system as it is
not reproduced by a set of normal modes from the null system. A similar trend
in Pij values is obtained regardless of the window size used in the
Pij calculation.
It is interesting that visual inspection of the
 trajectory shows that it
deforms the binding pocket and as a result facilitates the potential contact
of V2.61(91)F with the ligand (Fig. 6, A
and B). The trajectory describes a concerted TM2 kinking motion at
the proline kink (P2.59) along with the lateral TM3/TM4 motion away from the
cleft in the plane of the bilayer, which could potentially disrupt the TM2-TM3
interhelical packing to expose V2.61(91)F for interaction with the ligand.
Therefore, sodium induction of this particular mode must be considered
relevant to the observed enhancement of L-745,870 affinity in the
D2-V2.61(91)F mutant receptor.
trajectory shows that it
deforms the binding pocket and as a result facilitates the potential contact
of V2.61(91)F with the ligand (Fig. 6, A
and B). The trajectory describes a concerted TM2 kinking motion at
the proline kink (P2.59) along with the lateral TM3/TM4 motion away from the
cleft in the plane of the bilayer, which could potentially disrupt the TM2-TM3
interhelical packing to expose V2.61(91)F for interaction with the ligand.
Therefore, sodium induction of this particular mode must be considered
relevant to the observed enhancement of L-745,870 affinity in the
D2-V2.61(91)F mutant receptor.
Fig. 6.

The sodium-sensitive normal mode vector
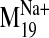 . A, motion along vector
. A, motion along vector
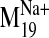 disrupts the hydrophobic
brace; red arrows, directionality of the open motion vectors for
Cα atoms (gray ribbon) in
disrupts the hydrophobic
brace; red arrows, directionality of the open motion vectors for
Cα atoms (gray ribbon) in
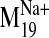 . The sodium ion in the
binding site is represented by a blue sphere. Green sticks, side chains of
V2.61(91)F and F3.28(110) at the TM2-TM3 interhelical junction (helices
identified by numbers). A small movement along the
. The sodium ion in the
binding site is represented by a blue sphere. Green sticks, side chains of
V2.61(91)F and F3.28(110) at the TM2-TM3 interhelical junction (helices
identified by numbers). A small movement along the
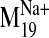 path from [0] (left) to
[+1] (middle) to [+2] (right) leads to disruption of interactions at this
TM2-TM3 junction. B, range of motions and flexibilities. Left and middle,
opening interval of
path from [0] (left) to
[+1] (middle) to [+2] (right) leads to disruption of interactions at this
TM2-TM3 junction. B, range of motions and flexibilities. Left and middle,
opening interval of 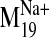 from
the initial (green = 0) to fully open conformation (red =+7). In this motion,
the kinking in TM2 becomes more pronounced as the intracellular segment of TM2
moves with TM3, whereas the extracellular segment moves with TM1. A
significant vertical movement (perpendicular to membrane plane) of TMs 6 and 7
upon opening pushes the extracellular loop 3 (e3) up and out, away from the
cleft; lateral motions (in the plane of the bilayer) of TMs 3 to 5 lead to a
clamping down of the e2 loop on the binding site
(Shi and Javitch, 2004). Note
that the sodium ion is fixed to reduce visual clutter. Right, extracellular
vantage with loop regions removed. The shearing motion at the extracellular
region of TMs 2 and 3 is apparent in the increased spacing between
Cα atoms of V2.61(91)F and F3.28(110) (spheres).
from
the initial (green = 0) to fully open conformation (red =+7). In this motion,
the kinking in TM2 becomes more pronounced as the intracellular segment of TM2
moves with TM3, whereas the extracellular segment moves with TM1. A
significant vertical movement (perpendicular to membrane plane) of TMs 6 and 7
upon opening pushes the extracellular loop 3 (e3) up and out, away from the
cleft; lateral motions (in the plane of the bilayer) of TMs 3 to 5 lead to a
clamping down of the e2 loop on the binding site
(Shi and Javitch, 2004). Note
that the sodium ion is fixed to reduce visual clutter. Right, extracellular
vantage with loop regions removed. The shearing motion at the extracellular
region of TMs 2 and 3 is apparent in the increased spacing between
Cα atoms of V2.61(91)F and F3.28(110) (spheres).
The dynamic response of the receptor to sodium binding, as measured by the
divergence between normal mode spectra from sodium-bound and null structures,
agrees with dynamic perturbation studies in other systems showing that
significant changes in conformational equilibrium can be triggered by what
would appear to be only small perturbations at “dynamical control
points” (Ming and Wall,
2005,
2006). In the D2
receptor, the allosteric site is likely coupled to instabilities near the
proline kink in TM2. Despite sequence divergence for the templates rhodopsin
and β2-adrenergic receptor in this region, the TM2s are
virtually superimposable. In the β2-adrenergic receptor, an
H-bond between W3.28 and the backbone carbonyl oxygen of V2.57 stabilizes the
Pro kink. In our homology model of the D2 receptor, the
corresponding interactions are missing because of differences in sequence, but
the dynamics of the TM2 region could still be coupled to the sodium binding
site near D2.50(80) via intrahelical H-bonding and/or local conformational
arrangements involving interhelical (TM2–3) side chain packing,
interhelical H-bonding, or structural waters near the sodium binding pocket.
To examine how dynamic flexibility expressed in the various receptor
conformations visited by the
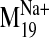 trajectory may influence
L-745,870 binding in the D2-V2.61(91)F mutant receptor, we
developed a docking protocol to explore a series of conformations determined
by this trajectory.
trajectory may influence
L-745,870 binding in the D2-V2.61(91)F mutant receptor, we
developed a docking protocol to explore a series of conformations determined
by this trajectory.
Ligand Docking Guided by Conformations from
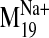 . Based on
preliminary results of L-745,870 docking into our initial model of the
D2 receptor (frame 0), the docking poses with the highest ranking
lacked the expected interactions: π-stacking between the
p-chlorophenyl moiety of L-745,870 and V2.61(91)F
(Fig. 6, A and B) and the
H-bond reinforcement of the ionic interaction between the piperazine ammonium
proton and D3.32(114). This is because in the “closed”
conformation of the D2 receptor model, V2.61(91)F is adjacent to
D2-F3.28(110), with which it can maintain stable interactions
(π-stacking or T-type interactions) that reduce the ability of the ligand
to interact with it. In addition, the cleft in which the protonatable amine of
the piperazine ring must fit to achieve this salt bridge is effectively
occluded by residues F3.28(110), V3.29(111), and F6.51(361). These
observations agree with our experimental data showing low affinity for
L-745,870 in wild-type D2 and D2-V2.61(91)F in the
absence of high concentrations of sodium.
. Based on
preliminary results of L-745,870 docking into our initial model of the
D2 receptor (frame 0), the docking poses with the highest ranking
lacked the expected interactions: π-stacking between the
p-chlorophenyl moiety of L-745,870 and V2.61(91)F
(Fig. 6, A and B) and the
H-bond reinforcement of the ionic interaction between the piperazine ammonium
proton and D3.32(114). This is because in the “closed”
conformation of the D2 receptor model, V2.61(91)F is adjacent to
D2-F3.28(110), with which it can maintain stable interactions
(π-stacking or T-type interactions) that reduce the ability of the ligand
to interact with it. In addition, the cleft in which the protonatable amine of
the piperazine ring must fit to achieve this salt bridge is effectively
occluded by residues F3.28(110), V3.29(111), and F6.51(361). These
observations agree with our experimental data showing low affinity for
L-745,870 in wild-type D2 and D2-V2.61(91)F in the
absence of high concentrations of sodium.
To explore the changes in ligand binding attributable to the dynamic
effects produced by sodium, we used the
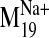 trajectory to construct
as described under Materials and Methods, 15 receptor conformers
(frames -7 to +7) representing the structure of the receptor along the motion
described by
trajectory to construct
as described under Materials and Methods, 15 receptor conformers
(frames -7 to +7) representing the structure of the receptor along the motion
described by 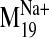 (Fig. 7A). Each of these
conformations was used to dock L-745,870. The protocol involves 50 separate
dockings into each of these conformations, and the resulting ligand poses
generated within each frame were binned into clusters based on similarity of
binding position and orientation (Fig.
7B). Receptor frames -1 and -2 in
Fig. 7A represent the closed
interval (-7 being maximally closed) (see
Fig. 6, A and B), and +1
through +7 represent the “opened” frames (+7 being maximally
opened) (Fig. 7B). Because the
amplitude of the
(Fig. 7A). Each of these
conformations was used to dock L-745,870. The protocol involves 50 separate
dockings into each of these conformations, and the resulting ligand poses
generated within each frame were binned into clusters based on similarity of
binding position and orientation (Fig.
7B). Receptor frames -1 and -2 in
Fig. 7A represent the closed
interval (-7 being maximally closed) (see
Fig. 6, A and B), and +1
through +7 represent the “opened” frames (+7 being maximally
opened) (Fig. 7B). Because the
amplitude of the 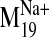 trajectory was set arbitrarily in the NMA calculation, we were most interested
in the smallest backbone movements away from the initial structure (±1,
2, 3) rather than the extremes (±7). Docking of L-745,870 into the
various frames of the receptor oscillating along the
trajectory was set arbitrarily in the NMA calculation, we were most interested
in the smallest backbone movements away from the initial structure (±1,
2, 3) rather than the extremes (±7). Docking of L-745,870 into the
various frames of the receptor oscillating along the
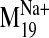 harmonic motion shows
that even a minimal excursion into the open phase of M19 better
accommodates the expected ligand binding poses (see
Kortagere et al., 2004) and,
hence, facilitates a direct interaction between either of the ligand's aryl
moieties and the phenyl ring in the V2.61(91)F mutant. Within these receptor
conformations visited by the
harmonic motion shows
that even a minimal excursion into the open phase of M19 better
accommodates the expected ligand binding poses (see
Kortagere et al., 2004) and,
hence, facilitates a direct interaction between either of the ligand's aryl
moieties and the phenyl ring in the V2.61(91)F mutant. Within these receptor
conformations visited by the
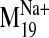 trajectory, L-745,870
can achieve reasonable binding geometries that are not available in the static
D2 receptor model. It is notable that proper ligand accommodation
in the open frames is because of the increase in the TM2 proline kink bend
angle and TM3 lateral translation and increased accessible depth of the
binding cleft that is noticeable in Fig.
7A (note the position of the gray regions that depict regions
favorable to ligand occupancy). As the receptor opens (frames +1 and +2), this
ligand occupancy region moves intracellularly into the vicinity of D3.32(114)
and W6.48(358). Thus, the movement provides increased space for the ligand to
access the entire binding cleft spanning from TM2,3,7 to TM3,5,6. Therefore,
while maintaining the previously identified contacts in the D2
receptor, L-745,870 can associate with a series of TM cleft-lining residues
including F2.61(91), L2.64(94), C3.25(107), F3.28(110), V3.29(111),
D3.32(114), V3.33(115), C3.36(118), F5.38(189), V5.39(190), S5.42(193),
F5.47(198), W6.48(358), F6.51(361), T7.39(386), G7.42(389), and Y7.43(390) and
potentially with e2 loop residues L143(171), E153(181), C154(182), I155(183),
and I156(184) (when the loop is replaced after docking). This enhancement of
the binding pocket by the effect of sodium binding on the dynamic properties
of the receptor makes some previously inaccessible binding sites available to
the ligand and results in the higher affinity observed in the presence of
sodium.
trajectory, L-745,870
can achieve reasonable binding geometries that are not available in the static
D2 receptor model. It is notable that proper ligand accommodation
in the open frames is because of the increase in the TM2 proline kink bend
angle and TM3 lateral translation and increased accessible depth of the
binding cleft that is noticeable in Fig.
7A (note the position of the gray regions that depict regions
favorable to ligand occupancy). As the receptor opens (frames +1 and +2), this
ligand occupancy region moves intracellularly into the vicinity of D3.32(114)
and W6.48(358). Thus, the movement provides increased space for the ligand to
access the entire binding cleft spanning from TM2,3,7 to TM3,5,6. Therefore,
while maintaining the previously identified contacts in the D2
receptor, L-745,870 can associate with a series of TM cleft-lining residues
including F2.61(91), L2.64(94), C3.25(107), F3.28(110), V3.29(111),
D3.32(114), V3.33(115), C3.36(118), F5.38(189), V5.39(190), S5.42(193),
F5.47(198), W6.48(358), F6.51(361), T7.39(386), G7.42(389), and Y7.43(390) and
potentially with e2 loop residues L143(171), E153(181), C154(182), I155(183),
and I156(184) (when the loop is replaced after docking). This enhancement of
the binding pocket by the effect of sodium binding on the dynamic properties
of the receptor makes some previously inaccessible binding sites available to
the ligand and results in the higher affinity observed in the presence of
sodium.
Fig. 7.

Docking of L-745,870 into D2 V2.61(91)F receptor conformers
visited by the 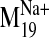 trajectory. A, frames shown from top to bottom correspond to closed (negative)
and open (positive) steps in the
trajectory. A, frames shown from top to bottom correspond to closed (negative)
and open (positive) steps in the
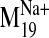 trajectory,
respectively. The D2 receptor structure is rendered as a ribbon,
and the TM segments are colored according to the rainbow spectrum: TM1, blue
to TM7, red. The transparent gray blob indicates the cumulative van der Waals
space occupied by the top five most favorable binding poses. Initial (0) and
closed frames -1 and -2 prevent ligand access for direct interactions with
V2.61(91)F/F3.28 and D3.32(114) and restrict occupancy to regions
extracellular of the presumed binding site. In contrast, in the frames
corresponding to the open phase of the harmonic motion of
trajectory,
respectively. The D2 receptor structure is rendered as a ribbon,
and the TM segments are colored according to the rainbow spectrum: TM1, blue
to TM7, red. The transparent gray blob indicates the cumulative van der Waals
space occupied by the top five most favorable binding poses. Initial (0) and
closed frames -1 and -2 prevent ligand access for direct interactions with
V2.61(91)F/F3.28 and D3.32(114) and restrict occupancy to regions
extracellular of the presumed binding site. In contrast, in the frames
corresponding to the open phase of the harmonic motion of
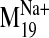 (frames +1, +2),
L-745,870 is accommodated in orientations that support experimentally
suggested interactions with V2.61(91)F, F3.28(110), and D3.32(114). Maroon
stick, L-745,870 bound in experimentally validated poses with the
p-chlorophenyl ring toward F2.61(91) and F3.28(110). B, histograms of
docking pose clusters. L-745,870 was docked into various conformers of the
D2 receptor based on the
(frames +1, +2),
L-745,870 is accommodated in orientations that support experimentally
suggested interactions with V2.61(91)F, F3.28(110), and D3.32(114). Maroon
stick, L-745,870 bound in experimentally validated poses with the
p-chlorophenyl ring toward F2.61(91) and F3.28(110). B, histograms of
docking pose clusters. L-745,870 was docked into various conformers of the
D2 receptor based on the
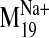 trajectory. Frame 0
represents the initial homology model structure. Frames -1 to -7 (top
histograms) represent various receptor conformers designated as closed, and
frames +1 to +7 (bottom histograms) represent open frames of the receptor. The
histogram collects results from 50 poses obtained from each independent
L-745,870 docking performed on each receptor conformation taken from the
trajectory and ranked with the AutoDock4 energy-based scoring function. The
poses are binned into clusters by similarity (3.0 Å RMSD), with the
vertical bar height indicating the number of cluster members (population).
Clusters in each docking run are also ranked most favorable to least favorable
(left to right) based on the lowest energy representative within each cluster.
Red bars highlight clusters that represent suitable binding conformations that
achieve the expected binding geometries defined for the 1,4-DAP class of
ligands docked to the D4 receptor
(Kortagere et al., 2004). Note
that proper binding geometries are only obtained in opened frames, indicating
the need for a conformation change in the D2 receptor to obtain the
high-affinity state.
trajectory. Frame 0
represents the initial homology model structure. Frames -1 to -7 (top
histograms) represent various receptor conformers designated as closed, and
frames +1 to +7 (bottom histograms) represent open frames of the receptor. The
histogram collects results from 50 poses obtained from each independent
L-745,870 docking performed on each receptor conformation taken from the
trajectory and ranked with the AutoDock4 energy-based scoring function. The
poses are binned into clusters by similarity (3.0 Å RMSD), with the
vertical bar height indicating the number of cluster members (population).
Clusters in each docking run are also ranked most favorable to least favorable
(left to right) based on the lowest energy representative within each cluster.
Red bars highlight clusters that represent suitable binding conformations that
achieve the expected binding geometries defined for the 1,4-DAP class of
ligands docked to the D4 receptor
(Kortagere et al., 2004). Note
that proper binding geometries are only obtained in opened frames, indicating
the need for a conformation change in the D2 receptor to obtain the
high-affinity state.
(-)-Raclopride was also docked into the wild-type D2 receptor
conformations visited by the
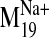 trajectory
(Fig. 8). We find that receptor
conformations generated along the opening path of this motion also increase
the likelihood of proper binding interactions with (-)-raclopride; an
H-bond-reinforced ionic interaction is achieved with D3.32, and the ligand's
phenyl substituent penetrates more deeply into the primary binding pocket
situated among TM 3, 5, and 6. These findings support our observation that
(-)-raclopride affinity for wild-type D2 receptor is enhanced in
the presence of sodium (Fig.
3).
trajectory
(Fig. 8). We find that receptor
conformations generated along the opening path of this motion also increase
the likelihood of proper binding interactions with (-)-raclopride; an
H-bond-reinforced ionic interaction is achieved with D3.32, and the ligand's
phenyl substituent penetrates more deeply into the primary binding pocket
situated among TM 3, 5, and 6. These findings support our observation that
(-)-raclopride affinity for wild-type D2 receptor is enhanced in
the presence of sodium (Fig.
3).
Fig. 8.

(-)-Raclopride docked into wild-type D2 receptor conformers
visited by the 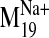 trajectory. A, initial (0) and closed frames (-1, -2) restrict (-)-raclopride
occupancy in the primary binding pocket (situated between TMs 4 and 6) that is
expected to accommodate the substituted benzamide ring moiety of
(-)-raclopride (Lan et al.,
2006). Although a few docking poses were observed in closed
conformers (-7 to -1) with the signature H-bond-reinforced ionic interaction
between the ligand's pyridyl ammonium group and the carboxylate side chain of
D3.32(114), the benzamide ring remained extracellular of the presumed primary
binding site, without deeper penetration into the primary cleft. In contrast,
the frames corresponding to the opened phase of the harmonic motion of
trajectory. A, initial (0) and closed frames (-1, -2) restrict (-)-raclopride
occupancy in the primary binding pocket (situated between TMs 4 and 6) that is
expected to accommodate the substituted benzamide ring moiety of
(-)-raclopride (Lan et al.,
2006). Although a few docking poses were observed in closed
conformers (-7 to -1) with the signature H-bond-reinforced ionic interaction
between the ligand's pyridyl ammonium group and the carboxylate side chain of
D3.32(114), the benzamide ring remained extracellular of the presumed primary
binding site, without deeper penetration into the primary cleft. In contrast,
the frames corresponding to the opened phase of the harmonic motion of
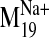 (frames +1, +2)
accommodate (-)-raclopride in orientations that satisfy the experimentally
suggested interactions, such as the H-bond-reinforced ionic interaction, an
H-bond interaction between the ligand's hydroxyl group and the side chain of
Y7.43(388) (not shown), and the deeper access of the benzamide ring into the
primary pocket (Lan et al.,
2006). B, histograms of docking poses for (-)-raclopride in
wild-type D2 receptor. (-)-Raclopride was docked into various
conformers of the D2 receptor based on the
(frames +1, +2)
accommodate (-)-raclopride in orientations that satisfy the experimentally
suggested interactions, such as the H-bond-reinforced ionic interaction, an
H-bond interaction between the ligand's hydroxyl group and the side chain of
Y7.43(388) (not shown), and the deeper access of the benzamide ring into the
primary pocket (Lan et al.,
2006). B, histograms of docking poses for (-)-raclopride in
wild-type D2 receptor. (-)-Raclopride was docked into various
conformers of the D2 receptor based on the
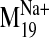 trajectory. Frame 0
represents the initial homology model structure. Frames -1 to -7 (top
histograms) represent various receptor conformers designated as closed, and
frames +1 to +7 (bottom histograms) represent opened frames of the receptor.
Fifty (-)-raclopride docking poses were obtained on each receptor conformation
taken from the trajectory and ranked with the AutoDock4 energy-based scoring
function. The poses are binned into clusters by similarity (3.0 Å RMSD),
with the vertical bar height indicating the number of cluster members
(population). Clusters in each docking run are also ranked most favorable to
least favorable (left to right) based on the lowest energy representative
within each cluster. Red bars highlight clusters that represent suitable
binding conformations that achieve the expected binding geometries defined for
the substituted benzamide class of ligands docked to the D2
receptor. Note that validated binding geometries are more frequently obtained
in opened frames (78 versus 22% in the closed frames), indicating a suitable
collective motion vector for conformation change in the D2 receptor
to obtain the high-affinity states for (-)-raclopride.
trajectory. Frame 0
represents the initial homology model structure. Frames -1 to -7 (top
histograms) represent various receptor conformers designated as closed, and
frames +1 to +7 (bottom histograms) represent opened frames of the receptor.
Fifty (-)-raclopride docking poses were obtained on each receptor conformation
taken from the trajectory and ranked with the AutoDock4 energy-based scoring
function. The poses are binned into clusters by similarity (3.0 Å RMSD),
with the vertical bar height indicating the number of cluster members
(population). Clusters in each docking run are also ranked most favorable to
least favorable (left to right) based on the lowest energy representative
within each cluster. Red bars highlight clusters that represent suitable
binding conformations that achieve the expected binding geometries defined for
the substituted benzamide class of ligands docked to the D2
receptor. Note that validated binding geometries are more frequently obtained
in opened frames (78 versus 22% in the closed frames), indicating a suitable
collective motion vector for conformation change in the D2 receptor
to obtain the high-affinity states for (-)-raclopride.
Discussion
Our finding that the affinity of L-745,870 for the rD2-V2.61(91)F mutant is drastically increased in the presence of high sodium concentration (140 mM) is significant for two main reasons. First, it explains the discrepancy (∼100-fold differences) in the reported affinities of L-745,870 and several other structurally similar D4-selective 1,4-DAPs for the D2-V2.61(91)F mutant and thus resolves an apparent contradiction in the literature (Simpson et al., 1999; Floresca et al., 2005). Second, it demonstrates how key molecular interactions between a ligand and a specific GPCR microdomain are influenced by occupancy of an allosteric site. In our case, the interactions that become accessible through the allosteric effect of sodium binding involve a π-stack or T-type interaction between the ligand's aryl moiety and F2.61(91) of the receptor as we proposed previously (Kortagere et al., 2004). However, we found that in the D2-V2.61(91)F receptor, this favorable interaction can occur only if an interhelical π-stack between the F2.61(91) and the adjacent F3.28(110), which forms a “hydrophobic brace,” is disrupted. We show that this disruption, achieved by sodium occupancy near D2.50(80), induces new dynamics that appear to widen the junction between TM2 and TM3 in the extracellular region of the receptor and thus disrupt the F2.61/F3.28 interaction. It is important that each of the 1,4-DAP compounds tested here (L-745,870, L-750,667, NGD 94-1, RBI-257, PD168,077, FAUC213, and Ro61-6270) is predicted from molecular models to engage in similar interactions when phenylalanine occupies position 2.61(91); in agreement, we found experimentally that all compounds tested display drastic sodium-dependent increases in affinity for the D2-V2.61(91)F mutant.
Although previous studies have revealed the affinity relationship between the discriminant structural features of 1,4-DAPs and the 1,4-DAP D4/D2 selectivity-conferring positions 2.61(91), 3.28(110), and 3.29(111), the effects of mutations in this microdomain on the activity of 1,4-DAP ligands had not been described previously (Simpson et al., 1999; Schetz et al., 2000; Kortagere et al., 2004; Floresca et al., 2005). Therefore, because the D2-V2.61(91)F receptor exhibits sodium-sensitive affinity changes to 1,4-DAPs, we examined here the functional properties of L-745,870, L-750,667, and NGD 94-1 at the wild-type and D2-V2.61(91)F receptor. In contrast to the reports showing reduction in agonist affinities as a result of allosteric modulation by sodium, we report here sodium-dependent enhancement of affinities for the 1,4-DAP agonists (and antagonists) similar to that observed for the substituted benzamide antagonists (Neve, 1991). Moreover, we report here that although the D2-V2.61(91)F mutant can be activated by dopamine and (-)-quinpirole, it cannot be activated by L-750,667, which exhibits weak partial agonist properties on the wild-type D2 receptor. Rather, L-750,667 acts as an antagonist of the D2-V2.61(91)F mutant. The relatively small influence of the D2-V2.61(91)F mutation on the binding affinity of (-)-quinpirole and methylspiperone demonstrated here suggests that neither ligand is likely to directly contact 2.61(91). Furthermore, this mutation had little effect on the activation of the receptor by (-)-quinpirole and its reversal by methylspiperone. This suggests that the 2.61(91)-3.28(110) hydrophobic brace can prevent the receptor from being activated by some agonists if their binding brings them near position 2.61(91). Because the affinities of other 1,4-DAPs, which retained their agonist properties at the D2-V2.61(91)F mutant receptor, were also enhanced in the presence of sodium, we reviewed the literature concerning the connection between agonist high- and low-affinity states, G protein coupling, and sensitivity to sodium and GTP.
In broken membrane preparations, high concentrations of either sodium ions (120 mM) or GTP and its related analogs (100 μM) decrease the affinity of agonists for catecholaminergic GPCRs (Paris et al., 1989; Neve, 1991; Neve et al., 2001; Schetz and Sibley, 2001). However, the similarities between the agonist affinity shift for sodium and GTP are coincidental because sodium binds an allosteric site on dopamine receptors accessible from the intracellular side (Neve, 1991; Neve et al., 2001; Schetz and Sibley, 2001), whereas GTP binds G proteins that then complex with the receptor at a different intracellular allosteric site. Under conditions (e.g., no GTP) in which a ternary complex is formed (receptor + G protein), kinetic studies of detergent-solubilized or broken membrane α adrenergic receptors demonstrate that both sodium ions and Gpp(NH)p accelerate the rate of agonist dissociation, and a synergistic increase in the rate is observed in the presence of both of these modulators (Limbird et al., 1982). In broken membrane equilibrium studies of α adrenergic receptors, agonist affinity decreases in the presence of either sodium or Gpp(NH)p, and a further decrease in affinity is observed in the presence of both of these modulators. These results were cited as evidence that sodium ions do not compete with GTP for its binding to G proteins (Limbird et al., 1982). Later studies by the same group demonstrated that substitution of the negative charge of the conserved aspartic acid in TM2 [D2.50(79)] to a neutral asparagine in the α2 adrenergic receptor resulted in a total loss of epinephrine's sensitivity to sodium (Horstman et al., 1990). When tested for its ability to stimulate GTPase activity, this same mutant receptor had a 7.5-fold decrease in potency for the agonist UK14304 with no change in efficacy (Ceresa and Limbird, 1994) and no change in affinity relative to the wild-type receptor. Increasing concentrations of Gpp(NH)p decrease the agonist p-[125I]iodoclonidine's binding to the wild-type but not the mutant receptor (Ceresa and Limbird, 1994). These data suggest that the sodium-insensitive D2.50(79)N mutant α adrenergic receptor can still couple to G proteins but that the state of coupling (coupled or uncoupled) no longer influences agonist high- and low-affinity states of the receptor. Thus, it would appear that, at least in the case of α2 adrenergic receptors, sodium ions control the agonist high- and low-affinity states of the receptor but do not uncouple G proteins from their receptor.
Broken membrane equilibrium studies demonstrate that dopamine's affinity for the D2 receptor is decreased when pertussis toxin or GTP uncouple G proteins from the receptor and that the addition of sodium ions further decreases dopamine's affinity (Neve et al., 1989). This was cited as evidence that sodium ions interact directly with dopamine receptors. Later studies by the same group demonstrated that substitution of the conserved aspartate in TM2 [D2.50(80)] for alanine results in a mutant receptor whose affinities for agonists and substituted benzamide antagonists are sodium insensitive (Neve et al., 1991); however, this same mutant receptor is unable to efficiently couple to G proteins, making it impossible to carry out the same types of studies as described above for α2 adrenergic receptors. Finally, the rate of chemical modification of D2 dopamine receptors by the thiolreactive agent N-ethylmaleimide is altered in the presence of sodium (Neve, 1991), and this is consistent with our finding [utilizing the D2-V2.61(91)F mutant receptor as a molecular probe of the 1,4-DAP binding pocket] that the binding of sodium ions to the allosteric site [D2-D2.50(80)] causes significant conformation changes in the receptor in the region of the orthosteric binding pocket.
The changes in dynamic properties of the receptor that can explain the observed pharmacological consequences of sodium binding are illustrated by our findings from NMA. Thus, the presence of sodium was found from this analysis to give rise to a unique, high-amplitude (low-frequency) normal mode that is not observed for the wild-type structure. Following the motions represented by this unique, sodium-related mode shows that it produces a wider cleft opening in the region responsible for ligand binding. These “opening dynamics” allow larger ligands to be accommodated in the pocket for ligand recognition and make room for better interaction of the ligand with the sites in the receptors at which it forms H-bonds, aromatic interactions, and hydrophobic matches. This leads to the measured improvement in the ligand's affinity for the receptor. Specifically, the effects of sodium binding near D2.50(80) on the dynamics in the microdomain surrounding the 2.61(91) position were shown by NMA to involve an increase in the scissors-like helical movements of the extracellular portions of TMs 2 and 3. This motion accounts for the enlarged opening and, consequently, for the large affinity changes observed at the D2-V2.61(91)F mutant, where these movements disrupt the hydrophobic brace formed by the interaction between V2.61(91)F and F3.28(110). The result of these local rearrangements is a receptor conformation more suitable for accommodating a ligand like L-745,870, which can now make stabilizing interactions as found in the optimal docking poses. Similar sodium-enhanced motions are predicted to occur in the wild-type receptor, which lacks a hydrophobic brace, but cleft opening would lead to better ligand access to the conserved aspartic acid at D3.32(114) for a reinforced ionic bond interaction with the protonatable piperizinyl amine portion of the docked 1,4-DAP. This prediction is supported experimentally by our findings for the wild-type D2 receptor whose affinities for L-745,870, L-750,667, and NGD 94-1 are somewhat increased (∼3–7-fold) in the presence of sodium. Moreover, the docking of the substituted benzamide (-)-raclopride, which belongs to a distinct structural class of D2 antagonists, into conformations along the sodium-induced normal mode produced very similar results. Again, suitable binding poses were more readily achieved in wild-type D2 receptor conformations visited along the open phase of the sodium-induced mode (Fig. 8), thus accounting for the enhanced affinity for (-)-raclopride (Fig. 3) and other substituted benzamides at the D2 receptor when modulated by sodium (Neve, 1991).
How these dynamic changes affect ligand binding is clearly demonstrated by
exploring the binding poses of L-745,870 in a series of receptor conformers
built along the characteristic vector of
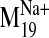 . It is notable that
involving NMA in a docking protocol to provide a basis for exploring
collective backbone movements in ligand docking has also been used
successfully to reproduce known conformations from ligand-bound crystal
structures (Lindahl and Delarue,
2005). Here, we found that docking into conformations generated
from the open interval of
. It is notable that
involving NMA in a docking protocol to provide a basis for exploring
collective backbone movements in ligand docking has also been used
successfully to reproduce known conformations from ligand-bound crystal
structures (Lindahl and Delarue,
2005). Here, we found that docking into conformations generated
from the open interval of
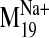 accommodates L-745,870
in the expected binding modes for 1,4-DAPs in D4 dopamine receptors
(Kortagere et al., 2004)
because the opening motion disrupts contact between F2.61(91) and F3.28(110),
which in turn creates access to these residues that appeared inaccessible to
the p-chlorophenyl (“mode 1”) or pyrrolopyridinyl group
(“mode 2”) of L-745,870 in the initial D2 receptor
homology model. The access is established by the dynamic changes and enables
favorable aromatic-aromatic interactions between the ligand moieties and the
receptor sites. This result suggests, therefore, that the specific
perturbation propagated from the sodium binding site influences ligand
affinity by dynamically changing the interactions in the orthosteric site. The
backbone movement observed in our model might capture the phenomenon of sodium
allostery by achieving the high-affinity receptor conformational state along
the
accommodates L-745,870
in the expected binding modes for 1,4-DAPs in D4 dopamine receptors
(Kortagere et al., 2004)
because the opening motion disrupts contact between F2.61(91) and F3.28(110),
which in turn creates access to these residues that appeared inaccessible to
the p-chlorophenyl (“mode 1”) or pyrrolopyridinyl group
(“mode 2”) of L-745,870 in the initial D2 receptor
homology model. The access is established by the dynamic changes and enables
favorable aromatic-aromatic interactions between the ligand moieties and the
receptor sites. This result suggests, therefore, that the specific
perturbation propagated from the sodium binding site influences ligand
affinity by dynamically changing the interactions in the orthosteric site. The
backbone movement observed in our model might capture the phenomenon of sodium
allostery by achieving the high-affinity receptor conformational state along
the 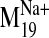 vector path and/or
by disrupting the brace that stabilizes the low-affinity state without a
significant, time-averaged, conformational change, i.e., through the
sodium-induced amplitude increase along this vector
(Cooper and Dryden, 1984;
Popovych et al., 2006). In any
case, the presence and effect of endogenous allosteric modulators (e.g.,
sodium, G protein, GPCR dimer partner, etc.) should be considered when
selecting a receptor representation for in silico library screening. It is
also important to point out that the use of target receptor conformations
based on normal mode trajectories could inform the design of drugs working
through allosteric mechanisms.
vector path and/or
by disrupting the brace that stabilizes the low-affinity state without a
significant, time-averaged, conformational change, i.e., through the
sodium-induced amplitude increase along this vector
(Cooper and Dryden, 1984;
Popovych et al., 2006). In any
case, the presence and effect of endogenous allosteric modulators (e.g.,
sodium, G protein, GPCR dimer partner, etc.) should be considered when
selecting a receptor representation for in silico library screening. It is
also important to point out that the use of target receptor conformations
based on normal mode trajectories could inform the design of drugs working
through allosteric mechanisms.
Acknowledgments
We thank Dr. Christina Z. Floresca and Shiuwei Chen for assistance with early phases of this work and the following people and their organizations for supplying samples of compounds that were not commercially available: Dr. Claus Riemer (Hoffman-La Roche, Nutley, NJ) for Ro 62-6170 and Dr. Andrew Thurkauf (Neurogen Corporation, Branford, CT) for NGD 94-1. In addition, we thank Xavier Rovira for providing NMA analysis scripts.
This study was supported by the National Institutes of Health [Grant R01-MH063162], the Cofrin Center for Biomedical Information in the HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine at Weill Medical College of Cornell University, and National Institutes of Health, National Institute on Drug Abuse [Postdoctoral Training Grant T32 DA007274].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.108.141531.
ABBREVIATIONS: 1,4-DAP, 1,4-disubstituted aromatic piperidine/piperazine; NMA, normal mode analysis; NET-856, [3H]methylspiperone; (-)-quinpirole, (4aR-trans)-4,4a,5,6,7,8,8a,9-octahydro-5-propyl-1H-pyrazolo[3,4-g]quinoline; TMS, transmembrane segment; HEK, human embryonic kidney; DMEM, Dulbecco's modified Eagle's media; MSP, methylspiperone, 8-[4-(4-fluorophenyl)-4-oxobutyl]-(3-methyl-1-phenyl)-1,3,8-triazaspiro[4,5]decan-4-one hydrochloride; Ro 20-1724, 4-[(3-butoxy-4-methoxyphenyl)-methyl]-2-imidazolidinone; ANOVA, analysis of variance; GROMACS, Groningen machine for chemical simulations; TM, transmembrane; NOMAD-Ref, normal mode analysis deformation and refinement; RMSD, root mean square deviation; L-745,870, 3-{[4-(4-chlorophenyl) piperazin-1-yl]methyl}-1H-pyrrolo[2,3-b]pyridine or CPPMA, chlorophenylpiperazinyl methylazaindole; L-750,667, 3-{[4-(4-iodophenyl) piperazin-1-yl]methyl}-1H-pyrrolo[2,3-b]pyridine; NGD 94-1, 2-phenyl-4(5)-[4–92-pyrimidinyl)-piperazin-1-yl)-methyl]-imidazole; (-)-raclopride, 3,5-dichloro-N-(1-ethylpyrrolidin-2-ylmethyl)-2-hydroxy-6-methoxybenzamide; RBI-257, 1-[4-iodobenzyl]-4-[N-(3-isopropoxy-2-pyridinyl)-N-methyl]-aminopiperidine; PD168,077, N-[[4-(2-cyanophenyl)-1-piperazinyl]methyl]-3-methylbenzamide; FAUC213, 2-[4-(4-chlorophenyl)piperazin-1-ylmethyl]pyrazolo[1,5-a]pyridine; Gpp(NH)p, 5′-guanylylimidodiphosphate; Ro61-6270, 2-amino-benzoic acid 1-benzyl-piperidin-4-yl ester; UK14304, 5-bromo-N-(4,5-dihydro-1H-imidazol-2-yl)-6-quinoxalinamine; p-iodoclonidine, 2-[(2,6-dichloro-4-iodophenyl)imino]imidazoline.
References
- Ballesteros JA and Weinstein H (1995) Integrated methods for modeling G-protein coupled receptors. Methods Neurosci 25 366-428. [Google Scholar]
- Ceresa BP and Limbird LE (1994) Mutation of an aspartate residue highly conserved among G-protein-coupled receptors results in nonreciprocal disruption of alpha 2-adrenergic receptor-G-protein interactions: A negative charge at amino acid residue 79 forecasts alpha 2A-adrenergic receptor sensitivity to allosteric modulation by monovalent cations and fully effective receptor/G-protein coupling. J Biol Chem 269 29557-29564. [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318 1258-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A and Dryden DT (1984) Allostery without conformational change: A plausible model. Eur Biophys J 11 103-109. [DOI] [PubMed] [Google Scholar]
- Cui Q and Bahar I (2006) Normal Mode Analysis: Theory and Applications to Biological and Chemical Systems, Chapman & Hall/CRC Press, New York.
- Essman U, Perela L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103 8577-8592. [Google Scholar]
- Floresca CZ and Schetz JA (2004) Dopamine receptor microdomains involved in molecular recognition and the regulation of drug affinity and function. J Recept Signal Transduct Res 24 207-239. [DOI] [PubMed] [Google Scholar]
- Floresca CZ, Chen S, Kortagere S, and Schetz JA (2005) Reciprocal mutations in TM2/TM3 in a D2 dopamine receptor background confirms the importance of this microdomain as a selective determinant of para-halogenated 1,4-disubstituted aromatic piperazines. Arch Pharm (Weinheim) 338 268-275. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, et al. (2004) Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT.
- Horstman DA, Brandon S, Wilson AL, Guyer CA, Cragoe EJ Jr, and Limbird LE (1990) An aspartate conserved among G-protein receptors confers allosteric regulation of alpha 2-adrenergic receptors by sodium. J Biol Chem 265 21590-21595. [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, and Tirado-Rives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118 11225-11236. [Google Scholar]
- Kortagere S, Gmeiner P, Weinstein H, and Schetz JA (2004) Certain 1,4-disubstituted aromatic piperidines and piperazines with extreme selectivity for the dopamine D4 receptor interact with a common receptor microdomain. Mol Pharmacol 66 1491-1499. [DOI] [PubMed] [Google Scholar]
- Lan H, DuRand CJ, Teeter MM, and Neve KA (2006) Structural determinants of pharmacological specificity between D1 and D2 dopamine receptors. Mol Pharmacol 69 185-194. [DOI] [PubMed] [Google Scholar]
- Li J, Edwards PC, Burghammer M, Villa C, and Schertler GF (2004) Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 343 1409-1438. [DOI] [PubMed] [Google Scholar]
- Limbird LE, Speck JL, and Smith SK (1982) Sodium ion modulates agonist and antagonist interactions with the human platelet alpha 2-adrenergic receptor in membrane and solubilized preparations. Mol Pharmacol 21 609-617. [PubMed] [Google Scholar]
- Lindahl E, Azuara C, Koehl P, and Delarue M (2006) NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res 34 W52-W56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E and Delarue M (2005) Refinement of docked protein-ligand and protein-DNA structures using low frequency normal mode amplitude optimization. Nucleic Acids Res 33 4496-4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming D and Wall ME (2005) Quantifying allosteric effects in proteins. Proteins 59 697-707. [DOI] [PubMed] [Google Scholar]
- Ming D and Wall ME (2006) Interactions in native binding sites cause a large change in protein dynamics. J Mol Biol 358 213-223. [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, and Olson AJ (1998) Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J Comp Chem 19 1639-1662. [Google Scholar]
- Neve KA (1991) Regulation of dopamine D2 receptors by sodium and pH. Mol Pharmacol 39 570-578. [PubMed] [Google Scholar]
- Neve KA, Cox BA, Henningsen RA, Spanoyannis A, and Neve RL (1991) Pivotal role for aspartate-80 in the regulation of dopamine D2 receptor affinity for drugs and inhibition of adenylyl cyclase. Mol Pharmacol 39 733-739. [PubMed] [Google Scholar]
- Neve KA, Cumbay MG, Thompson KR, Yang R, Buck DC, Watts VJ, DuRand CJ, and Teeter MM (2001) Modeling and mutational analysis of a putative sodium-binding pocket on the dopamine D2 receptor. Mol Pharmacol 60 373-381. [DOI] [PubMed] [Google Scholar]
- Neve KA, Henningsen RA, Bunzow JR, and Civelli O (1989) Functional characterization of a rat dopamine D-2 receptor cDNA expressed in a mammalian cell line. Mol Pharmacol 36 446-451. [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Tronq I, Teller DC, Okada T, Stenkamp RE, et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 298 739-745. [DOI] [PubMed] [Google Scholar]
- Paris H, Galitzky J, and Senard JM (1989) Interactions of full and partial agonists with HT29 cell alpha 2-adrenoceptor: comparative study of [3H]UK-14,304 and [3H]clonidine binding. Mol Pharmacol 35 345-354. [PubMed] [Google Scholar]
- Popovych N, Sun S, Ebright RH, and Kalodimos CG (2006) Dynamically driven protein allostery. Nat Struct Mol Biol 13 831-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A and Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234 779-815. [DOI] [PubMed] [Google Scholar]
- Schetz JA (2005) Allosteric modulation of dopamine receptors. Mini Rev Med Chem 5 555-561. [DOI] [PubMed] [Google Scholar]
- Schetz JA, Benjamin PS, and Sibley DR (2000) Nonconserved residues in the second transmembrane-spanning domain of the D(4) dopamine receptor are molecular determinants of D(4)-selective pharmacology. Mol Pharmacol 57 144-152. [PubMed] [Google Scholar]
- Schetz JA, Chu A, and Sibley DR (1999) Zinc modulates antagonist interactions with D2-like dopamine receptors through distinct molecular mechanisms. J Pharmacol Exp Ther 289 956-964. [PubMed] [Google Scholar]
- Schetz JA and Sibley DR (2001) The binding-site crevice of the D4 dopamine receptor is coupled to three distinct sites of allosteric modulation. J Pharmacol Exp Ther 296 359-363. [PubMed] [Google Scholar]
- Shi L and Javitch JA (2004) The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A 101 440-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MM, Ballesteros JA, Chiappa V, Chen J, Suehiro M, Hartman DS, Godel T, Snyder LA, Sakmar TP, and Javitch JA (1999) Dopamine D4/D2 receptor selectivity is determined by a divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol Pharmacol 56 1116-1126. [DOI] [PubMed] [Google Scholar]
- Tama F and Sanejouand YH (2001) Conformational change of proteins arising from normal mode calculations. Protein Eng 14 1-6. [DOI] [PubMed] [Google Scholar]
- Tirion MM (1996) Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett 77 1905-1908. [DOI] [PubMed] [Google Scholar]
- van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, and Berendsen HJC (2005) GROMACS: fast, flexible, and free. J Comp Chem 26 1701-1718. [DOI] [PubMed] [Google Scholar]
- Vivo M, Lin H, and Strange PG (2006) Investigation of cooperativity in the binding of ligands to the D2 dopamine receptor. Mol Pharmacol 69 226-235. [DOI] [PubMed] [Google Scholar]