

Abstract
Chronic cocaine exposure is associated with severe cardiac complications,
but the mechanisms of cocaine cardiotoxicity remain unclear, and current
therapies are unsatisfactory. We investigated the hypothesis of oxidative
stress-mediated cardiotoxicity and the role of NADPH oxidase in this process
in a mouse model of chronic escalating “binge” cocaine
administration (milligrams per kilogram): days 1 to 4 at 3 × 15 mg, days
5 to 8 at 3 × 20 mg, days 9 to 12 at 3 × 25 mg, and days 13 to 14
at 3 × 30 mg. Compared with vehicle controls, chronic binge cocaine
administration significantly increased the cardiac NADPH-dependent
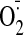 production (1.96- ± 0.4-fold) as detected by tiron (an
production (1.96- ± 0.4-fold) as detected by tiron (an
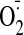 scavenger)-inhibitable lucigenin chemiluminescence and dihydroethidium
fluorescence. Cocaine-induced reactive oxygen species (ROS) production was
associated with significant increases (∼2-fold) in the protein expressions
of Nox2 (an isoform of NADPH oxidase) and its regulatory subunits:
p22phox, p67phox, p47phox,
p40phox, and Rac1, and in p47phox phosphorylation as
detected by immunoblotting (all p < 0.03). Increased Nox2 activity was
accompanied by the activation of extracellular signal-regulated kinase 1/2,
p38 mitogen-activated protein kinase (MAPK), and c-Jun NH2-terminal
kinase, notably in the cardiomyocytes. Cell culture experiments revealed that
cocaine-induced ROS production was primarily a direct action of cocaine on
cardiac myocytes, which caused severe oxidative damage to myocytes and cell
death as detected by terminal deoxynucleotidyl transferase dUTP nick-end
labeling assay. These could be inhibited by inhibitors to protein kinase C
(bisindolymaleimide) or by depletion of Nox2 using small interfering RNA. In
conclusion, chronic cocaine administration directly causes severe myocardial
oxidative stress through the activation of Nox2 oxidase. Increased ROS
production contributes to MAPK activation and the subsequent myocyte damage.
Inhibitors to NADPH oxidase or antioxidants may have therapeutic potential in
the treatment of cocaine cardiotoxicity.
scavenger)-inhibitable lucigenin chemiluminescence and dihydroethidium
fluorescence. Cocaine-induced reactive oxygen species (ROS) production was
associated with significant increases (∼2-fold) in the protein expressions
of Nox2 (an isoform of NADPH oxidase) and its regulatory subunits:
p22phox, p67phox, p47phox,
p40phox, and Rac1, and in p47phox phosphorylation as
detected by immunoblotting (all p < 0.03). Increased Nox2 activity was
accompanied by the activation of extracellular signal-regulated kinase 1/2,
p38 mitogen-activated protein kinase (MAPK), and c-Jun NH2-terminal
kinase, notably in the cardiomyocytes. Cell culture experiments revealed that
cocaine-induced ROS production was primarily a direct action of cocaine on
cardiac myocytes, which caused severe oxidative damage to myocytes and cell
death as detected by terminal deoxynucleotidyl transferase dUTP nick-end
labeling assay. These could be inhibited by inhibitors to protein kinase C
(bisindolymaleimide) or by depletion of Nox2 using small interfering RNA. In
conclusion, chronic cocaine administration directly causes severe myocardial
oxidative stress through the activation of Nox2 oxidase. Increased ROS
production contributes to MAPK activation and the subsequent myocyte damage.
Inhibitors to NADPH oxidase or antioxidants may have therapeutic potential in
the treatment of cocaine cardiotoxicity.
Cocaine is one of the most common illicitly used drugs in the world and causes the most frequent drug-related deaths in young adults (<40 years) (Vasica and Tennant, 2002; Darke et al., 2006; Afonso et al., 2007). Chronic cocaine consumption is associated with serious cardiovascular complications such as hypertension, cardiac hypertrophy, and sudden death (Vasica and Tennant, 2002; Darke et al., 2006; Afonso et al., 2007). Conventionally, cocaine cardiotoxicity has been thought to be mediated indirectly through its sympathomimetic effect, i.e., by inhibiting the reuptake and increasing the levels of neuronal catecholamines to work on adrenoceptors (Afonso et al., 2007). However, clinical therapies with adrenoceptor antagonists are problematic and not always effective (Vasica and Tennant, 2002; Afonso et al., 2007).
It has been discovered recently that cocaine administration is associated with severe oxidative stress in the heart (Moritz et al., 2003; Pacifici et al., 2003; Kovacic, 2005; Ren et al., 2006; Isabelle et al., 2007). Although there are several potential enzymatic sources of ROS existing in the heart, in an animal model of chronic cocaine administration, treatment with apocynin (an NADPH oxidase inhibitor) has been reported to be effective in reducing ROS generation and to restore the cardiac output, stroke volume, and fractional shortening, suggestive of the involvement of an NADPH oxidase (Isabelle et al., 2007). However, apocynin has been reported recently to be not a selective NADPH oxidase inhibitor but an antioxidant (Heumüller et al., 2008). Therefore, the enzymatic sources of cocaine-induced cardiac oxidative stress remain to be elucidated.
NADPH oxidase comprises a cytochrome b, which can be further divided into one catalytic subunit (a member of the Nox family) and one p22phox and at least four regulatory subunits (p47phox, p67phox, p40phox, and rac1). To date, five members of the Nox family have been identified (Nox1–5), each encoded by a separate gene with a different function (Sumimoto et al., 2005). Cardiac tissue expresses both Nox2 and Nox4 (Byrne et al., 2003; Ribé et al., 2008).
In this study, we used an experimental mouse model of chronic escalating dose “binge” cocaine administration to mimic the common pattern of human repeated cocaine consumption, where steady increases in drug dose are required to achieve the desired drug effect because of an increase in cocaine tolerance. We have investigated: 1) the effect of chronic cocaine administration on cardiac ROS production and the role of Nox2 and Nox4 in this process and 2) the mechanisms and the downstream signaling pathways of cocaine-induced NADPH oxidase activation. We have also examined the direct effect of cocaine on Nox2 activation and subsequent mitogen-activated protein kinase (MAPK) activation in cultured cardiac myocytes.
Materials and Methods
Reagents. Dihydroethidium (DHE) was purchased from Invitrogen (Carlsbad, CA). Polyclonal antibodies against p22phox, Nox4, p40phox, p47phox, p67phox, rac1, and cardiac-troponin I were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal antibodies against a 30-amino acid C-terminal fragment of Nox2 (Pep37), were kindly provided by Dr. F. Wientjes (University College London, UK) (Li and Shah, 2002). Antibodies to phospho-ERK1/2, phospho-p38 MAPK, phospho-JNK, and phosphoserine were from Cell Signaling Technology Inc. (Danvers, MA). Bisindolymaleimide (Bis) was from Calbiochem (San Diego, CA). All other chemicals were from Sigma Chemical (Poole, Dorset, UK), unless stated otherwise.
Animal Model of Chronic Escalating Dose Binge Cocaine Consumption. A
mouse model (C57BL/6J, male, 5 weeks old) of chronic escalating dose binge
cocaine administration was generated as described previously
(Bailey et al., 2007). All
studies were performed in accordance with protocols approved by the Home
Office under the Animals (Scientific Procedures) Act 1986 UK. The control
group was given intraperitoneal injections of saline (10 ml/kg), and the
cocaine group was given cocaine dissolved in saline (10 ml/kg) three times
daily at 1-h intervals. The first injection was given ∼1 h after the
lights were switched on. The cocaine doses were escalated in a pattern of 3
× 15 mg/kg for days 1 to 4, 3 × 20 mg/kg for days 5 to 8, 3
× 25 mg/kg for days 9 to 12, and 3 × 30 mg/kg for days 13 to 14 to
mimic a common pattern of human cocaine abuse. Animals were killed 30 min
after the last injection. Left ventricular tissues were used for measuring
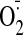 generation, immunoblotting, and immunocytochemistry. Twelve mice from each
group were used for the study.
generation, immunoblotting, and immunocytochemistry. Twelve mice from each
group were used for the study.
Cardiac Myocyte Culture, Nox2 siRNA Transfection, and Cocaine Stimulation. The rat cardiac myocyte cell line (H9C2) was obtained from the American Type Culture Collection (Manassas, VA) and grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. For the stimulation with cocaine, cells were seeded the day before experiment at 8 × 104/ml to achieve ∼90% confluence and stimulated with cocaine (0–100 μM) in culture medium for 24 h in the absence or presence of a pan-PKC inhibitor, Bis (10 μM), or tiron (2.5 mM). Cells were washed three times with PBS and detached by scraping and snap-freezing in liquid nitrogen. In some experiments, cells were transfected with Nox2 siRNA or a random negative control siRNA with the nucleotide sequence exactly as described previously (Hingtgen et al., 2006) using Lipofectamine 2000 plus (Invitrogen, Paisley, UK) as transfection reagent (Li et al., 2007). Forty-eight hours after the transfection, cells were stimulated with cocaine (25 μM) for 24 h.
Measurement of ROS Production.
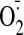 production by tissue or cell homogenate was measured using lucigenin (5
μM)-chemiluminescence (BMG Labtech GmbH, Offenburg, Germany)
(Ribé et al., 2008).
The specificity of the assay was confirmed by adding superoxide dismutase (100
units/ml) or tiron (10 mM). Other enzymatic sources of
production by tissue or cell homogenate was measured using lucigenin (5
μM)-chemiluminescence (BMG Labtech GmbH, Offenburg, Germany)
(Ribé et al., 2008).
The specificity of the assay was confirmed by adding superoxide dismutase (100
units/ml) or tiron (10 mM). Other enzymatic sources of
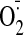 production were identified using inhibitors such as l-NAME (100
μM), rotenone (50 μM), oxypurinol (100 μM), and diphenyleneiodonium
(20 μM). All studies were performed in triplicate. ROS generation in
cardiac section was measured in situ using DHE (1 μM) fluorescence in the
presence or absence of tiron (Ribé
et al., 2008). Fluorescence intensity was quantified under
confocal microscopy from at least five random fields (1024 × 1022
pixels; 269.7 × 269.2 μm) per slide, three slides per animal and six
animals per group.
production were identified using inhibitors such as l-NAME (100
μM), rotenone (50 μM), oxypurinol (100 μM), and diphenyleneiodonium
(20 μM). All studies were performed in triplicate. ROS generation in
cardiac section was measured in situ using DHE (1 μM) fluorescence in the
presence or absence of tiron (Ribé
et al., 2008). Fluorescence intensity was quantified under
confocal microscopy from at least five random fields (1024 × 1022
pixels; 269.7 × 269.2 μm) per slide, three slides per animal and six
animals per group.
Immunoprecipitation and Immunoblotting. Immunoprecipitation and immunoblotting was performed as described previously (Ribé et al., 2008). For p47phox serine phosphorylation, the p47phox was firstly immunoprecipitated down with antibodies to p47phox coupled to protein G agarose beads overnight at 4°C. Normal rabbit IgG was used as a negative control. p47phox serine phosphorylation was detected by immunoblotting using a phosphoserine-specific monoclonal antibody.
Confocal Microscopy. Confocal microscopy were performed as described previously (Ribé et al., 2008). Biotin-conjugated anti-rabbit or anti-goat IgG were used as secondary antibodies and detected by extravidin-fluorescein isothiocyanate or streptavidin-Cy3. Normal rabbit or goat IgG (5 μg/ml) were used instead of primary antibody as a negative control. Images were acquired on a Zeiss LS510 confocal microscopy system (Carl Zeiss GmbH, Jena, Germany). Optical sections were taken at 1-μm intervals, and images were captured digitally.
TUNEL Assay. H9C2 cells were cultured onto the chamber slide and stimulated with cocaine (25 μM) for 24 h. The cells were then washed and fixed in 4% methanol-free formaldehyde/PBS solution and treated with 0.2% Triton X-100/PBS solution for 5 min. The TUNEL assay was performed using the DeadEnd fluorometric technique (Promega, Madison, WI) as described by the company and visualized under confocal microscopy.
Statistics. Data were presented as mean ± S.D. Animal data were from 12 mice per group. Comparisons were made by unpaired Student's t test, with Bonferroni correction for multiple testing. p < 0.05 was considered statistically significant.
Results
Changes in the Levels of Cardiac ROS Production. The
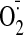 production in the cardiac homogenate was detected by lucigenin
chemiluminescence (Fig. 1, A and
B). There was no significant difference in the basal levels
(without adding NADPH) of ROS production between saline-treated (control) and
cocaine groups. However, the levels of NADPH-dependent
production in the cardiac homogenate was detected by lucigenin
chemiluminescence (Fig. 1, A and
B). There was no significant difference in the basal levels
(without adding NADPH) of ROS production between saline-treated (control) and
cocaine groups. However, the levels of NADPH-dependent
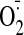 production were significantly increased in cocaine-treated hearts up to 1.96-
± 0.4-fold of the controls (p < 0.03). The
production were significantly increased in cocaine-treated hearts up to 1.96-
± 0.4-fold of the controls (p < 0.03). The
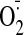 production thus measured was virtually abolished by tiron (a superoxide
scavenger), significantly inhibited by apocynin (69 ± 4%, NADPH oxidase
inhibitor), diphenyleneiodonium (82 ± 2%, flavoprotein inhibitor), and
superoxide dismutase (69 ± 4%) (all p < 0.03) but not by
rotenone (mitochondrial complex 1 enzyme inhibitor) or oxypurinol (xanthine
oxidase inhibitor) (Fig. 1B).
There was a slight (26 ± 3%) but significant (p < 0.05)
reduction in NADPH-dependent ROS production in the presence of
l-NAME (NOS inhibitor) in cocaine-treated hearts. Although NOS
uncoupling may account for a small proportion of increase in
production thus measured was virtually abolished by tiron (a superoxide
scavenger), significantly inhibited by apocynin (69 ± 4%, NADPH oxidase
inhibitor), diphenyleneiodonium (82 ± 2%, flavoprotein inhibitor), and
superoxide dismutase (69 ± 4%) (all p < 0.03) but not by
rotenone (mitochondrial complex 1 enzyme inhibitor) or oxypurinol (xanthine
oxidase inhibitor) (Fig. 1B).
There was a slight (26 ± 3%) but significant (p < 0.05)
reduction in NADPH-dependent ROS production in the presence of
l-NAME (NOS inhibitor) in cocaine-treated hearts. Although NOS
uncoupling may account for a small proportion of increase in
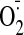 generation in cocaine group, NADPH oxidase certainly represents a major source
of chronic binge cocaine administration-induced
generation in cocaine group, NADPH oxidase certainly represents a major source
of chronic binge cocaine administration-induced
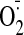 generation in the hearts.
generation in the hearts.
Fig. 1.

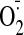 production in saline- and cocaine-treated hearts. A, kinetic detection of
production in saline- and cocaine-treated hearts. A, kinetic detection of
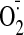 production by lucigenin chemiluminescence. NADPH was added at 20 min of
measurement. RLU, relative light unit(s). B, effects of inhibitors on the
levels of
production by lucigenin chemiluminescence. NADPH was added at 20 min of
measurement. RLU, relative light unit(s). B, effects of inhibitors on the
levels of
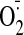 production by cocaine-treated hearts. *, p < 0.05 for indicated values
versus the NADPH (without inhibitor) values. n = 12 hearts. C,
tiron-inhibitable DHE fluorescence detection of ROS production by confocal
microscopy. D, fluorescence intensities were quantified and expressed as mean
± S.D. from 18 sections of six mice/per group. *, p < 0.05
for indicated values versus the controls.
production by cocaine-treated hearts. *, p < 0.05 for indicated values
versus the NADPH (without inhibitor) values. n = 12 hearts. C,
tiron-inhibitable DHE fluorescence detection of ROS production by confocal
microscopy. D, fluorescence intensities were quantified and expressed as mean
± S.D. from 18 sections of six mice/per group. *, p < 0.05
for indicated values versus the controls.
The increase in
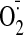 production was further examined by DHE fluorescence on cardiac sections in the
presence or absence of tiron (Fig.
1C). Compared with saline controls, the tiron-inhibitable DHE
fluorescence intensity in the myocardium of the cocaine-treated group was
significantly increased to 1.83- ± 0.6-fold compared with control
values (p < 0.05) as quantified by confocal microscopy
(Fig. 1D).
production was further examined by DHE fluorescence on cardiac sections in the
presence or absence of tiron (Fig.
1C). Compared with saline controls, the tiron-inhibitable DHE
fluorescence intensity in the myocardium of the cocaine-treated group was
significantly increased to 1.83- ± 0.6-fold compared with control
values (p < 0.05) as quantified by confocal microscopy
(Fig. 1D).
Changes in the Levels of NADPH Oxidase Expression. We then investigated the changes in the protein expression of NADPH oxidase subunits by immunoblotting (Fig. 2A). Results were normalized to the expression of cardiac troponin I detected in the same sample and quantified (Fig. 2B). Both Nox2 and Nox4 were detected in the hearts. In cocaine-treated hearts, there was a significant increase in Nox2 expression (2.34- ± 0.6-fold compared with controls, p < 0.05) but not in Nox4 expression. Accompanying the increase in Nox2 expression, there were significant increases in p22phox (2.5- ± 0.29-fold), p67phox (2.1- ± 4.1-fold), p47phox (2.6- ± 0.4-fold), p40phox (1.66- ± 0.32-fold), and Rac1 (2.1- ± 0.37-fold) compared with the control values (all p < 0.05). Because p47phox, p67phox and rac1 are only required for Nox2 but not Nox4 oxidase activation (Martyn et al., 2006), it is likely that Nox2 plays a prominent role in mediating cardiac oxidative stress in the cocaine group.
Fig. 2.

Immunoblotting for the expression of NADPH oxidase subunits in saline- and cocaine-treated hearts. A, representative examples of immunoblots. Cardiac troponin I was used as a loading control. B, protein bands were quantified densitometrically and normalized to the expression of cardiac troponin I in the same sample. The results were expressed as arbitrary units. *, p < 0.05 for cocaine value versus saline controls. n = 12 animals.
Changes in p47phox Phosphorylation and Translocation. p47phox phosphorylation has been shown to be a prerequisite for Nox2 oxidase activation (Li and Shah, 2003). Therefore, we examined the serine phosphorylation of p47phox by immunoblotting using a phosphoserine-specific monoclonal antibody after immunoprecipitation of p47phox (Fig. 3A). We found that chronic cocaine administration increased the levels of p47phox serine phosphorylation by 3.2- ± 0.5-fold compared with control levels (p < 0.03). The increase in p47phox expression (Cy3-labeled) was notably in the cardiac myocyte sarcolemmal membrane area (fluorescein isothiocyanate-labeled laminin) (Fig. 3B), and this was further highlighted by the yellow fluorescence in the superposed image of the cocaine group (Fig. 3B).
Fig. 3.

p47phox phosphorylation and myocyte expression of p47phox in saline- and cocaine-treated hearts. A, immunoblotting. Phos-p47phox bands were quantified densitometrically and normalized to the total p47phox level in the same sample and expressed as arbitrary units. n = 12 hearts. *, p < 0.05 for cocaine values versus saline control values. B, confocal microscopy for the expression of p47phox in cardiac myocytes. Cardiac myocytes were outlined by laminin (green fluorescence) and detected for the expression of the p47phox (red fluorescence).
Changes in MAPK Activation. MAPK are important redox-signaling molecules involved in the pathogenesis of many cardiac diseases such as hypertrophy and heart failure (Bueno and Molkentin, 2002; Li et al., 2002a). We investigated the changes in the levels of ERK1/2, p38 MAPK and JNK activation using phosphorylation-specific monoclonal antibodies. The expression of total ERK1/2, p38 MAPK, and JNK in the same samples was used as loading controls (Fig. 4A). We found that the levels of phosphorylated ERK1/2, p38 MAPK, and JNK in cocaine-treated hearts were significantly increased by 2.7- ± 0.4-, 2.5- ± 0.25-, and 2.6- ± 0.36-fold, respectively, compared with saline controls (all p < 0.05).
Fig. 4.

Changes in MAPK activation in saline- and cocaine-treated hearts. A, immunoblotting. The phosphoprotein bands were quantified densitometrically and the levels of phos-ERK1/2, phos-p38 MAPK, and phos-JNK were normalized to the total protein levels of these molecules in the same samples and expressed as arbitrary units. n = 12 hearts. *, p < 0.05 for cocaine values versus saline values. B, confocal microscopy detection of phos-ERK1/2 in cardiac sections.
Cocaine-induced ERK1/2 activation was further investigated by confocal microscopy on cardiac cryosections (Fig. 4B). Compared with saline-treated controls, there was a remarkable increase in phos-ERK1/2 expression in the sarcolemmal membrane area of the cardiomyocytes in the cocaine-treated group.
The Action of Cocaine on Cultured Cardiomyocytes. Conventionally,
cocaine was believed to exert cardiotoxicity through an indirect
sympathomimetic action on α-adrenergic receptors. However, cocaine also
has been found to act directly on cardiomyocytes and damage cell function. To
address the question of whether the action of cocaine on myocardial oxidative
stress was direct or indirect, we stimulated H9C2 cells in culture with
different concentrations of cocaine (0–100 μM) for 24 h and then
examined the NADPH-dependent
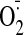 production (Fig. 5A). Compared
with control cells (without cocaine), cocaine used at a dose as low as 5 μM
significantly increased
production (Fig. 5A). Compared
with control cells (without cocaine), cocaine used at a dose as low as 5 μM
significantly increased
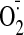 production up to 1.8- ± 0.3-fold compared with control value
(p < 0.05). At a dose of 25 μMor above, cocaine markedly
increased the levels of
production up to 1.8- ± 0.3-fold compared with control value
(p < 0.05). At a dose of 25 μMor above, cocaine markedly
increased the levels of
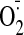 production up to a maximum of 3-fold compared with controls. Cocaine (25
μM)-induced ROS production by H9C2 cells was not significantly changed in
the presence of a β-adrenergic receptor antagonist (propanolol, 1 μM)
(Fig. 5B). However,
cocaine-induced
production up to a maximum of 3-fold compared with controls. Cocaine (25
μM)-induced ROS production by H9C2 cells was not significantly changed in
the presence of a β-adrenergic receptor antagonist (propanolol, 1 μM)
(Fig. 5B). However,
cocaine-induced
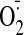 production was accompanied by a remarkable increased in Nox2 protein
expression and was virtually abolished by in vitro knockout of Nox2 expression
using Nox2 siRNA (Fig. 5C). We
then examined the effect of cocaine on myocyte death by TUNEL assay
(Fig. 5D). Compared with
control cells, cocaine (25 μM) caused severe myocyte damage and cell death
as indicated by TUNEL-positive nuclei (yellow color), plus nuclear
disintegration and fragmentation (arrow in
Fig. 5D). Cocaine-induced
myocyte death was effectively inhibited by knockout of Nox2 using siRNA.
production was accompanied by a remarkable increased in Nox2 protein
expression and was virtually abolished by in vitro knockout of Nox2 expression
using Nox2 siRNA (Fig. 5C). We
then examined the effect of cocaine on myocyte death by TUNEL assay
(Fig. 5D). Compared with
control cells, cocaine (25 μM) caused severe myocyte damage and cell death
as indicated by TUNEL-positive nuclei (yellow color), plus nuclear
disintegration and fragmentation (arrow in
Fig. 5D). Cocaine-induced
myocyte death was effectively inhibited by knockout of Nox2 using siRNA.
Fig. 5.

Direct effects of cocaine on cultured H9C2 cardiomyocytes. A,
cocaine-induced
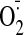 mean light unit(s). B, effect of propanolol on
mean light unit(s). B, effect of propanolol on
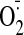 production with or with out cocaine (25 μM). C, effect of Nox2 siRNA on
cocaine-induced
production with or with out cocaine (25 μM). C, effect of Nox2 siRNA on
cocaine-induced
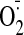 production and Nox2 protein expression. α-Tubulin was used as loading
control. *, p < 0.05 for indicated values versus control values in the same
group. D, effect of Nox2 siRNA on cocaine-induced myocyte death. Arrow,
nuclear fragmentation and TUNEL-positive nucleus.
production and Nox2 protein expression. α-Tubulin was used as loading
control. *, p < 0.05 for indicated values versus control values in the same
group. D, effect of Nox2 siRNA on cocaine-induced myocyte death. Arrow,
nuclear fragmentation and TUNEL-positive nucleus.
The Effects of the PKC Inhibitor on Cocaine-Induced Nox2 Activation.
To investigate the possible mechanisms involved in the direct effect of
cocaine on Nox2 activation, we examined the phosphorylation of
p47phox (a key regulator of Nox2 oxidase), p47phox/Nox2
complex formation, and Nox2 oxidase activity
(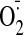 production) in cultured cardiac myocytes in the presence or absence of a
pan-PKC inhibitor (Bis, 10 μM) (Fig.
6A). Cocaine (25 μM) significantly increased p47phox
serine phosphorylation, which was accompanied by a significant increase in
p47phox/Nox2 complex formation and in Nox2 oxidase activity
(
production) in cultured cardiac myocytes in the presence or absence of a
pan-PKC inhibitor (Bis, 10 μM) (Fig.
6A). Cocaine (25 μM) significantly increased p47phox
serine phosphorylation, which was accompanied by a significant increase in
p47phox/Nox2 complex formation and in Nox2 oxidase activity
(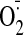 production). All of these cocaine-induced effects could be significantly
inhibited in the presence of Bis.
production). All of these cocaine-induced effects could be significantly
inhibited in the presence of Bis.
Fig. 6.

The effects of PKC inhibitor and
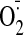 scavenger on cocaine (25 μM)-induced p47phox serine
phosphorylation and ERK1/2 activation in H9C2 cells. A, p47phox was
immunoprecipitated down (IP) and detected by immunoblotting (IB) using
antibodies specific to phosphoserine or to Nox2. p47phox IB was
used as loading control. B, cocaine-induced ERK1/2 phosphorylation in H9C2
cells in the presence and absence of Bis or tiron. The phos-ERK1/2 bands were
quantified densitometrically and normalized to the total ERK1/2 levels in the
same samples and expressed as arbitrary units. *, p < 0.05 for cocaine
values versus control (without cocaine) values. †, p < 0.05
for the values of cocaine plus Bis or tiron versus the cocaine values.
n = 4 independent cell cultures.
scavenger on cocaine (25 μM)-induced p47phox serine
phosphorylation and ERK1/2 activation in H9C2 cells. A, p47phox was
immunoprecipitated down (IP) and detected by immunoblotting (IB) using
antibodies specific to phosphoserine or to Nox2. p47phox IB was
used as loading control. B, cocaine-induced ERK1/2 phosphorylation in H9C2
cells in the presence and absence of Bis or tiron. The phos-ERK1/2 bands were
quantified densitometrically and normalized to the total ERK1/2 levels in the
same samples and expressed as arbitrary units. *, p < 0.05 for cocaine
values versus control (without cocaine) values. †, p < 0.05
for the values of cocaine plus Bis or tiron versus the cocaine values.
n = 4 independent cell cultures.
We then examined the effects of PKC inhibitor (Bis) and
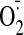 scavenger (tiron, 2.5 mM) on cocaine-induced ERK1/2 phosphorylation
(Fig. 6B). Compared with cells
cultured in the medium (control), cocaine (25 μM) significantly increased
the ERK1/2 phosphorylation, and this was significantly inhibited to the
control levels in the presence of Bis or completely abolished in the presence
of tiron. Taken together, these results implicated a critical role of PKC in
mediating the direct effect of cocaine on Nox2 activation and further
confirmed the role of Nox2-derived ROS in cocaine-induced MAPK activation.
scavenger (tiron, 2.5 mM) on cocaine-induced ERK1/2 phosphorylation
(Fig. 6B). Compared with cells
cultured in the medium (control), cocaine (25 μM) significantly increased
the ERK1/2 phosphorylation, and this was significantly inhibited to the
control levels in the presence of Bis or completely abolished in the presence
of tiron. Taken together, these results implicated a critical role of PKC in
mediating the direct effect of cocaine on Nox2 activation and further
confirmed the role of Nox2-derived ROS in cocaine-induced MAPK activation.
Discussion
Chronic cocaine consumption has been shown to be associated with
life-threatening cardiac abnormalities including left ventricular hypertrophy,
arrhythmias, heart failure, myocarditis, and sudden cardiac death
(Vasica and Tennant, 2002;
Darke et al., 2006;
Afonso et al., 2007). However,
our understanding of the underlying mechanisms of cocaine cardiotoxicity is
far from complete and pharmacological management of such patients is
problematic (Vasica and Tennant,
2002; Afonso et al.,
2007). In this study, we used an experimental mouse model of
chronic escalating dose of cocaine binge administration to mimic the common
pattern of human repeated cocaine consumption. We found that chronic cocaine
consumption significantly increased the cardiac NADPH-dependent
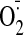 generation because of an increase in the expression of Nox2 components and the
p47phox phosphorylation.
generation because of an increase in the expression of Nox2 components and the
p47phox phosphorylation.
Emerging evidence has revealed that cardiac oxidative stress is a prominent
early event of cocaine administration, which severely compromises the cardiac
antioxidative system and causes cardiac damage
(Boess et al., 2000;
Kovacic, 2005). Evidences of
oxidative damage such as peroxidation of membrane phospholipids and depletion
of nonenzymatic antioxidants such as GSH have been found in the myocardium of
chronic cocaine-treated animals (Pacifici
et al., 2003) and in patients
(Darke et al., 2006;
Afonso et al., 2007). In the
present study, using two independent assays, we have provided direct evidence
that chronic cocaine binge consumption doubles the cardiac
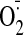 production. A small but significant proportion (∼25%) of the
NADPH-dependent ROS production was inhibited by an NOS inhibitor
(l-NAME), indicating that dysfunctional NOS activity might also
partly contribute to ROS production in cocaine group
(Li and Shah, 2004). This is
not surprising because ROS production from any source may induce dysfunctional
production. A small but significant proportion (∼25%) of the
NADPH-dependent ROS production was inhibited by an NOS inhibitor
(l-NAME), indicating that dysfunctional NOS activity might also
partly contribute to ROS production in cocaine group
(Li and Shah, 2004). This is
not surprising because ROS production from any source may induce dysfunctional
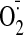 -generating
NOS activity, as a consequence of ROS-dependent degradation of the essential
NOS cofactor tetrahydrobiopterin
(Vásquez-Vivar et al.,
2003).
-generating
NOS activity, as a consequence of ROS-dependent degradation of the essential
NOS cofactor tetrahydrobiopterin
(Vásquez-Vivar et al.,
2003).
During the last decade, it has become clear that the myocardium expresses constitutively both Nox2 and Nox4 (Byrne et al., 2003). In general, Nox2 activity is dependent on the phosphorylation of p47phox (a major regulatory subunit) and requires the presence of other cytosolic components, such as rac1 and p67phox (Li et al., 2002b; Li and Shah, 2004; Sumimoto et al., 2005), whereas Nox4 seems not to exhibit a binding site to p47phox (Sumimoto et al., 2005) and is not regulated by p47phox or any other known regulatory components (Martyn et al., 2006). In the present study, we demonstrated by immunoblotting that chronic binge cocaine consumption significantly up-regulated the protein transcription of Nox2 and its components: p22phox, p47phox, p67phox, and rac1. The role of Nox2 in mediating cocaine oxidative stress was further supported by our data that cocaine treatment indeed increased p47phox serine phosphorylation (up to 3-fold) in the cardiomyocytes as demonstrated by confocal microscopy.
ROS have multiple effects on cell function depending on the amount and subcellular location of ROS generated. Controlled intracellular ROS production from NADPH oxidase has been shown to be necessary for normal cellular development and function (Biswas et al., 2006), whereas excessive ROS generation is implicated in myocardial hypertrophy and heart failure (Li et al., 2002a). The mitogenic effects of ROS may involve the modulation of redox-sensitive signaling pathways such as the MAPK superfamily, which lead to the activation of transcriptional factors (Li et al., 2002a, 2004). MAPKs, in particular ERK1/2, have been shown to be activated by cocaine administration and represent an important cellular mechanism in cocaine addition (Lu et al., 2006). In accordance with previous reports, we found significant increases in the levels of ERK1/2, p38 MAPK, and JNK phosphorylation in cocaine-treated hearts. In addition, we also found, by confocal microscopy, that the increases in p47phox expression and ERK1/2 phosphorylation were both in the sarcolemmal membrane area of cardiac myocytes in cocaine-treated hearts. Therefore, MAPKs can serve as intermediates that couple Nox2 oxidase to its downstream signaling molecules involved in cocaine cardiotoxicity.
Cocaine-induced cardiac oxidative stress has been regarded previously as an
indirect effect of cocaine on cardiomyocytes via its sympathomimetic action on
adrenoceptors (Afonso et al.,
2007; Isabelle et al.,
2007). Although NADPH oxidase can also be activated by
catecholamines, the severity of myocardial damage seen in cocaine patients
could not be fully addressed by a simple sympathomimetic action. Moreover, the
cardiac β-adrenoceptor signaling pathways are impaired by cocaine
exposure (Sun, 2000), which is
opposite to the conventional view of cocaine sympathomimetic action. The
current study provides the first evidence that in the absence of
catecholamines, cocaine used at a dose of 5 μM, which is far below the
plasma levels of cocaine found in cocaine abusers, has direct effects on
cultured cardiac myocytes and strongly promotes ROS production from Nox2
oxidase. Cocaine, used at 25 μM, a concentration that is relevant to the
mean plasma concentration found in patients
(Wu et al., 2006), caused a
3-fold increase in
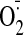 generation and severe cell death in cultured cardiac myocyte. All these
effects were abolished or effectively inhibited by in vitro knockout of Nox2
using siRNA. Furthermore, using inhibitor of PKC and an
generation and severe cell death in cultured cardiac myocyte. All these
effects were abolished or effectively inhibited by in vitro knockout of Nox2
using siRNA. Furthermore, using inhibitor of PKC and an
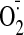 scavenger (tiron), we showed that the effect of cocaine on cardiac myocyte ROS
production is PKC-dependent and an increase in
scavenger (tiron), we showed that the effect of cocaine on cardiac myocyte ROS
production is PKC-dependent and an increase in
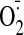 generation from Nox2 oxidase is a prerequisite for cocaine-induced ERK1/2
activation. It is well known that PKC-mediated p47phox
phosphorylation is a key step in Nox2 oxidase activation. Therefore, cocaine
may act through PKC to cause p47phox phosphorylation and to
activate Nox2 oxidase rather than indirectly through the actions on
adrenoceptors. However, we have not examined yet whether other PKC downstream
signaling pathways are involved in cocaine cardiac toxicity. It is clear from
the current study that cocaine can cause myocyte oxidative damage without
adrenergic signals. However, in the in vivo situation, adrenergic signals can
further escalate the severity of cocaine-related oxidative stress and toxicity
to the heart. Nevertheless, it is important to know that knockout of Nox2 can
effectively reduce cocaine-induced myocardial oxidative stress and damage.
generation from Nox2 oxidase is a prerequisite for cocaine-induced ERK1/2
activation. It is well known that PKC-mediated p47phox
phosphorylation is a key step in Nox2 oxidase activation. Therefore, cocaine
may act through PKC to cause p47phox phosphorylation and to
activate Nox2 oxidase rather than indirectly through the actions on
adrenoceptors. However, we have not examined yet whether other PKC downstream
signaling pathways are involved in cocaine cardiac toxicity. It is clear from
the current study that cocaine can cause myocyte oxidative damage without
adrenergic signals. However, in the in vivo situation, adrenergic signals can
further escalate the severity of cocaine-related oxidative stress and toxicity
to the heart. Nevertheless, it is important to know that knockout of Nox2 can
effectively reduce cocaine-induced myocardial oxidative stress and damage.
In conclusion, we report for the first time that chronic cocaine administration causes severe cardiac oxidative stress through the activation of Nox2 oxidase. The mechanisms involved are the transcriptional up-regulation of Nox2 oxidase components and the post-translational modification of p47phox phosphorylation. We also found that cocaine is able to cause p47phox phosphorylation in cardiac myocytes through PKC, in the absence of catecholamines. Further investigation into the inhibition of p47phox phosphorylation and Nox2 activation may provide a potential therapeutic strategy for cocaine cardiotoxicity.
Acknowledgments
We thank Professor S. Hourani for constructive advice and help with manuscript preparation.
This work was supported by the British Heart Foundation [Grant PG/06/073]; by the Biotechnology and Biological Sciences Research Council, UK [Grant BB/D009510/1]; and by the EC GENADDICT program [Grant PL005166].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.108.145201.
ABBREVIATIONS: ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase; DHE, dihydroethidium; ERK, extracellular signal-regulated kinase; JNK, c-Jun NH2-terminal kinase; Bis, bisindolymaleimide; siRNA, small interfering RNA; PBS, phosphate-buffered saline; PKC, protein kinase C; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; l-NAME, N-ω-nitro-l-arginine methyl ester.
References
- Afonso L, Mohammad T, and Thatai D (2007) Crack whips the heart: a review of the cardiovascular toxicity of cocaine. Am J Cardiol 100 1040-1043. [DOI] [PubMed] [Google Scholar]
- Bailey A, Yoo JH, Racz I, Zimmer A, and Kitchen I (2007) Preprodynorphin mediates locomotion and D2 dopamine and μ-opioid receptor changes induced by chronic “binge” cocaine administration. J Neurochem 102 1817-1830. [DOI] [PubMed] [Google Scholar]
- Biswas S, Chida AS, and Rahman I (2006) Redox modification of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol 71 551-564. [DOI] [PubMed] [Google Scholar]
- Boess F, Ndikum-Moffor FM, Boelsterli UA, and Roberts SM (2000) Effects of cocaine and its oxidative metabolites on mitochondrial respiration and generation of reactive oxygen species. Biochem Pharmacol 60 615-623. [DOI] [PubMed] [Google Scholar]
- Bueno OF and Molkentin JD (2002) Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res 91 776-781. [DOI] [PubMed] [Google Scholar]
- Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, and Shah AM (2003) Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 93: 802-805. [DOI] [PubMed] [Google Scholar]
- Darke S, Kaye S, and Duflou J (2006) Comparative cardiac pathology among deaths due to cocaine toxicity, opioid toxicity and non-drug-related causes. Addiction 101 1771-1777. [DOI] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, and Brandes RP (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51 211-217. [DOI] [PubMed] [Google Scholar]
- Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, Sharma RV, Engelhardt JF, and Davisson RL (2006) Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics 26 180-191. [DOI] [PubMed] [Google Scholar]
- Isabelle M, Vergeade A, Moritz F, Dautréaux B, Henry JP, Lallemand F, Richard V, Mulder P, Thuillez C, and Monteil C (2007) NADPH oxidase inhibition prevents cocaine-induced up-regulation of xanthine oxidoreductase and cardiac dysfunction. J Mol Cell Cardiol 42 326-332. [DOI] [PubMed] [Google Scholar]
- Kovacic P (2005) Role of oxidative metabolites of cocaine in toxicity and addiction: oxidative stress and electron transfer. Med Hypotheses 64 350-356. [DOI] [PubMed] [Google Scholar]
- Li JM, Fan LM, George VT, and Brooks G (2007) Nox2 regulates endothelial cell cycle arrest and apoptosis via p21cip1 and p53. Free Radic Biol Med 43 976-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Gall NP, Grieve DJ, Chen M, and Shah AM (2002a) Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40 477-484. [DOI] [PubMed] [Google Scholar]
- Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, and Shah AM (2002b) Essential role of the NADPH oxidase subunit p47phox in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α. Circ Res 90 143-150. [DOI] [PubMed] [Google Scholar]
- Li JM and Shah AM (2004) Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 287 R1014-R1030. [DOI] [PubMed] [Google Scholar]
- Li JM and Shah AM (2003) Mechanism of endothelial cell NADPH oxidase activation by angiotensin II: Role of the p47phox subunit. J Biol Chem 278 12094-12100. [DOI] [PubMed] [Google Scholar]
- Li JM and Shah AM (2002) Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem 277 19952-19960. [DOI] [PubMed] [Google Scholar]
- Li JM, Wheatcroft S, Fan LM, Kearney MT, and Shah AM (2004) Opposing roles of p47phox in basal versus angiotensin II-stimulated alterations in vascular O2- production, vascular tone, and mitogen-activated protein kinase activation. Circulation 109 1307-1313. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, and Shaham Y (2006) Role of ERK in cocaine addiction. Trends Neurosci 29 695-703. [DOI] [PubMed] [Google Scholar]
- Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, and Knaus UG (2006) Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18 69-82. [DOI] [PubMed] [Google Scholar]
- Moritz F, Monteil C, Isabelle M, Bauer F, Renet S, Mulder P, Richard V, and Thuillez C (2003) Role of reactive oxygen species in cocaine-induced cardiac dysfunction. Cardiovasc Res 59 834-843. [DOI] [PubMed] [Google Scholar]
- Pacifici R, Fiaschi AI, Micheli L, Centini F, Giorgi G, Zuccaro P, Pichini S, Di Carlo S, Bacosi A, and Cerretani D (2003) Immunosuppression and oxidative stress induced by acute and chronic exposure to cocaine in rat. Int Immunopharmacol 3 581-592. [DOI] [PubMed] [Google Scholar]
- Ren S, Tong W, Lai H, Osman NF, Pannu H, and Lai S (2006) Effect of long term cocaine use on regional left ventricular function as determined by magnetic resonance imaging. Am J Cardiol 97 1085-1088. [DOI] [PubMed] [Google Scholar]
- Ribé D, Sawbridge D, Thakur S, Hussey M, Ledent C, Kitchen I, Hourani S, and Li JM (2008) Adenosine A2A receptor signaling regulation of cardiac NADPH oxidase activity. Free Radic Biol Med 44 1433-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Miyano K, and Takeya R (2005) Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun 338 677-686. [DOI] [PubMed] [Google Scholar]
- Sun LS (2000) Perinatal cocaine exposure impairs myocardial beta-adrenoceptor signaling in the neonatal rat. Anesth Analg 90 50-56. [DOI] [PubMed] [Google Scholar]
- Vasica G and Tennant CC (2002) Cocaine use and cardiovascular complications. Med J Aust 177 260-262. [DOI] [PubMed] [Google Scholar]
- Vásquez-Vivar J, Kalyanaraman B, and Martásek P (2003) The role of tetrahydrobiopterin in superoxide generation from eNOS: Enzymology and physiological implications. Free Radic Res 37 121-127. [DOI] [PubMed] [Google Scholar]
- Wu SN, Chang HD, and Sung RJ (2006) Cocaine-induced inhibition of ATP-sensitive K+ channels in rat ventricular myocytes and in heart-derived H9c2 cells. Basic Clin Pharmacol Toxicol 98 510-517. [DOI] [PubMed] [Google Scholar]