

Abstract
Epilepsy remains a devastating neurological disorder associated with recurrent, unprovoked, spontaneous epileptic seizures. Current treatments involve seizure suppression using antiepileptic drugs (AEDs); however, many patients remain refractory to current treatments or suffer serious side effects. In view of this continued need for more effective and safer AEDs, we have designed a novel compound, 3-hydroxy-3-(4-methoxyphenyl)-1-methyl-1,3-dihydro-indol-2-one (YWI92), based on a lactam structural class, and evaluated its modulation of human neuronal sodium channel isoform (hNav)1.2 currents and hippocampal neuron action potential firing. Furthermore, we have tested its AED activity using a chronic and acute rat seizure model. In a similar manner to lamotrigine, a clinically used AED, YWI92 exhibited tonic block of hNav1.2 channels and caused a hyperpolarizing shift in the steady-state inactivation curve when using a 30-s inactivating prepulse. YWI92 also delayed the time constants of channel repriming after a 30-s inactivating prepulse and exhibited use-dependent block at 20-Hz stimulation frequency. In membrane excitability experiments, YWI92 inhibited burst firing in CA1 neurons of animals with temporal lobe epilepsy at concentrations that had little effect on CA1 neurons from control animals. These actions on neuronal activity translated into AED activity in the maximal electroshock acute seizure model (ED50 = 22.96 mg/kg), and importantly, in a chronic temporal lobe epilepsy model, in which the mean number of seizures was reduced. Notably, YWI92 exhibited no sedative/ataxic side effects at concentrations up to 500 mg/kg. In summary, greater affinity for inactivated sodium channels, particularly after long depolarizing prepulses, may be important for both anticonvulsant activity and drug tolerability.
Epilepsy is a devastating neurological disorder, characterized by recurrent spontaneous seizures either in both brain hemispheres (general seizures) or localized in one or more parts of one or both hemispheres (partial seizures). Current treatment options for patients with epilepsy involve seizure suppression through the use of a myriad of currently available antiepileptic drugs (AEDs). Unfortunately, a substantial proportion of patients (∼30%) continues to experience seizures even in the presence of optimal doses of AEDs, and they are considered pharmaco-resistant (Remy and Beck, 2006). In addition, many patients that achieve seizure control with medications suffer from medication-induced neurotoxicity, sedation, and cognitive side effects (Brodie, 2001).
Since voltage-gated sodium (Nav) channels play a critical role in the initiation and propagation of action potentials in excitable cells, they remain a promising target for the development of new AEDs. The sodium channel consists of a pore-forming α subunit, which is sufficient to induce sodium currents in heterologous expression systems, and a variable number of auxiliary β subunits that modulate the gating properties of the channel (Catterall, 2000). At resting membrane potentials, sodium channels are closed, but they open upon depolarization, giving rise to a fast (transient) inward current, and in certain cells, a slowly inactivating (persistent) current (Goldin, 2003). The modulation of both the transient and persistent sodium currents is believed to be a primary mechanism of action for many clinically used AEDs such as phenytoin, carbamazepine, and lamotrigine (Catterall, 2002). An underlying property of several AEDs is their state-dependent inhibition of the sodium channel. Thus, these agents selectively bind to open and inactivated states visited at depolarized potentials. This feature is best described by the modulated receptor hypothesis (Hille, 1977), and it allows AEDs to preferentially inhibit action potential (AP) bursts that occur during seizures with minimal affects on normal neuronal activity (Rogawski and Löscher, 2004).
Inactivation of sodium channels has at least two kinetic time courses: fast and slow. Fast channel inactivation and recovery from inactivation occur within milliseconds and are best described by the “hinged lid” mechanism. Slow inactivation develops and recovers over a much longer time course (i.e., seconds to minutes) and is a more complex process involving structural rearrangement of the pore and other regions of the sodium channel (Ulbricht, 2005). Disruptions to these inactivation processes have been associated with the incidence of generalized epilepsy with febrile seizures plus (GEF+) and severe myoclonic epilepsy of infancy (Spampanato et al., 2001). Further evaluation of these disease-causing mutations has revealed subtle changes in the steady-state inactivation properties, including entry into and recovery from slow inactivated states, which likely account for the increased persistent sodium current observed in almost all of the mutations examined (Meisler and Kearney, 2005). Preferential targeting of slow inactivated states, or states present as a result of prolonged depolarization, may represent a new molecular target for AEDs in reducing sodium channel activity and suppressing seizures, as has been recently proposed as a contributing mechanism of action for the novel AED lacosamide (Vimpat) (Errington et al., 2008).
Previous studies have identified lactams as a structural class of effective AEDs, providing protection in either the acute maximal electric shock (MES) or the subcutaneous metrazole animal model of acute seizures (Grimm et al., 2003). Lactams can displace [3H]batrachotoxinin A 20-α-benzoate binding from rat brain cerebral cortex synaptasomes, suggesting that they share a similar binding site to many of the clinically used AEDs (Clare et al., 2000). In this study, we designed a novel benzolated lactam, YWI92, based on a simplified lactam motif (Scheme 1A). Synthesis was accomplished in high yield (Scheme 1B), and its effects were determined on the human neuronal sodium channel isoform Nav1.2, in comparison with lamotrigine (LTG). The ability of YWI92 to selectively suppress epileptiform AP discharges in CA1 neurons from chronic temporal lobe epilepsy (TLE) animals over control nonepileptic animals was also assessed. YWI92 exhibited a greater affinity for channels inactivated during long prepulses, shifting their steady-state inactivation curves to greater hyperpolarized potentials and accelerating the development, and slowing of their recovery. Furthermore, YWI92 was more effective at suppressing action potential firing in TLE CA1 neurons over control CA1 neurons. Consistent with these in vitro results, YWI92 inhibited seizures evoked in the MES acute seizure model with no sedative or ataxic side effects, and it reduced the frequency of spontaneous seizures in the TLE rat model of chronic epilepsy. This study suggests that the lactam moiety may represent an important structural feature for the development of state-selective sodium channel blockers with anticonvulsant activity.
Scheme 1.

A, design strategy for the synthesis of YWI92. B, synthesis of YWI92. YWI92 was designed using a progression strategy based on a lactam pharmacophore. The synthesis was accomplished by addition of the methoxyphenyl through a nucleophilic addition to commercially available methylisatin.
Materials and Methods
Synthesis of YWI92
1H and 13C NMR spectra were measured at 300 MHz on a GE 300-MHz NMR spectrometer. Chemical shifts were reported relative to internal CDCl3 (1H, 7.26 ppm and 13C, 77.0 ppm) and CD3OD (1H, 3.30 ppm and 13C, 49.2 ppm). Flash column chromatography was performed on silica gel 60 (35–75 μm) and thin layer chromatography on silica gel 60 F254 aluminum sheets. Melting points were determined on an electrothermal melting apparatus. High-resolution mass spectrometry spectra were recorded using electrospray ionization or matrix-assisted laser desorption ionization/time of flight techniques at the University of Illinois Mass Spectrometry Core Facility (Urbana-Champaign, IL).
YWI92. 4-Methoxyphenylmagnesium bromide (6 ml; 2.5 mmol) was added dropwise to a solution of N-methylisatin (0.40 g; 2.5 mmol) in 10 ml of anhydrous tetrahydrofuran in a 100-ml two-neck round-bottomed flask under N2 atmosphere at 0°C (Scheme 1B). The reaction mixture was warmed to room temperature, stirred for 24 h, and quenched by addition of 10 ml of 1 N HCl. The mixture was extracted with CH2Cl2 (20 ml × 3). The organic layers were combined, dried over anhydrous magnesium sulfate, and filtered, and the solvent was evaporated. The crude product was purified by column chromatography on silica gel eluting with ethyl acetate/hexane to give a light yellow solid (0.58 g; 86%). m.p. = 148–150°C, 1H NMR (300 MHz, CDCl3), d 7.38–7.24 (m, 4H), 7.13–7.02 (t, J = 7.5 Hz, 1 H), 6.91–6.77 (m, 3 H), 3.80–3.71 (s, 3 H), 3.22–3.14 (s, 3 H). 13C NMR (300 MHz, CDCl3) d 178.3, 160.0, 144.0, 132.7, 132.3, 130.2, 127.5, 125.4, 124.0, 114.4, 109.1, 78.1, 55.8, 27.0. High-resolution mass spectrometry: calculated for C16H15NO3 269.11, found 269.1019.
LTG was obtained from Sigma-Aldrich (St. Louis, MO). Compounds were prepared as stock solutions in dimethyl sulfoxide and diluted to desired concentration in perfusion solution. The maximal dimethyl sulfoxide concentration used was 0.1% and had no effect on sodium current amplitude or action potential firing.
Sodium Channel Electrophysiology
Human embryonic kidney cells stably expressing human Nav1.2 were a kind gift from H. A. Hartmann (University of Maryland, College Park, MD) and were grown in Dulbecco's modified Eagle's medium/F-12 media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), and Geneticin (G418, 500 μg/ml; Sigma-Aldrich). Cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
Sodium currents were recorded using the whole-cell configuration of the patch-clamp recording technique with an Axopatch 200 amplifier (Molecular Devices, Sunnyvale, CA). All voltage protocols were applied using pClamp 9 software (Molecular Devices) and a Digidata 1322A (Molecular Devices). Currents were amplified, low pass filtered (2 kHz), and sampled at 33 kHz. Borosilicate glass pipettes were pulled using a Brown-Flaming puller (model P97; Sutter Instruments Company, Novato, CA) and heat-polished to produce electrode resistances of 0.5 to 1.5 MΩ when filled with the following electrode solution: 130 mM CsCl, 1 mM MgCl2, 5 mM MgATP, 10 mM BAPTA, and 5 mM HEPES (pH adjusted to 7.4 with CsOH). Cells were plated on glass coverslips and superfused with solution containing the following composition: 130 mM NaCl, 4 mM KCl, 1 mM CaCl2, 5 mM MgCl2, 5 mM HEPES, and 5 mM glucose (pH adjusted to 7.4 with NaOH). All sodium channel current experiments were performed at room temperature (20–22°C). After establishing whole-cell configuration, a minimum series resistance compensation of 75% was applied, and cells were held at -100 mV for 5 min to account for any equilibrium gating shifts. Capacitive and leak currents were corrected for using standard P/4 protocols except during steady-state inactivation and use-dependent block protocols. After control recordings, compound solutions were applied for 5 min to allow for bath equilibration. Tonic block was assessed by comparing peak sodium current in drug-free conditions with peak current when drug was present. Dose response data were fitted using the Hill equation:
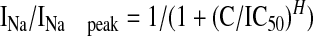 |
(1) |
where C is the drug concentration, IC50 is the concentration that blocks 50% of the current, and H is the Hill coefficient.
Conductance as a function of voltage was derived from the current-voltage relationship using the equation g = INa/(V - ENa), where V is the test potential and ENa is the reversal potential. The voltage dependence of activation and steady-state inactivation data were fitted by the following equation:
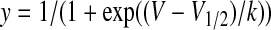 |
(2) |
where y is the normalized conductance (g/gmax) or the normalized current for activation and inactivation, respectively, V1/2 is voltage of half-maximal activation or inactivation, and k is the slope factor. The difference between the V1/2 value in the presence and absence of compound is shown as ΔV1/2 (millivolts). Time constants for recovery from inactivation and development of inactivation were obtained using either a single or a double exponential function:
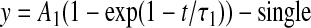 |
(3) |
where A1 is the coefficients for the exponential, t is time (milliseconds), and τ1 is the time constant, and
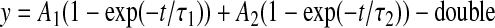 |
(4) |
where A1 and A2 are the coefficients for the fast and slow exponentials, t is time (milliseconds), and τ1 and τ2 are the fast and slow time constants, respectively. The percentage of the current represented by the fast time constant was calculated from the equation 100% × A1/(A1 + A2), where A1 and A2 are the amplitudes of the fast and slow gating modes, respectively.
The apparent affinity (Ki) for the inactivated state of the channel was calculated using the steady-state inactivation and dose-response curves (Kuo and Bean, 1994). Data were fit using a Boltzmann function, and the V1/2 and k values were calculated. This method uses the following equation:
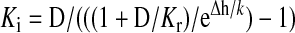 |
(5) |
where Δh is the shift of the steady-state curve, k is the slope of the steady-state inactivation curve, D is the drug concentration tested, and Kr is the affinity for the resting state (IC50) taken from the dose-response curve (Bean et al., 1983).
Brain Slice Preparation
Coronal hippocampal slices (250–300 μm) were prepared from animals (300–450 g; Sprague-Dawley rats) with TLE and age-matched controls. TLE animals used were documented as having four or more spontaneous seizures per day by EEG recordings, 2 to 3 months after the induction of status epilepticus. Animals were euthanized with halothane and then decapitated. The brains were rapidly removed and placed in chilled (4°C) artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, 0.5 mM l-ascorbic acid, 10 mM glucose, 25 mM NaHCO3, and 2 mM pyruvate (oxygenated with 95% O2 and 5% CO2). Slices were prepared using a sectioning system (Vibratome 1000 Plus; Vibratome, St. Louis, MO) and transferred to a chamber containing oxygenated ACSF, incubated at 37°C for 20 to 35 min, and then stored at room temperature. For recording, slices were held in a small chamber perfused with heated (32°C) oxygenated ACSF at 2 ml/min. CA1 hippocampal neurons were visually identified by infrared videomicroscopy using an Axioscope microscope (Carl Zeiss, Oberkochen, Germany). Whole-cell current-clamp recordings were performed using borosilicate glass pipettes with resistances of 3.5 to 4.0 MΩ when filled with an intracellular recording solution containing 120 mM potassium gluconate, 10 mM NaCl, 2 mM MgCl2, 0.5 mM K2EGTA, 10 mM HEPES, 4 mM Na2ATP, and 0.3 mM NaGTP (pH adjusted to 7.2 with KOH). Action potentials were evoked with a series of depolarizing current injection steps.
In Vivo Experiments
MES Acute Seizure Tests. Adult male Sprague-Dawley rats (100–150 g) were obtained from Charles River Laboratories (Raleigh, NC) and were fed, handled, and housed in a matter consistent with the recommendations of the National Research Council's Guide for the Care and Use of Laboratory Animals. No insecticides capable of altering hepatic drug metabolism were used. All animals had free access to food (Prolab RMH 3000; W.F. Fisher, Somerville, NJ) and water except during the times they were used in experimental procedures. At the conclusion of each experiment, animals were euthanized in accordance with the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Institute of Laboratory Resources, and the University of Utah's policy on the humane care and use of laboratory animals. For the MES acute seizure test, 60 Hz of alternating current was delivered via corneal electrodes for a period of 0.2 s. A drop of anesthetic solution containing 0.5% tetracaine hydrochloride in normal saline was placed on each eye of each animal before placement of the corneal electrodes for stimulation. Supramaximal seizures were elicited at a current intensity of 150 mA, five times that necessary to evoke maximal threshold seizures. Immediately after the stimulation, rats were observed for elicited seizure activity characterized by ataxia manifested by evidence of abnormal or uncoordinated gait and stance. Since individual animals may have peculiarities in gait, equilibrium, and placing response, all rats used for evaluating toxicity were examined before the test drug was administered for evidence of pre-existing impairment. Protection was defined as the absence of these behaviors. All drugs were either dissolved or suspended in 0.5% (w/v) methylcellulose or 0.9% sodium chloride (saline). The time of peak effect (TPE) for YWI92 was determined at 0.25, 0.5, 1, 2, 4, 6, and 8 h following oral administration. Toxicity was determined at TPE using the above-noted criteria at time points 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h. Rats were observed by trained and experienced technicians observing for motor impairment, ataxia, or other signs of behavioral toxicity. For each compound, the TPE was determined and then used to calculate an ED50 and TD50.
Spontaneous Temporal Lobe Seizures (TLE) Model. YWI92 was assessed in a rat model of spontaneous temporal lobe seizures, a model that has many features in common with human temporal lobe epilepsy. The following protocol was used.
Surgery. Adult male Sprague-Dawley rats weighing 250 to 300 g received bipolar twisted pair stainless steel electrodes to either hemisphere bilaterally in the posterior ventral hippocampus for stimulation and recording (coordinates from bregma AP, -5.3 mm; ML, -4.9 mm; DV, -5.0 mm; and bite at -3.3 mm) (Paxinos and Watson, 1996). Additional monopolar indifferent reference and ground electrodes were placed over the cerebellum. All electrodes, intracerebral and reference, were attached to Amphenol connectors and secured to the skull with jeweler's screws and dental acrylic.
Induction of Status Epilepticus. One week following surgery, rats were stimulated through the hippocampal electrode to induce limbic status epilepticus using a protocol described by us previously (Lothman et al., 1989). In brief, animals were stimulated for 90 min with 10-s trains of 50-Hz, 1-ms biphasic square wave with a maximal intensity of 400 μA peak to peak delivered every 11 s. After 90 min, stimulation was stopped, and hippocampal activity was recorded for a minimum of 8 h to ensure that a prolonged period of continuous EEG seizure activity was maintained. Animals that exhibited continuous electrographic seizure activity for at least 8 h after stimulation were at uniform risk for development of limbic epilepsy. Animals (approximately 15%) that did not meet the EEG criteria of minimal continuous seizure activity were not maintained, because their chance of developing chronic epilepsy was extremely low.
Following the induction of and recovery from limbic status epilepticus, rats were placed in standard laboratory housing. Three months after the induction of status epilepticus, animals were evaluated for the presence and frequency of spontaneous temporal lobe seizures, because the seizure pattern and frequency will have plateaued by this time (Bertram and Cornett, 1994). During the monitoring phase, rats were placed in specially designed cages, which allowed full mobility of the animals, good visualization for video monitoring, and a stable recording environment. Animals had free access to food and water and were kept under a standard 12-h light/dark cycle. Seizures were recorded and documented using a commercial computerized EEG program (Harmonie; Stellate Systems, Westmount, QC, Canada). All data are reviewed at an offline reading station connected to the vivarium computers via a local area network. The time of occurrence, behavioral severity (Racine 5 point scale), and duration for all seizures were noted.
Seizure Determination. Electrographic seizures in the rats were characterized by the paroxysmal onset of high frequency (greater than 5 Hz) increased amplitude discharges that showed an evolutionary pattern of a gradual slowing of the discharge frequency and subsequent post-ictal suppression. Seizure duration was measured from the onset of the high-frequency activity or initial spike to the cessation of the terminal regular electrographic clonic activity. Testing of YWI92 was carried out on animals exhibiting a regular pattern of a minimum of four seizures over a 6-h period from 10:00 AM to 4:00 PM every day. This choice was made to be certain that any effect following the administration of YWI92 was a true drug effect and not the result of random variation in seizure frequency.
Drug Administration. To provide stable and consistent levels, YWI92 was administered intraperitoneally. Under halothane anesthesia, a silicon tube was placed in the abdomen through a small puncture in the midline. The tube was sewn into place and then tunneled subcutaneously to the back, where it exited just behind the electrode headset. The tube was secured to the headset through a small tube that was built into the headset. The line was used so that the animals would not be disturbed by an intraperitoneal injection of YWI92. All containers and materials for drug injection were sterilized. YWI92 was suspended in normal saline using a high-speed shear mixer, and the injections were made within 30 min of mixing.
Data Analysis
Electrophysiology data analysis was performed using Clampfit software version 9 (Molecular Devices) and Origin version 6 (OriginLab Corp, Northampton, MA). Statistical analyses were performed using the standard one-way analysis of variance followed by Tukey's or Dunn's post hoc test (SigmaStat; SPSS Inc., Chicago, IL). Averaged data are presented as means ± S.E.M. Statistical significance was set at P < 0.05.
Results
YWI92 Inhibits hNav1.2 Channel Currents in a Voltage-Dependent Manner. To determine the affinity of LTG and YWI92 for the resting state of Nav1.2 channels stably expressed in human embryonic kidney 293 cells, a single pulse voltage protocol was used. Depolarization of cells from a holding potential of -100 to +10 mV for a duration of 12 ms evoked transient inward sodium currents that inactivated within a few milliseconds. Both LTG and YWI92 inhibited the recorded sodium currents in a concentration-dependent manner, which was reversible on washout (Fig. 1, A and B). The calculated IC50 values for LTG and YWI92 using the Hill equation were 2.6 mM with a Hill slope of 0.9 and 1.1 mM with a Hill slope of 1.0, respectively. At a more depolarized holding potential (-60 mV), both LTG and YWI92 yielded lower IC50 values of 172 and 57.1 μM. The Hill slope for YWI92 remained unchanged, whereas the Hill slope for LTG was reduced to 0.7. These results suggest that both LTG and YWI92 have a higher affinity for sodium channels in the inactivated state over those in the resting state. At a holding potential of -60 mV, 30 μM YWI92 and LTG exhibited similar tonic block (33.2 ± 3.5 and 27.8 ± 3.7%, respectively). We compared the effects of both compounds on Nav1.2 channel gating at this concentration.
Fig. 1.

Concentration-dependent curves for LTG and YWI92. Sodium currents were elicited by a step depolarization to +10 mV for 12 ms from a holding potential of either -60 mV or -100 mV. Dose-response relationships for LTG (A) and YWI92 (B) are shown along with example current traces of drug block at 100 μM at a holding potential of -100 or -60 mV. The IC50 values at a holding potential of -100 and -60 mV for LTG were 2.6 mM and 172 μM, respectively, and 1.2 mM and 57.1 μM, respectively, for YWI92. The chemical structures of LTG and YWI92 are shown within the respective dose-response curves. Data represent mean ± S.E.M. Smooth lines represent the least-squares fit when data were fitted with the Hill equation INa/INa peak = 1/(1 + (C/IC50)H), where C is the drug concentration, IC50 is the concentration that blocks 50% of the current, and H is the hill coefficient.
Modulation of Sodium Channel Activation. To study activation parameters, sodium currents were elicited by applying a 25-ms voltage step ranging from -80 to +20 mV in 5-mV increments from a holding potential of -120 mV (Fig. 2). In the presence of YWI92 (30 μM), a small hyperpolarizing shift in half-activation voltage (V1/2) by -6.2 mV was observed (from V1/2 =-19.0 ± 1.2 mV, n = 14 in control to -25.2 ± 2.1 mV, n = 6 in the presence of YWI92; P < 0.005). Slope factors (k) were unchanged (k =-5.1 ± 0.3 mV in control and -5.7 ± 0.5 mV in presence of YWI92). LTG (30 μM) had no effect on activation parameters.
Fig. 2.

Voltage dependence of channel conductance was derived from the current-voltage relationship as described under Materials and Methods. Under drug-free conditions, the V1/2 and k values were -19.0 ± 1.2 mV and -5.1 ± 0.3 (n = 14), respectively. LTG (30 μM) had no effect on channel activation parameter values (V1/2 =-20.0 ± 1.6 mV; k =-5.4 ± 0.4; n = 8). In contrast, YWI92 (30 μM) caused a significant hyperpolarizing shift of the half-activation voltage (V1/2 =-25.2 ± 2.1 mV; k = -5.7 ± 0.5; n = 6; P < 0.005). Slope values were unchanged between control and drug conditions. Data represent mean ± S.E.M. Smooth lines correspond to the average of the least-squares fits when data were fitted with the Boltzmann equation.
YWI92 Has Greater Affinity for Channels Inactivated during Long Depolarizing Prepulses. Since both LTG and YWI92 exhibited greater tonic block at depolarized potentials, their effects on inactivation equilibrium gating were determined. Inactivation was examined using either a short 10-ms prepulse, or a longer 30-s prepulse as described previously (Xie et al., 2001). Cells were initially held at -120 mV and then subjected to a prepulse at voltages ranging from -135 to +20 mV. This was followed by a test pulse to +10 mV to establish the extent of channel inactivation (Fig. 3, A and B; Table 1). LTG (30 μM) had no effect on inactivation when using a short 10-ms inactivating prepulse. In contrast, YWI92 (30 μM) caused a small, but significant (P < 0.05), hyperpolarizing shift in the inactivation curve by -10 mV and increased the slope value (k).
Fig. 3.

Steady-state fast and slow inactivation. Steady-state inactivation was determined using either a short conditioning pulse of 10 ms (LTG; A) and (YWI92; B) or a longer 30-s pulse (C) from a holding potential of -120 mV. Data were recorded under drug-free conditions or in the presence of LTG or YWI92 (30 μM). D, example traces for the effects of LTG and YWI92 at a 30-s prepulse voltage of -50 mV. Horizontal bar, 0.5 ms. Data represent mean ± S.E.M. Smooth lines correspond to the average of the least-squares fits when data were fitted with the Boltzmann equation.
TABLE 1.
Steady-state inactivation parameters
|
Fast Inactivation
|
Slow Inactivation
|
|||||
|---|---|---|---|---|---|---|
| V1/2 | k | n | V1/2 | k | n | |
| mV | mV | |||||
| Control | –41.7 ± 1.8 | 9.2 ± 0.5 | 10 | –47.5 ± 0.6 | 9.9 ± 0.5 | 9 |
| YWI92 (30 μM) | –51.7 ± 4.2a | 11.7 ± 0.8a,b | 5 | –62.9 ± 1.7c | 10.6 ± 0.4 | 5 |
| LTG (30 μM) | –43.8 ± 2.9 | 8.2 ± 0.2 | 5 | –58.3 ± 3.9c | 9.9 ± 0.3 | 4 |
P < 0.05 vs. control
P < 0.01 vs. LTG
P < 0.001 vs. control
To evaluate the affinity of LTG and YWI92 for channels inactivated by a longer 30-s inactivating prepulse, a similar protocol was used except that a 50-ms pulse at -100 mV was applied following the longer depolarizing prepulse to recover unblocked fast inactivated channels (Sandtner et al., 2004). In contrast to its effects on inactivation using a short 10-ms inactivating prepulse, LTG (30 μM) significantly shifted the inactivation curve resulting from the longer depolarizing prepulse by -10.8 mV. A similar effect was seen with YWI92 (30 μM), resulting in a shift of -15.4 mV. Slope factors remained unchanged (Fig. 3C; Table 1).
From the inactivation and dose-response curves, the apparent affinity for the inactivated state (Ki) was calculated using the following equation: Ki = D/(((1 + D/Kr)/eΔh/k) - 1) first described by Bean et al. (1983). Using this equation, YWI92 was calculated to have an apparent IC50 value of 7.4 μM for the channels inactivated by a long 30-s prepulse (Ki). Given that the affinity for the resting state of the channel (Kr) was determined previously to be 1.2 mM (holding potential of -100 mV), a ratio of Kr/Ki suggests that YWI92 has a 135-fold greater affinity for channels inactivated during long depolarizing prepulses than over the resting state of the channel. Using the same assumption, the apparent affinity for inactivated channels for LTG was calculated to be 15.9 μM, yielding a Kr/Ki ratio value of 163.
Delay of Recovery from Inactivation at -60 mV. During epileptic seizures neuronal firing is thought to increase in frequency. Compounds that delay recovery from inactivation ultimately reduce the number of channels available to open and pass current during prolonged depolarizations. We examined the effects of LTG and YWI92 on recovery from both short and long inactivating prepulses (Fig. 4; Table 2). To evaluate the effects of each compound on recovery after short inactivating prepulses, cells were held at -120 mV for 1 s and inactivated with a depolarizing step to 0 mV for 10 ms. Sodium channels were subsequently recovered at -60 mV, a typical resting membrane potential for neurons, for a variable length of time (1 ms to 100 s) and then subjected to a test pulse of +10 mV to determine the extent of recovery. Data were normalized to the peak current amplitude under control, drug-free conditions and best fitted using a single exponential function. In the presence of LTG (30 μM), but not YWI92 (30 μM), the fast time constant (τ1) was significantly (P < 0.05) increased, suggesting a delay in the recovery kinetics of channels inactivated using a short 10-ms inactivating prepulse.
Fig. 4.

Recovery from inactivation at -60 mV is delayed. Modulation of recovery from fast and slow inactivation was examined for LTG and YWI92 at a concentration of 30 μM. Recovery from fast and slow inactivation was assessed using a two-pulse protocol. From a holding potential of -120 mV, a step to 0 mV for 10 ms was used to assess recovery from fast inactivation (LTG, A; YWI92, B), whereas a longer step of 30 s was applied to assess recovery from slow inactivation (C). Recovery was determined at -60 mV using a variable period of 1 ms to 100 s. The proportion of recovered channels assessed with a final voltage step to +10 mV. D, example traces for the effects of LTG and YWI92 on recovery from slow inactivation at 100 s. Horizontal bar, 1 ms. Data points represent the mean ± S.E.M. Smooth lines correspond to the average of the least-squares fits when data were fitted by either a single or a double exponential function.
TABLE 2.
Recovery from inactivation
|
Recovery from Fast Inactivation
|
Recovery from Slow Inactivation
|
||||||
|---|---|---|---|---|---|---|---|
| τ1 | n | τ1 | τ2 | Recovery | A1 | n | |
| Ms | s | % | |||||
| Control | 17.5 ± 1.5 | 8 | 2.6 ± 0.9 | 22.0 ± 2.8 | 100 | 43.8 ± 10 | 13 |
| YWI92 (30 μM) | 19.6 ± 0.8 | 4 | 16.6 ± 1.3b | 61.7 ± 3b | 100 | 5 | |
| LTG (30 μM) | 21.2 ± 1.8a | 4 | 17.8 ± 1.8b | 24.3 ± 3b,c | 100 | 4 | |
P < 0.05 vs. control
P < 0.001 vs. control
P < 0.001 vs. LTG
To determine whether LTG and YWI92 exhibited a preference for channels inactivated by short or long prepulses, we increased the duration of the depolarization step used to 30 s. This longer inactivating prepulse allows channels to occupy additional inactivated states that have been referred to as slow-inactivated states. Recovery from these states requires longer times of several seconds. Data were normalized to the peak current amplitude recorded under control, drug-free conditions. Under control conditions, the rates of recovery were best fit with a double exponential function (Fig. 4, C and D; Table 2). In the presence of YWI92 and LTG (both at 30 μM), channel recovery was best fit to a single exponential function. The most profound effect was a substantial decrease in the proportion of channels that had recovered after 100 s at -60 mV in the presence of both test compounds.
Development of Inactivation at 0 mV. To determine whether LTG and YWI92 could modulate the rate at which sodium channels entered the inactivated state, a development of inactivation protocol was developed (Fig. 5). From a holding potential of -120 mV, an inactivating prepulse to 0 mV was applied ranging from 1 ms to 100 s, followed by a step to -100 mV for 50 ms and a final step to +10 mV for 12 ms to assess the extent of sodium channel inactivation. Data were normalized to the peak current amplitude and fitted using a double exponential function. Under control conditions, the time constants for the development of inactivation were τ1 = 2.38 ± 0.61 s, τ2 = 16.7 ± 2.35 s, and A1 = 64 ± 7% (n = 7). YWI92 significantly reduced both time constants, accelerating the rate at which sodium channels developed inactivation and significantly reduced the percentage of development represented by the fast time constant τ1 compared with both control and LTG (30 μM YWI92: τ1 = 0.41 ± 0.02 s, τ2 = 6.40 ± 0.62, and A1 = 42 ± 4%; n = 4; P < 0.05). Time constants were also reduced in the presence of LTG but were not different from control (30 μM LTG: τ1 = 1.58 ± 0.24 s, τ2 = 12.2 ± 1.76, and A1 = 65 ± 4%; n = 5).
Fig. 5.

Development of slow inactivation at 0 mV. Development of inactivation was assessed using a two-pulse protocol. From a holding potential of -120 mV, a prepulse to 0 mV was applied, ranging from 1 ms to 100 s followed immediately by a step to -100 mV for 50 ms to recover fast inactivated channel. Availability of sodium channels was assessed using a step to +10 mV for 12 ms. Data points represent the mean ± S.E.M. Smooth lines correspond to the average of the least-squares fits when data were fitted by a double exponential function.
Block by YWI92 Is Use-Dependent. In addition to voltage-dependent block, AEDs often exhibit use-dependent block. This characteristic is considered important since it allows the enhanced block of high-frequency action potential discharges that occur during epileptic seizures (Catterall, 1999; Rogawski and Löscher, 2004). The effects of LTG and YWI92 on a train of depolarizing voltage steps from a holding potential of -120 to +10 mV for 12 ms at a frequency of 20-Hz pulse are shown in Fig. 6. Under control, drug-free conditions, accumulation of inactivated channels as a result of the protocol resulted in an 8.8 ± 1.3% (n = 11) reduction in peak current. In the presence of LTG and YWI92 (30 μM), current amplitudes were further reduced by 15.6 ± 4.0% (n = 4; P = 0.051) and 21.2 ± 2.7% (n = 7; P < 0.001), respectively.
Fig. 6.

Use-dependent block by YWI92 and LTG (30 μM) was assessed using a depolarizing pulse to +10 mV for 12 ms at a 20-Hz pulse frequency for 30 pulses. Peak current amplitude was normalized to the first pulse in each experiment to account for tonic block. Data points represent the mean ± S.E.M.
YWI92 Inhibits Burst Firing in CA1 Neurons of Chronic Limbic Epilepsy Animals. Because sodium channels play a critical role in the generation of action potentials, the actions of YWI92 on sodium channel currents should affect action potential generation in neurons. To explore this idea, we examined the actions of YWI92 on membrane excitability of CA1 neurons from both control brain slices and brain slices prepared from animals with chronic TLE (Fig. 7). To standardize our tests the resting membrane potential was recorded under current-clamp conditions and then maintained at -65 mV (indicated by arrow) by injection of a DC current before stimulation. Depolarizing current injections (range, 20–260 pA, for 300 ms) were used to elicit AP discharges from control CA1 neurons (Fig. 7A) and TLE CA1 neurons (Fig. 7B). Resting membrane potentials between control (-64.4 ± 0.4 mV; n = 22) and TLE CA1 neurons (-63.7 ± 0.8 mV; n = 10) were not significantly different. For comparison, the number of APs elicited by a depolarizing current injection of 140 pA was used to determine the inhibitory actions of YWI92. Under these conditions, the most profound observation was the significantly greater number of APs that were evoked in TLE neurons compared with control neurons (6.0 ± 2.1, n = 6 for TLE compared with 1.3 ± 0.2, n = 16 for control; P < 0.05). In control CA1 neurons, YWI92 at 10 μM(n = 3) was without effect but did decrease the number of APs evoked by 16.5 ± 10.5% at 30 μM(n = 5) and 67.4 ± 12.3% at 100 μM(n = 7). In contrast, YWI92 caused a profound inhibition of AP firing in CA1 neurons from TLE animals, reducing the number of APs evoked by 76.6 ± 15.8% (n = 4) and 88.8 ± 6.7% (n = 5) for 10 and 30 μM, respectively. At 100 μM, a complete inhibition of AP firing was observed (n = 4; Fig. 7C). These effects of YWI92 on AP firing were fully reversible on washout.
Fig. 7.

YWI92 selectively inhibits action potential discharges in TLE CA1 neurons. To standardize our tests the resting membrane potential was recorded and then maintained at -65 mV (indicated by arrow) by injection of a DC current before stimulation. Action potentials were evoked using a series of DC injection steps from -20 to 260 pA in 10-pA steps for 300 ms, with a 5-s interpulse interval. A, effects of 100 μM YWI92 on control CA1 neurons. B, effects of both 30 and 100 μM YWI92 on CA1 neurons from TLE animals is shown. In both cases only current steps of -20-, 0-, 30-, 60-, and 140-pA DC current injections are shown for clarity. Concentration-dependent effects of YWI92 on both control (stripped columns: 10 μM, n = 3; 30 μM, n = 5; 100 μM, n = 7) and TLE (filled columns: 10 μM, n = 4; 30 μM, n = 5; 100 μM, n = 4) neurons is shown in C. Data points represent the mean ± S.E.M.
YWI92 Exhibits Anticonvulsant Activity in an Acute Seizure Model and a Chronic Limbic Epilepsy Model. To determine whether the inhibition of epileptiform burst firing in TLE CA1 neurons would translate into AED activity, YWI92 was tested in both the MES acute seizure model and the TLE chronic seizure model. In the MES model, orally administered YWI92 yielded an ED50 value of 22.96 mg/kg. In the Rotarod toxicity test, a TD50 value for YWI92 could not be determined since no signs of sedation or ataxia were observed at doses up to 500 mg/kg.
In the TLE model of chronic epilepsy intraperitoneal administration of 100 mg/kg YWI92 significantly reduced the frequency of spontaneous seizures to less than 25% baseline frequency in the 6 h following administration of the drug (Fig. 8; n = 4; P < 0.01). Although there were also reductions in seizure duration and behavioral severity, these changes did not achieve significance.
Fig. 8.

YWI92 (100 mg/kg; n = 4) reduces the mean number of seizures in animals with chronic TLE over the 6 h following drug administration (vertical frequency scale is seizures/6 h). Although there were reductions in mean seizure duration (seconds) and in behavioral severity (Racine 5 point scale; BSS), these changes did not achieve significance. Data points represent the mean ± S.E.M. *, P < 0.01.
Discussion
Lactams represent an important structural class of compounds that exhibit anticonvulsant activity (Grimm et al., 2003). In this study, we have used the lactam structural motif and designed a novel compound, YWI92, incorporating a rigid analog of the α-hydroxyamide moiety, a structural feature previously shown to be important for increased affinity for the inactivated state of the sodium channel (Grimm et al., 2003; Jones et al., 2007).
Lactams have been shown to displace [3H]batrachotoxinin A 20-α-benzoate binding from rat brain cerebral cortex synaptosomes, suggesting that they share the same binding site as many clinically used AEDs (Brouillette et al., 1988; Clare et al., 2000). Here, we have compared the actions of YWI92 and LTG, a clinically useful AED, for inhibition of the sodium channel isoform Nav1.2. This particular isoform represents a reasonable target for the evaluation of potential AEDs since it is abundantly expressed within the hippocampus, with specific expression along fine axonal fibers and varicosities, indicating close localization to presynaptic release sites (Jarnot and Corbett, 2006). Furthermore, mutations in SCN2A, the gene encoding Nav1.2, have been associated with epilepsy, supporting its role in controlling neuronal activity (Meisler and Kearney, 2005).
A common feature of many AEDs is their ability to bind with greater affinity to open and inactivated channels over channels at rest (Kuo and Bean, 1994; Kuo et al., 1997). This voltage- and frequency-dependent block is considered critical for the selective suppression of epileptiform activity prevalent during epileptic seizures, while sparing normal channel function (Rogawski and Löscher, 2004). This characteristic is best described by the modulated receptor hypothesis that states that drug affinity for the channel receptor is dependent on the channel state, transitioning from a low- to high-affinity site during inactivation of the channel (Hondeghem and Katzung, 1977). In agreement with the modulated receptor hypothesis, block of Nav1.2 by both YWI92 and LTG was voltage dependent, with greater block at more depolarized holding potentials, suggesting a greater affinity for inactivated channels. However, sodium channels can exist in multiple inactivated states, including fast, slow, and even an ultraslow inactivated state (Goldin, 2003), and it is still unclear whether selective affinity for a particular inactivated state is predictive of AED activity. We determined the effects of LTG and YWI92 on channels inactivated using a short 10-ms depolarizing prepulse and on channels inactivated using a 30-s depolarizing prepulse (Sandtner et al., 2004). Using short depolarizing prepulses, channels are thought to enter a fast inactivation state, involving the short intracellular loop between domains III and IV (West et al., 1992). This state is important for the voltage-dependent block by the local anesthetic lidocaine but not for its use-dependent block (Vedantham and Cannon, 1999). Fast inactivated channels are also thought to be targeted by the anticonvulsants LTG and phenytoin, although the rate of binding is considered to be extremely slow (Kuo and Bean, 1994; Kuo and Lu, 1997). For this to occur, the channel must continue to occupy the fast inactivated conformation even after prolonged depolarizations, leading others to conclude that it may preferentially bind to slow inactivated channels rather than fast (Xie et al., 1995). Slow inactivated channels are targets for mibefradil and lidocaine (Chen et al., 2000; McNulty and Hanck, 2004), and more recently, for the AED lacosamide (Errington et al., 2008). Slow inactivation is distinct from fast inactivation and does not involve the fast inactivation gate. Instead, mutational and sulfhydryl modification studies point toward a rearrangement of the channel pore, involving the P-segment and the S6 segments (Vilin and Ruben, 2001).
Our studies suggest that YWI92 has affinity for channels inactivated during both a short and long depolarizing prepulse. YWI92 caused a small hyperpolarizing shift in the inactivation curve when using a short 10-ms prepulse and shifted the activation curve, although recovery from inactivation was not affected. When using a longer depolarizing prepulse, YWI92 caused a hyperpolarizing shift in inactivation and attenuated the proportion of channels recovered after a 30-s depolarizing prepulse. Furthermore, YWI92's acceleration of the development of inactivation was only apparent following inactivating prepulses that were longer than 30 ms. Although the effects of YWI92 on inactivation and recovery from inactivation were more pronounced at longer prepulses, it is difficult to conclude that YWI92 preferential associates with slow inactivated channels over fast channels since slow association with fast inactivated channels cannot be ruled out.
In contrast to YWI92, LTG had no effect on inactivation parameters when using a short 10-ms prepulse, but it did cause a hyperpolarizing shift in the inactivation curve when using a longer 30-s depolarizing prepulse. Again, these differences can be accounted for by either slow binding of LTG to fast inactivated channels (Kuo and Lu, 1997) or greater affinity for slow inactivated channels (Xie et al., 1995). Interestingly, LTG did delay recovery from fast inactivation, suggesting that it can bind to channels inactivated during a short 10-ms prepulse, delaying their recovery.
Several studies have now highlighted the importance of specific sodium channel inactivation states in regulating neuronal activity, synaptic integration, and neuronal spiking. For example, in cortical pyramidal neurons, sodium channel slow inactivation accounted for the reduction in spike rate during high-frequency stimulation and the eventual failure to evoke a spike (Fleidervish et al., 1996). Reduction in sodium channel activity by either protein kinase C/protein kinase A phosphorylation or activation of G protein-coupled receptors has been attributed to the increased development of slow inactivated channels (Carr et al., 2003). Development of slow inactivation during high-frequency action potential discharges is also thought to play a major role in attenuating action potential back propagation into dendritic regions in CA1 hippocampal neurons, controlling synaptic integration and postsynaptic firing in the axon (Jung et al., 1997). In view of the importance of slow inactivation in modulating synaptic integration and activity, the ability of YWI92 to bind to channels inactivated by prolonged a depolarization could be important for the suppression of action potential firing in CA1 hippocampal neurons. It is unclear why YWI92 was more effective at inhibiting epileptiform burst firing in chronically epileptic rats. However, sodium channel isoform expression patterns and gating kinetics are distinct in epileptic neurons and could exist in a state that is preferentially targeted by AEDs (Ketelaars et al., 2001).
Protection against both acute and spontaneous seizures were assessed using the MES model of acute seizures and an electrical stimulation model of TLE with spontaneous seizures. The TLE model has similar pathology to that of humans with mesial temporal lobe epilepsy (Margerison and Corsellis, 1966) and has many common parallels with human drug-resistant epilepsy (Stables et al., 2003). In the MES model, orally administered YWI92 afforded profound protection against the acutely evoked generalized tonic-clonic seizures (ED50 value of 22.96 mg/kg) without any sedative or ataxic side effects (Rotarod toxicity test; TD50 > 500 mg/kg). These actions of YWI92 are encouraging considering that phenytoin, a clinically used AED, had a similar ED50 value of 27.5 mg/kg but a TD50 of 35.6 mg/kg (Lenkowski et al., 2007). Importantly, YWI92 significantly decreased the number of seizures in the TLE model, providing protection in a chronic model of epilepsy with spontaneous seizures.
In summary, we propose that a greater affinity for the inactivated state of a sodium channel is important for the inhibition of epileptiform discharges observed in neurons from animals with chronic TLE and also for anticonvulsant activity in animal models of TLE. This molecular state of sodium channels may be an important target for the development of new anticonvulsant medication with tolerable side effect profiles. Furthermore, the lactam moiety itself may offer an important structural lead from which future development work can begin.
Acknowledgments
We thank the Department of Anesthesiology at the University of Virginia (M.K.P) and acknowledge John M. Williamson for generating the TLE animals. In addition, we acknowledge Drs. Misty Smith and H. Steve White for providing the initial in vivo evaluations of YWI92.
This work was supported by the National Institutes of Health [Grants NIH R01 CA 105435-01, 5 T32 GM008328]; and the United Negro College Fund/Merck Science Initiative.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.108.144709.
ABBREVIATIONS: AED, antiepileptic drug; Nav, voltage-gated sodium; AP, action potential; MES, maximal electric shock; YWI92, 3-hydroxy-3-(4-methoxyphenyl)-1-methyl-1,3-dihydro-indol-2-one; LTG, lamotrigine; TLE, temporal lobe epilepsy; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; ACSF, artificial cerebrospinal fluid; TPE, time of peak effect.
References
- Bean BP, Cohen CJ, and Tsien RW (1983) Lidocaine block of cardiac sodium channels. J Gen Physiol 81 613-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EH and Cornett JF (1994) The evolution of a rat model of chronic spontaneous limbic seizures. Brain Res 661 157-162. [DOI] [PubMed] [Google Scholar]
- Brodie MJ (2001) Do we need any more new antiepileptic drugs? Epilepsy Res 45 3-6. [DOI] [PubMed] [Google Scholar]
- Brouillette WJ, Brown GB, DeLorey TM, Shirali SS, and Grunewald GL (1988) Anticonvulsant activities of phenyl-substituted bicyclic 2,4-oxazolidinediones and monocyclic models. Comparison with binding to the neuronal voltage-dependent sodium channel. J Med Chem 31 2218-2221. [DOI] [PubMed] [Google Scholar]
- Carr DB, Day M, Cantrell AR, Held J, Scheuer T, Catterall WA, and Surmeier DJ (2003) Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron 39 793-806. [DOI] [PubMed] [Google Scholar]
- Catterall WA (1999) Molecular properties of brain sodium channels: an important target for anticonvulsant drugs. Adv Neurol 79 441-456. [PubMed] [Google Scholar]
- Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26 13-25. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2002) Molecular mechanisms of gating and drug block of sodium channels. Novartis Found Symp 241 206-218; discussion 218–232. [PubMed] [Google Scholar]
- Chen Z, Ong BH, Kambouris NG, Marbán E, Tomaselli GF, and Balser JR (2000) Lidocaine induces a slow inactivated state in rat skeletal muscle sodium channels. J Physiol 524 37-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare JJ, Tate SN, Nobbs M, and Romanos MA (2000) Voltage-gated sodium channels as therapeutic targets. Drug Discov Today 5 506-520. [DOI] [PubMed] [Google Scholar]
- Errington AC, Stöhr T, Heers C, and Lees G (2008) The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol 73 157-169. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Friedman A, and Gutnick MJ (1996) Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocortical neurones in slices. J Physiol 493 83-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL (2003) Mechanisms of sodium channel inactivation. Curr Opin Neurobiol 13 284-290. [DOI] [PubMed] [Google Scholar]
- Grimm JB, Stables JP, and Brown ML (2003) Design, synthesis, and development of novel caprolactam anticonvulsants. Bioorg Med Chem 11 4133-4141. [DOI] [PubMed] [Google Scholar]
- Hille B (1977) Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol 69 497-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM and Katzung BG (1977) Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta 472 373-398. [DOI] [PubMed] [Google Scholar]
- Jarnot M and Corbett AM (2006) Immunolocalization of NaV1.2 channel subtypes in rat and cat brain and spinal cord with high affinity antibodies. Brain Res 1107 1-12. [DOI] [PubMed] [Google Scholar]
- Jones PJ, Wang Y, Smith MD, Hargus NJ, Eidam HS, White HS, Kapur J, Brown ML, and Patel MK (2007) Hydroxyamide analogs of propofol exhibit state-dependent block of sodium channels in hippocampal neurons: implications for anticonvulsant activity. J Pharmacol Exp Ther 320 828-836. [DOI] [PubMed] [Google Scholar]
- Jung HY, Mickus T, and Spruston N (1997) Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci 17 6639-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketelaars SO, Gorter JA, van Vliet EA, Lopes da Silva FH, and Wadman WJ (2001) Sodium currents in isolated rat CA1 pyramidal and dentate granule neurones in the post-status epilepticus model of epilepsy. Neuroscience 105 109-120. [DOI] [PubMed] [Google Scholar]
- Kuo CC and Bean BP (1994) Slow binding of phenytoin to inactivated sodium-channels in rat hippocampal-neurons. Mol Pharmacol 46 716-725. [PubMed] [Google Scholar]
- Kuo CC and Lu L (1997) Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol 121 1231-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Chen RS, Lu L, and Chen RC (1997) Carbamazepine inhibition of neuronal Na+ currents: quantitative distinction from phenytoin and possible therapeutic implications. Mol Pharmacol 51 1077-1083. [DOI] [PubMed] [Google Scholar]
- Lenkowski PW, Batts TW, Smith MD, Ko SH, Jones PJ, Taylor CH, McCusker AK, Davis GC, Hartmann HA, White HS, et al. (2007) A pharmacophore derived phenytoin analogue with increased affinity for slow inactivated sodium channels exhibits a desired anticonvulsant profile. Neuropharmacology 52 1044-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Bekenstein JW, and Perlin JB (1989) Self-sustaining limbic status epilepticus induced by `continuous' hippocampal stimulation: electrographic and behavioral characteristics. Epilepsy Res 3 107-119. [DOI] [PubMed] [Google Scholar]
- Margerison JH and Corsellis JA (1966) Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain 89 499-530. [DOI] [PubMed] [Google Scholar]
- McNulty MM and Hanck DA (2004) State-dependent mibefradil block of Na+ channels. Mol Pharmacol 66 1652-1661. [DOI] [PubMed] [Google Scholar]
- Meisler MH and Kearney JA (2005) Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 115 2010-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G and Watson G (1996) The Rat Brain in Stereotaxic Coordinates, Academic Press, San Diego, CA.
- Remy S and Beck H (2006) Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 129 18-35. [DOI] [PubMed] [Google Scholar]
- Rogawski MA and Löscher W (2004) The neurobiology of antiepileptic drugs. Nat Rev Neurosci 5 553-564. [DOI] [PubMed] [Google Scholar]
- Sandtner W, Szendroedi J, Zarrabi T, Zebedin E, Hilber K, Glaaser I, Fozzard HA, Dudley SC, and Todt H (2004) Lidocaine: a foot in the door of the inner vestibule prevents ultra-slow inactivation of a voltage-gated sodium channel. Mol Pharmacol 66 648-657. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH, and Goldin AL (2001) Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci 21 7481-7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stables JP, Bertram E, Dudek FE, Holmes G, Mathern G, Pitkanen A, and White HS (2003) Therapy discovery for pharmacoresistant epilepsy and for disease-modifying therapeutics: summary of the NIH/NINDS/AES models II workshop. Epilepsia 44 1472-1478. [DOI] [PubMed] [Google Scholar]
- Ulbricht W (2005) Sodium channel inactivation: molecular determinants and modulation. Physiol Rev 85 1271-1301. [DOI] [PubMed] [Google Scholar]
- Vedantham V and Cannon SC (1999) The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J Gen Physiol 113 7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilin YY and Ruben PC (2001) Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys 35 171-190. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, and Catterall WA (1992) A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A 89 10910-10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Dale TJ, John VH, Cater HL, Peakman TC, and Clare JJ (2001) Electrophysiological and pharmacological properties of the human brain type IIA Na+ channel expressed in a stable mammalian cell line. Pflugers Arch 441 425-433. [DOI] [PubMed] [Google Scholar]
- Xie X, Lancaster B, Peakman T, and Garthwaite J (1995) Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native Na+ channels in rat hippocampal neurones. Pflugers Arch 430 437-446. [DOI] [PubMed] [Google Scholar]