

Introduction
The 2004 rules for regulating clinical research and, much worse, their implementation have been a disaster that threatens patients' lives, damages the economy of the UK and compromises the careers of our academic trainees. Despite prior warnings from similar changes in medical education and training, the medical profession has again yielded control over a world-renowned asset to middle-ranking officials with little or no relevant experience, but absolute power to obstruct. That medical educationalists and undergraduate Deans now put more premium on communication of what it feels like to take a pill than on an understanding of dose–response or volume of distribution, is unfortunate. That postgraduate Deans could turn blind eyes to the discrimination of the Medical Training Application Service (MTAS) against gifted trainees with prizes or degrees in pharmacology and clinical pharmacology, was careless. But these little local difficulties do not compare, for impact, to the surrender of clinical research governance to individuals and bodies who are not guided by the principles of the Hippocratic oath, do not care for patients, and, with rare exceptions, have little idea – far less first-hand knowledge – of conducting clinical trials. Clinical research reduces morbidity and mortality worldwide, and for decades the UK had been second only to the USA. Clinical trials contributed to Pharma becoming one of only three net earners for UKplc. All this is now under threat (Figures 1 and 2), at the very time when largesse and enthusiasm for clinical translational research has been at an all time high, and the UK depends more than ever on a thriving pharmaceutical industry [1,2].
Figure 1.
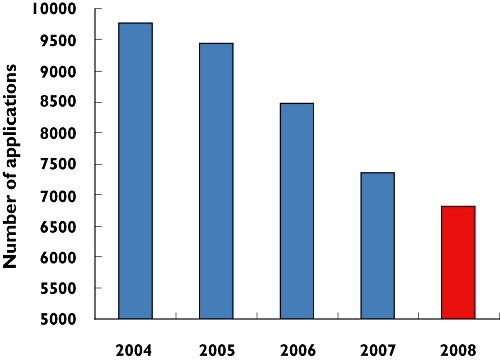
Annual decline in UK submissions for ethical approval since the implementation of the EU directive. Data redrawn from http://www.nres.npsa.nhs.uk/news-and-publications/news/nres-year-in-review/ (year's run April-to-March; 2008 projected from data until October)
Figure 2.
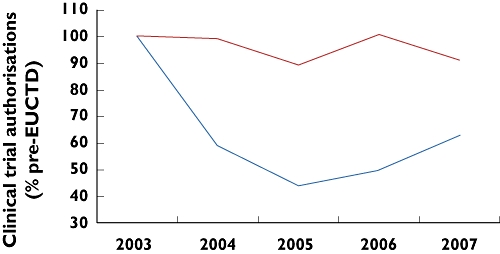
Annual decline in UK Clinical Trial Authorizations (noncommercial sponsors). Data redrawn from http://www.efgcp.be/Downloads/confPresentations/ICRELWebDoc2/1300-3-Break_out-CA-RH.pdf. Competent Authorities in every EU country were surveyed. ‘Rest of Europe’ = seven Western European countries with >50 clinical trial agreements p.a. over the survey period. United Kingdom ( ); Rest of Europe (
); Rest of Europe ( )
)
The need for research governance
Most professions have governance procedures, although few are entirely external. The need for a minimum of three independent bodies to pontificate on each protocol, even after expert peer-review, is unique, inevitably obstructive, and almost certain to be counterproductive. Most governance is periodic and retrospective; for individual doctors assessment of research competence should be incorporated into forthcoming re-validation, whereas annual licensing might be reasonable for facilities. Prospective research governance for individual projects should be triaged according to prior expert review (e.g. during funding) and likely risk; for the vast majority, non-expert review should be a light-touch failsafe measure to protect the public from the occasional bad penny or well-intentioned error that has escaped the notice of both the investigators and expert peer reviewers. It is the latters' role to assess the benefit–risk of any proposed research, and this assessment that is fundamental to an ethical appreciation of the proposal. It is in large part the need for research to define exactly what will happen in advance – the protocol – that renders clinical research so much better and safer than everyday medicine. Failing to appreciate this intrinsically high benefit–risk of research, and lacking the expert knowledge required for individual assessments, Medicines & Healthcare products Regulatory Agency (MHRA), Ethics Committees (REC) and R&D fall back on a fixation with process, form-filling and punctuation that cannot determine whether a proposed piece of research will be beneficial or safe. As amply demonstrated recently in other arenas (financial, social care), obsession with forms is inversely related to disaster-aversion or rogue-detection: what might be called Madoff's law. The result in medicine was Northwick Park. Everything done by the 2004 book, all the boxes ticked, all forms present and correct in the trial master file – but no one among investigators, committees or inspectors had time or incentive to think beyond the forms [3]
Last October I wrote in the British Medical Journal (BMJ) with colleagues from Cambridge and Birmingham, stimulated by a series of atrocious regulatory decisions and complaints from clinician scientists coming to their reviews at grants committees [4]. No longer was it possible within 1 year to have any clinical data to analyse or present. The purpose of the article, describing the many obstacles to starting research, was to stimulate others to register their experiences, and open a dialogue with regulators on a pathway to change. However, despite the chorus of horror stories well summarized by the Editor, and a recent meeting of European regulators and users in Brussels confirming that all has not been harmonious with the introduction of the EU clinical trials directive, UK regulators refuse to acknowledge that we have a major problem [5]. Out of touch, and in denial, MHRA leaders assure me that their assault on academic research is justified by the absence of ethical approval in the majority of trials (20 years behind the times?); cite unpublished or selective data belying the reduction in UK clinical research (a contradictory protest, if they believed their first assertion); and criticise the attractive Dutch model for being inconsistent with European harmonization. Yet their own visible contribution to harmonization is to leave submissions at the mercy of a third-party European database that goes off-air without warning, and after 5 years still requires parchment-and-quill repetitive data entry instead of the trivial IT task of reading the XML output from other national databases. The National Research Ethics Service (NRES) is a paralegal edifice that does not feature in the 2004 Act. It claims a post bag of fans for their 80-page applications, and Alice-like regards its own documentation of year-on-year reduction in submissions (Figure 1) as evidence of procedural success that justifies its mission to protect (from us?) the dignity and well-being of our patients.
Need for a campaign to reform research governance
This personal review will therefore go beyond the descriptive mode of our BMJ piece to document the damage being done; to explain why this is not an inevitable consequence of the EU directive or secondary UK legislation; and to propose the urgent measures and radical solutions now required. As I learned during the MTAS debacle, oppressive bureaucracy often results when middle grades of authority indulge in rule inflation, unchecked by either the evangelists who created the initial rules, or the professional leaders who are too lofty and busy to devote time to consequences of regulation [6]. Those to whom the lot of form-filling usually falls feel too lowly to mount serious resistance. Yet one of the wonders of a medical career is that it allows me to be on the front line of both routine and research clinical care, 30 years after my first study, and very much in touch with those on the starting line. If I am regularly depressed at the endless delays to my own research, I owe it to trainees whose careers are blighted by bureaucracy to expose what is happening. Recently, the Wellcome Trust entrusted me (and three other Clinical Pharmacologists in the UK) with £2.75M each to introduce training in Translational Medicine and Therapeutics. If our subject is to be regenerated by this largesse, the new trainees must be spared sagas such as I relate below (Box 1).
BOX 1 Crazy governance: a tale
In early 2008, I received on behalf of the British Hypertension Society (BHS) research network a £1.9M grant for a programme of three related trials using 25–50-year-old generic drugs. The research questions were agreed over many months by eight senior investigators sharing some 200 years' worth of clinical experience, and authorship of several landmark trials that have shaped National Institute for Clinical Excellence guidance and current practice. The protocols underwent several rounds of international peer review before the award, and are comparing options already practised. In short, it is hard to imagine in what way the year or more of regulatory hurdles could improve quality of the trials, far less provide sufficient improvement to justify morally and financially the cost of delay, during which thousands of people with suboptimally controlled hypertension will have strokes and die. Indeed, one of the ironies of this saga is that the reason for applying as a network for the grant was precisely so that acknowledged leaders in hypertension could agree in advance the important questions and best methods of answering them. By contrast, the multiple reviews since funding have been by groups with no experience of hypertension or clinical research.
If one believed the promotions on the Integrated Research Application Service (IRAS) website, our task would comprise completion of a simple online form, which is then shared by any regulator whose approval is required – namely Ethics, Medicines & Healthcare products Regulatory Agency (MHRA), R&D. The reality has been very different. The Ethics application has some 80 sections, and although almost all information is in the existing protocol on the BHS website, it takes 12-20 h to cut and paste, box by box, into the form, with the most troublesome section being the new questions on drugs that are the only visible evidence so far of IRAS – typically, it has added not subtracted labour. Why does any regulator need to know anatomical therapeutic chemical (ATC) and chemical abstracts service (CAS) numbers of drugs: I had never heard of the latter, and took them from Google since neither ATC or CAS is on the summary of product characteristics (SpC) of drugs as the form claims. But worst of all is the discriminatory question added by the National Research Ethics Service (NRES) to REC forms asking whether Consultants hold a substantive or honorary contract, and requiring the latter to take out separate insurance before being permitted to submit an application. Even modestly sized trials require a premium of many thousands of pounds –£30K in this case – to ‘insure the protocol, written in non-NHS time, therefore not covered by NHS indemnity’. Only after months of stand-off was I permitted to apply for REC approval.
A further abuse by NRES is to require an R&D signature prior to REC submission. R&D receives no mention in the 2004 Act, and sponsorship is dependent on REC approval, not the other way round. In our case, R&D refused to sign the form on the grounds that the 30-page protocol was in the wrong format. Our BHF grant included a large data-management fee for jobs that included production of the electronic case record forms and full protocol, but these steps awaited both the agreeing of third-party contracts (below) and other logistical details such as identification of a manufacturing pharmacy. I was thus in no position to provide the ‘full’ protocol – demanded 12 h before REC's last submission deadline before its 2-month Christmas break. I appealed to NRES, who agreed with me, and asked the local REC to receive the application without R&D signature. However, the bind now is that REC will not issue a favourable opinion without Cambridge agreeing to sponsor the trial, while Cambridge refuses to agree this until I have a favourable opinion! Unless some steps in the regulatory hurdle race happen in parallel tracks, rather than in series, some catch 22s are inevitable. REC's decision letter added further delay with ‘administrative issues’: a euphemism for demanding that another two pages be added to the 6-page patient information sheet. Among the extra questions of supposed concern to patients was ‘For males who have female partners of a child bearing age, there should be some mention of what effects the drugs might have on an unborn baby’. When I alerted NRES to the folly of this question for safe, licensed drugs, I was told ‘REC considers the application from the role of the participant. However, in this instance there is considerable expertise and experience within the REC membership, and the issues raised will improve the standard of the participant information’. Really? A little knowledge may be dangerous; in the hands of regulators, it becomes lethal.
Meanwhile, a major headache was proving identification of a manufacturing pharmacy who would re-encapsulate the drugs to enable double-blinding. Despite this being an essential component of trials that will change practice and be published in top journals, there are few NHS pharmacies licensed to do large-volume work, because of the oppressive MHRA requirements, and their identity was hard to discover. Eventually, by sacrificing the planned double-dummy design, agreement was reached. The next bind was that MHRA submission could not occur without pharmacy agreement (including design of the proposed label), but manufacture cannot occur without MHRA approval. Having set aside a weekend for this next task, I found that the IRAS ‘1-stop’ claims are a mirage. MHRA submissions are passed to a third party, the European Medicines Agency (EMEA), which on this particular weekend was off-air for planned engineering. I contacted MHRA, to draw a ‘sorry not me, guv’ response. They insist that the third-party arrangement is required by law, in order to populate the Eudract database. Strangely, I find no mention of EMEA or Eudract in the 2004 Act, and not all EU countries submit for application via EMEA. I again found myself having to cut and paste information already entered countless times into other forms. After 30 days, I received a non-acceptance letter from MHRA – because the drug details imported from IRAS did not include a field to mention that the re-encapsulation would use gelatine capsules back-filled with lactose. For this expert improvement to the protocol, MHRA sent an invoice for an extra £4040.
Two out of the three studies still await submission, and I await outcome from months of negotiation between University of Cambridge, as grant-holder, with each of the other seven Universities; and Addenbrookes, as co-sponsor, must then agree that the University has satisfactorily taken into account responsibilities of Trusts at each site. Why am I doing all this rather than staff appointed on the grant? Because one of the knock-on consequences of the delays is that clocks on grants start running from the first appointment; nothing is more anxiogenic than finding staff being paid for a year or more while awaiting R&D and others to allow them to do any research.
Evidence of adverse impact from abuse of governance
Given the hours which applicants spend entering data into databases as part of regulatory submissions, it is disappointing that regulators and researchers cannot access a single public data source to answer the key question: what has happened to trial recruitment since 2004. The nearest to an unselected public domain answer is that presented to the recent Conference on the Impact on Clinical Research of European Legislation (http://www.efgcp.be/Conference_details.asp?id=204&L1=7&L2=2&TimeRef=2) [5]. Competent authorities in all member states were asked to report annual clinical trial authorizations since 2000. Whereas these were steady or increased in every other country, in the UK the number of noncommercial trials fell from an average of 600 between 2000 and 2003 to <300 in the period since 2004 (Figure 2). Given that clinical trials in healthy subjects did not require authorization clinical trial agreements (CTAs) prior to 2004, this fall is likely to be an underestimate. Commercial CTAs held steady, but the Director-General of the Association of British Pharmaceutical Industries reports a reduction in the percentage of UK patients in international trials from 6 to 2%, and individual companies report a similar experience, contrasting with sustained or increased recruitment elsewhere. Cancer Research UK-supported cancer trials have bucked the trend, perhaps reflecting the better organization and funding of cancer trials than in other disease areas. However, despite cancer dominating the research networks, their director has commented on the slow recruitment in UK compared with other countries [7].
The obstacles responsible for the decline in clinical research are partly the multiple jeopardy of parallel and serial applications for approval, and partly the knock-on effects of the oppressive MHRA inspections. Even MHRA leaders privately admit that inspectors can be over-zealous, and acknowledge that the recommendations of the International Conference on Harmonisation Good Clinical Practice (GCP) are just that, recommendations not mandatory. Yet inspectors terrorize under-resourced R&D departments, which then use threats to close down a Trust's research as an excuse to introduce yet another piece of time-taking research prevention. To convey a flavour of a current obstacle course, I relate in Box 1 an ongoing experience where current regulation will have delayed by at least 1 year a programme of three British Heart Foundation (BHF)-funded trials – for no conceivable gain, but at immense financial and human loss.
Absence of legal basis for current red tape
It is easy to blame the EU directive for all our woes, but this is unfair. Nor even does it seem that the gold-plating of directives during secondary legislation is to blame. The 2004 Act asks the Secretary of State to appoint committees who will consider 13 points (Box 2) [8]. Except for patient information, and indemnity, all other points are much better considered during expert peer review. Indemnity should be as standard as for any routine NHS care, and taken for granted for any NHS research; and paragraph (g) does not require the detailed six pages of information, which are often the main excuse for delaying decisions. So there is simply no case for the edifice of NRES and its inflated applications.
BOX 2 Whence NRES and its 80 pages? – what the 2004 Act says about the role of RECs
‘In preparing its opinion, the committee shall consider, in particular, the following matters –
the relevance of the clinical trial and its design
whether the evaluation of the anticipated benefits and risks as required under paragraph 2 of Part 2 of Schedule 1 is satisfactory and whether the conclusions are justified
the protocol
the suitability of the investigator and supporting staff
the investigator's brochure
the quality of the facilities for the trial
the adequacy and completeness of the written information to be given, and the procedure to be followed, for the purpose of obtaining informed consent to the subjects' participation in the trial
if the subjects are to include persons incapable of giving informed consent, whether the research is justified having regard to the conditions and principles specified in Part 5 of Schedule 1
provision for indemnity or compensation in the event of injury or death attributable to the clinical trial
any insurance or indemnity to cover the liability of the investigator or sponsor
the amounts, and, where appropriate, the arrangements, for rewarding or compensating investigators and subjects
the terms of any agreement between the sponsor and the owner or occupier of the trial site which are relevant to the arrangements referred to in sub-paragraph (k); and
the arrangements for the recruitment of subjects.'
As for the MHRA, the Act requires documents to be submitted (to the national competent authority) that largely duplicate those submitted to REC, with the addition of information about investigational medicinal products (IMPs). Where these are licensed, their summary of product characteristics needs to be copied from the Monthly Index of Medical Specialties Compendium and scanned into pdfs. Are there really no practising medics at MHRA who qualify for a free copy – perhaps the clinical section of British Pharmacological Society could find the money to purchase them one! If the MHRA were seriously interested in the paramount needs of database completion (over helping applicants), there should be a simple drop-down list of licensed IMPs that automatically inserts the relevant page from the e-Compendium. The MHRA also requires information about certification of a manufacturing pharmacy, e.g. doing the re-encapsulation. Since MHRA will have granted the certification in the first place, and there are few such pharmacies in the NHS, a single drop-down field should replace the delays and paper required to satisfy current demands.
If MHRA were an efficient organization, and their submission could be made concurrently with REC, I would mind less. But on my last three attempts at approval, I have suffered: (i) a 2-month delay because MHRA denied receiving the application, but managed to bank the cheque sent with it; (ii) rejection of the application for not giving shelf-life – of a 11C-PET agent with a 20-min half-life; (iii) a demand for £3283.00 as fee for this high-quality decision-making on a BHF-funded project – on top of the previously denied but banked cheque; (iv) website for application out of action for 5 days; (v) a demand for £4040 for reviewing ‘a phase 1 study of spironolactone, an unknown drug’. Governance should surely start at home!
As for R&D, this receives no mention in the Act. Sponsors and NRES excuse R&D as the guardians of GCP, but the Act does not mandate any new principles beyond those in the Declaration of Helsinki. Clearly there was and remains scope for academic trials to move closer to commercial trials in the quantity and quality of documentation and monitoring. However, R&D departments which, pre-2004, played some role in facilitating research, now act predominantly as MHRA policemen. Numerous researchers, commercial and noncommercial, cite R&D as the principal source of delay. Even after investigators have obtained all the approvals in Box 1, each site is hit by section 23 of the local REC (‘site specific’) application:
’Authorizations required prior to R&D approval. This section should be signed in accordance with the guidance provided by the NHS organization. This may include authorization by clinical supervisors, line managers, service managers, support department managers, pharmacy, data protection officers or finance managers, etc. Managers completing this section should confirm in the text what the authorization means, in accordance with the guidance provided by the NHS organisation.’
In short, a license, fully utilized, for an army of jobsworth individuals to do their bit to protect patients and save the world. Yet there is nothing in the Act to preclude ‘automatic’ sponsorship of a licensed investigator, i.e. an experienced clinical researcher who, under the new General Medical Council (GMC) rules, will be annually licensed and quinquennially re-validated as someone who observes GCP rules laid down by the Declaration of Helsinki; it is this observation of GCP, no more no less, for which the Sponsor is responsible. There is no excuse in the legislation for R&D to impede research while inventing individual requirements for each trial, nor for MHRA to demand of the sponsor more evidence than provided by the chief investigator's annual GMC license that GCP is observed. Indeed, inspections of the sponsor's premises appear discretionary under the Act.
Solution
In proposing a solution for the UK's self-inflicted recession in clinical research (Figures 1 and 2), I am optimistically influenced by the green shoots in Box 3. My urgent recommendation is for an open review of current regulation, undertaken by a body representing all those funding, practising and benefiting from clinical research. The Office for Strategic Co-ordination of Health Research (OSCHR) is the obvious body [9]. The aim of the review should be to consider the model of the Netherlands and devise a way, compatible with the 2004 Act, in which investigators of peer-reviewed trials can start recruitment within 3 months at most of the grant decision. OSCHR will need to peel away nonmandatory accretions to the legislation, and realise that the Act holds no bar to the NHS and universities developing seamless and automatic methods for indemnifying and sponsoring licensed clinical investigators. In 2004, Lord Warner as Minister of Health told the Academy of Medical Sciences that the new legislation ‘does not change the underlying liabilities in clinical trials. NHS Indemnity for clinical negligence continues as it did before 1 May. A big safety net for clinical researchers’ (http://www.dh.gov.uk/en/News/Speeches/Speecheslist/DH_4085207).
BOX 3 Grounds for optimism
The recent establishment of a single body designed to facilitate clinical research: Office for Strategic Co-ordination of Health Research
The one-stop procedure in the Netherlands, requiring only protocol and summary of product characteristics
The National Institute of Health Research's recognition that a trial protocol should be a unitary document, web-based, used by all granting and regulatory bodies [1]
The imminent introduction of individual licensing and re-validation
In this article, I have mainly considered peer-reviewed research, with or without licensed IMPs. The proposed review must also address the urgent resuscitation of ‘registrar research’, which takes place before major grant submissions; the facilitation of experimental medicine investigating novel IMPs; and the rescue of biotechs and pharma. I do not believe it is realistic or necessary as a first measure to seek change in the 2004 Act, or the EU directive, and indeed it is important that perceived or real barriers to such changes are not used as an excuse for doing nothing. What may instead be necessary is an expert legal opinion of what exactly are the minimum requirements of the 2004 Act (and any other relevant legislation), and how best judicial review could be mounted if any of the bodies continue to obstruct research.
The concept of Integrated Research Application Service suggests that someone, possibly the National Institute of Health Research (NIHR) Director, has the right idea, but (as Box 1 illustrates) is sabotaged partly by apparatchiks who use any change to elaborate rather than simplify the process, and partly by the snail's pace of implementation. ‘Give us more time’ is not accepted by MHRA inspectors from investigators as an excuse for non-implementation of non-existent GCP rules, and is inexcusable from regulators when public money is being squandered and patients' lives jeopardized.
Who should arbitrate benefit–risk and the Ethics of Clinical Research
Good research is good medicine
Symbolic of the gulf between research and medicine opened since 2004 is the takeover of Ethics by the National Patient Safety Office (NPSA), and by the Licensing Division at MHRA of all research using medicines. It takes considerable sleuthing to identify the names of those in charge, and it is almost impossible to discover their relevant qualifications or experience: a striking contrast with the repeated demands for investigators' life history and CV to accompany regulatory documents. A marginal but damaging cost of permitting clinical research to be dictated by anonymous individuals in back rooms at the NPSA and MHRA is that real questions of right and wrong are ignored, artificial wedges are driven between research and medicine, and medical leaders are silent on the virtues, benefits and importance of patient participation. The compensation for increased regulation was meant to be harmonization of EU procedures. Why process should be king was never clear, but harmonization has not happened, nor is there greater safety.
Whenever offered participation in a research study, patients are effectively offered a choice between research and everyday medicine. What the ‘transparent and honest’ information sheets are not allowed to tell participants is that research is the safer option (no unexpected deaths in the UK in the last 30 years), and often likely to improve the health of man present as well as mankind future. In part this is because of all the extra care and monitoring intrinsic to a study. But as the cancer community have known and practised for years, there are only two alternatives we should be offering patients. One is the best known treatment (or diagnostic procedure, etc); the other is a treatment that is potentially superior (on the basis of pilot and/or theoretical evidence). In everyday medicine, variable resources and competency mean that many patients are not even offered best available treatment, whereas in research there is no point in comparing ‘possibly better’ with second best. Although not all trials, far less nontrial research, fall into this comparative mould, there is little doubt that the ‘average’ patient would fare better if standard practice were for 50% of patients to receive best and 50% to receive ‘may be even better’ treatment. The only bar to this being a statistical certainty is the element of risk that ‘better’ might turn out to be ‘worse’. Clearly, during developmental stages of new drugs the licensing authority should have an expert role in assessing toxicology and other safety parameters, but if one considers the ongoing ‘cetrapib’ and ezetimibe trials, for example, does anyone seriously believe that MHRA or RECs would have been, or will be, more clairvoyant than expert peer review in predicting the fate of torcetrapib and other surviving agents [10,11]?
Yet the examples of drugs which have failed after faring worse than control in long-term studies do not undermine the argument that over-regulation is bound to increase overall risk–benefit. In part this is because patients on the ‘worse’ treatment, even placebo, may fare better than patients outside trials. Regulators need reminding that the securest way to reduce risk in trials is to close them down altogether: no trials, no risk – but no benefit. However, in part, and paradoxically, it is the regulators who increase risk by permitting, even sometimes obliging, long-term outcome studies to be performed before a drug is licensed. Perhaps this is defensible for an indication, like heart failure, where we know that short-term benefit can carry long-term risk, but it is wrong when regulators require 25 000 patients (as in the OCTAVE trial of omapatrilat) to take part in a safety-dressed-up-as-efficacy pseudo-trial, rather than grant a controlled license for use in individual patients where there is potential benefit as well as risk [12].
Lessons from medicine for research
Cutting off benefit–risk decision making from medical researchers and their funders and ceding this to regulators is multiply perverse. First, we tacitly concede the higher moral ground to the bodies and individuals who obstruct advances in medicine while still failing to anticipate the ‘unexpected’ risks. Regulators do not care for patients; they have little concept of how much patients like the opportunity to take part in research, and do not understand how the recruitment process actually works. They may imagine that it starts and finishes with patients receiving a 6-page anonymous document in the post, with 24 h to read what the researcher will do with his records in 10 years' time. The reality is that as in any doctor–patient relationship the giving and taking of information is dependent on the trust that develops, and recruitment frequently occurs over several clinic visits.
Calculation and discussion of benefit–risk, far from being a peculiarity of research that requires expert external guidance and monitoring, is an everyday part of medicine. Every administration of a drug with known side-effects is a benefit–risk calculation requiring assessment of the strength of clinical indication (the ‘inclusion criteria’) for the drug, and an individual patient's likelihood of adverse reaction (‘exclusion criteria’). Whether to administer an antibiotic that risks Clostridium difficile infection; whether to start anticonvulsant therapy without a definitive diagnosis of epilepsy and necessitate loss of the patient's driving license; whether to sign a not-to-be-resuscitated form – these are daily decisions in wards and clinics, and when the decision is unclear or depends on patient choices, we involve patients (or relatives) and expert colleagues. We do not turn to REC, MHRA or R&D for advice.
From Ethics Committees to ethics
The auto-apotheosis of NRES does not need my help. ‘The change in name’ (from COREC to NRES) ‘symbolises the change to a more responsive and robust research ethics review process, and makes a shift away from the concept of a system composed of only Research Ethics Committees (RECs) to one of a service that will provide robust ethical review to protect the safety, dignity and well being of research participants as well as ensuring through the delivery of a professional service that it is also able to promote and facilitate ethical research within the NHS’ (http://www.orecni.org.uk/display/corec). Both claims in their ‘mission statement’ are extraordinary. How can a group of 24 individuals, only two with medical degrees, claim to protect safety and well-being of patients, far less claim superiority in this respect over the tens to hundreds of years of specialist experience provided by the clinical investigators? And what is the professional experience of most committee members in either ethics or clinical research? The Clinical Trials office of the MHRA is similarly bereft of ascertainable expertise in clinical research.
Rights and dignity are a limited selection of ethical issues, and will not stem from promoting amateurism and ignorance above the professionalism, knowledge and skill of clinical researchers. NRES's own statistics show that it is NRES, their 80-page application form and 6-page patient information sheets from which patients truly need protection. Of course, there are ethical issues on which expert advice is required. Having read Ethics for my first degree at Cambridge, I recognize the difficulties. It is an even harder skill to acquire than clinical research because it lacks the knockout force of either empirical observation or the strict logical argument that serves many other branches of philosophy. There is no way of arguing from what is to what ought to be. The right answer is not necessarily the majority view, or one that appears to serve the largest number of individuals. Professional moral philosophers wrestle with definitions of justice and goodness, and with the logical inconsistencies between simultaneously held views. However, such debate lies well outside the remit or expertise of the NPSA; therefore the real ethical questions are ignored, especially if deemed politically incorrect.
Research is central to the ethos, for staff and patients, at a teaching hospital
For example, there are questions of responsibility on those benefiting from previous research to make their own contribution, especially when (as is usually the case) this involves no increase in risk. Patients coming to Teaching Hospitals know that the default – from which they can ask to be excused – is for students to be present at many inpatient and outpatient consultations. Sensible patients soon realise that the presence of students is a win–win for them. Students have more time to hear patients' problems; often these days they are the only ‘staff’ who can follow patients from door to needle without disruption by changes in firm or shift; above all, teaching gains patients more attention than usual, and less corner cutting, from their doctors. Nowadays, some teaching hospitals are receiving even more support from NIHR for research than they do for teaching, and this raises the question whether patients enjoying the higher staff–patient ratio and other facilities of a teaching hospital have some obligation to consider taking part in the research that helps pay for these benefits. As with teaching, of course patients have an absolute right to say no. The sensible ones, especially those who have previously participated, appreciate the extra attention and other benefits of participation. Should not all patients coming to a teaching hospital be informed of both the hospital's need to teach and to undertake research, and of available research projects? When patients do not have a choice whether they can take part in research, because none is available, that is unethical and a critical finding.
Competing interests
None to declare.
REFERENCES
- 1.Cookson C. £1bn for new health research body. [accessed 21 February 2009]. Financial Times 2006; 6 Dec. Available at http://www.ft.com/cms/s/0/467338c8-8538-11db-b12c-0000779e2340,dwp_uuid=4d1501ce-6a81-11db-83d9-0000779e2340.html.
- 2.Jack A. Big drugs companies shift trials overseas. [accessed 21 February 2009]. Financial Times 26 June 2008. Available at http://www.ft.com/cms/s/0/0c102bce-4318-11dd-81d0-0000779fd2ac.html.
- 3.Mitchell P. Critics pan timid European response to TeGenero disaster. Nat Biotechnol. 2007;25:485–6. doi: 10.1038/nbt0507-485. [DOI] [PubMed] [Google Scholar]
- 4.Stewart PM, Stears A, Tomlinson JW, Brown MJ. Regulation – the real threat to clinical research. BMJ. 2008;337:a1732. doi: 10.1136/bmj.a1732. [DOI] [PubMed] [Google Scholar]
- 5.Godlee F. It's time to change how Europe regulates research. BMJ. 2008;337:a2986. [Google Scholar]
- 6.Brown M, Boon N, Brooks N, Brown E, Camm J, Caulfield M, Chilvers E, Gibson J, Griffin G, Grossman A, Hall A, Hart G, Heagerty G, Home P, Hodgson H, Horton R, Hughes R, Khaw KT, Lazarus J, Leaper D, McCollum P, Monson J, O'Rahilly S, Rowlands B, Scott J, Sutton R, Taylor R, Watkins H, Wright N. Modernising medical careers, medical training application service, and the postgraduate medical education and training board: time for the emperors to don their clothes. Lancet. 2007;369:967–8. doi: 10.1016/S0140-6736(07)60459-0. [DOI] [PubMed] [Google Scholar]
- 7.UKCRC Board Subgroup for the UKCRN. [accessed 21 February 2009]. Minutes. 12 May 2006. Available at http://www.ukcrc.org/PDF/BSG_12_05_06.pdf.
- 8.The medicines for human use (clinical trials) regulations. [accessed 21 February 2009]. 2004. Available at http://www.opsi.gov.uk/si/si2004/20041031.htm. [PubMed]
- 9.The Office for Strategic Co-ordination of Health Research (OSCHR) Background on the Review of Health Research in the UK and OSCHR. [accessed 21 February 2009]. 2008. Available at http://www.nihr.ac.uk/about/Pages/about_oschr.aspx.
- 10.Nicholls SJ, Tuzcu EM, Brennan DM, Tardif J-C, Nissen SE. Cholesteryl ester transfer protein inhibition, high-density lipoprotein raising, and progression of coronary atherosclerosis: insights from ILLUSTRATE (Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation) Circulation. 2008;118:2506–14. doi: 10.1161/CIRCULATIONAHA.108.790733. [DOI] [PubMed] [Google Scholar]
- 11.Kastelein JJP, Akdim F, Stroes ESG, Zwinderman AH, Bots ML, Stalenhoef AFH, Visseren SL, Sijbrands EJ, Trip MD, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E. Simvastatin with or without Ezetimibe in Familial Hypercholesterolemia. N Engl J Med. 2008;358:1431–43. doi: 10.1056/NEJMoa0800742. ENHANCE Investigators. [DOI] [PubMed] [Google Scholar]
- 12.Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens. 2004;17:103–11. doi: 10.1016/j.amjhyper.2003.09.014. [DOI] [PubMed] [Google Scholar]