

Abstract
CaVβ subunits of voltage-gated calcium channels contain two conserved domains, a src-homology-3 (SH3) domain and a guanylate kinase-like (GK) domain with an intervening HOOK domain. We have shown in a previous study that, although Gβγ-mediated inhibitory modulation of CaV2.2 channels did not require the interaction of a CaVβ subunit with the CaVα1 subunit, when such interaction was prevented by a mutation in the α1 subunit, G protein modulation could not be removed by a large depolarization and showed voltage-independent properties (Leroy et al., J Neurosci 25:6984–6996, 2005). In this study, we have investigated the ability of mutant and truncated CaVβ subunits to support voltage-dependent G protein modulation in order to determine the minimal domain of the CaVβ subunit that is required for this process. We have coexpressed the CaVβ subunit constructs with CaV2.2 and α2δ-2, studied modulation by the activation of the dopamine D2 receptor, and also examined basal tonic modulation. Our main finding is that the CaVβ subunit GK domains, from either β1b or β2, are sufficient to restore voltage dependence to G protein modulation. We also found that the removal of the variable HOOK region from β2a promotes tonic voltage-dependent G protein modulation. We propose that the absence of the HOOK region enhances Gβγ binding affinity, leading to greater tonic modulation by basal levels of Gβγ. This tonic modulation requires the presence of an SH3 domain, as tonic modulation is not supported by any of the CaVβ subunit GK domains alone.
Keywords: Calcium channel, Beta subunit, Electrophysiology
Introduction
Voltage-gated calcium (CaV) channels play a major role in the physiology of all excitable cells. Three families have been identified, CaV1–3 (for review, see [17]). The high-voltage-activated (HVA) CaV1 and 2 classes are heteromultimers composed of the pore-forming α1 subunit, associated with auxiliary CaVβ and α2δ subunits (for review, see [12]). Four CaVβ subunit genes have been cloned, and these subunits are important for HVA calcium channel function (for review, see [16]), since they promote the expression of functional channels at the plasma membrane and modulate their biophysical properties [6, 8, 11, 29]. CaVβ subunits bind with high affinity to the α-interaction domain (AID) on the I–II loop of CaV1 and 2 channels [29], although other α1 subunit interaction sites are also likely to be important in mediating the actions of CaVβ subunits [35, 40].
In a previous study, we investigated the role of CaVβ subunits in the plasma membrane expression and G protein modulation of CaV2.2 calcium channels, by mutating the AID tryptophan (W391) in the I–II loop of CaV2.2, and thus disrupting the high-affinity interaction with CaVβ subunits [21]. One conclusion was that the CaV2.2W391A mutant channels lost all modulation by CaVβ1b and showed strongly reduced expression at the plasma membrane. While they still showed G protein modulation following the activation of a coexpressed dopamine D2 receptor, this modulation could not be reversed by depolarization. In contrast, for palmitoylated CaVβ2a, only the expression at the plasma membrane was affected when it was coexpressed with the mutant CaV2.2W391A channels, while all the biophysical properties of the expressed CaV2.2W391A channels remained normally modulated by CaVβ2a. Furthermore, they also showed voltage-dependent G protein modulation. We concluded that the continuing influence of β2a was dependent on its palmitoylation, which increased the local concentration of β2a near the plasma membrane sufficiently to allow lower-affinity interactions to occur between it and the mutant channel α1 subunit, which were effective in modulating the channel properties [21].
CaVβ subunits were originally predicted by structural modeling to contain a src-homology-3 (SH3) domain followed by a guanylate kinase-like (GK) domain [18]. The SH3 domain is split with its final (fifth) β-strand separated from the rest of the domain by an intervening sequence termed the HOOK domain, whose sequence varies between CaVβ subunits and which is encoded by either a short or an alternative long exon. X-ray crystallographic studies have now produced detailed information on the domain structure [13, 27, 39]. From these studies, it is clear that the fifth β-strand of the SH3 domains provides the interaction with the GK domain, being situated after the variable HOOK region, whose structure was not determined (for review, see [31]). The GK domain interacts with the AID motif and has since been shown to be an important determinant of function for the HVA channels [32, 37, 38].
The primary goal of the present study was to determine the minimal domain(s) of CaVβ subunits that is able to confer voltage dependence on G protein modulation of CaV2.2 channels.
Materials and methods
Materials
The cDNAs used in this study were CaV2.2 (D14157), CaVβ1b (X61394), CaVβ2a (M88751), α2δ-2 [2], and dopamine D2 receptor (X17458). When used, the green fluorescent protein (GFP-mut3b, U73901) was used to identify transfected cells. All cDNAs were subcloned into pMT2 vector. Transducin-α was used as described [5].
Construction of truncated β subunit domains
We have been guided by the structure in our choice of truncations and deletions in the present study (Fig. 1). In the case of the GK domains, we have used the exon boundary to determine the C-terminal end, since such boundaries often delimit a stable functional domain, and this marks the end of the second conserved domain, as originally identified (for review, see [7]). It was important that the GK domain constructs were stable since previous studies have examined the properties of several GK domain constructs with varying results, regarding their ability to mimic the functions of intact CaVβ subunits, and it is possible that these constructs have varying stabilities in different cell types [23, 37]. All constructs were made by standard molecular biological techniques and their sequences verified by sequencing both strands. The truncated constructs used for electrophysiology (with their amino acid residues) were β2a-Δ-vHOOK (Δ169–213), β2a-SH3 (1–135), β2a-(SH3+HOOK+ɛSH3) (1–225), β2a-GK (226–442), β2a-(HOOK+ɛSH3+GK) (136–442), β1b-GK (230–426), and β1b-(HOOK+ɛSH3+GK) (179–426).
Fig. 1.

Diagram of the main constructs used in the electrophysiological experiments in the present study
Yeast two-hybrid assays
Assays were carried out using the MATCHMAKER GAL4 two-hybrid kit (Clontech). Fragments of Cavβ2a (amino acids 5–442, 5–134, 5–224, 135–442, 214–442, or 225–442), the CaV2.2 I–II loop (360–483), and CaVβ1b were generated by polymerase chain reaction and subcloned in-frame into the vectors pACT2 and pAS2-1. Plasmids were cotransformed into the yeast strain Y190 and transformants were selected by plating onto minimal selective dropout (SD) -Leu, -Trp agar. Protein interactions were identified by restreaking colonies onto SD -Leu, -Trp plates and carrying out colony-lift β-galactosidase assays according to the supplied protocol.
Cell culture, heterologous expression, and whole cell recording
The tsA-201 cells were cultured in a medium consisting of D-MEM, 10% fetal bovine serum, 2 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The cDNAs (all at 1 μg/μl) for CaVα1 subunits, CaVβ, α2δ-2, and GFP (when used as a reporter of transfected cells) were mixed in a ratio of 3:2:2:0.4. The cells were transfected using Fugene 6 (Roche Diagnostics, Lewes, UK; DNA/Fugene 6 ratio of 2 μg in 3 μl). The tsA-201 cells were replated at low density on 35-mm tissue culture dishes on the day of recording. Whole-cell patch-clamp recordings were performed at room temperature (22–24°C). Only fluorescent cells expressing GFP were used for recording. The single cells were voltage-clamped using an Axopatch 200B patch-clamp amplifier (Molecular Devices). The electrode potential was adjusted to give zero current between pipette and external solution before the cells were attached. The cell capacitance varied from 10 to 40 pF. Patch pipettes were filled with a solution containing (in mM) 140 Cs-aspartate, 5 EGTA, 2 MgCl2, 0.1 CaCl2, 2 K2ATP, 10 HEPES, titrated to pH 7.2 with CsOH (310 mOsm) with a resistance of 2–4 MΩ. The external solution contained (in mM) 150 tetraethylammonium bromide, 3 KCl, 1.0 NaHCO3, 1.0 MgCl2, 10 HEPES, 4 glucose, 10 BaCl2, pH adjusted to 7.4 with Tris–Base (320 mOsm). The pipette and cell capacitance as well as the series resistance were compensated by 80%. Leak and residual capacitance current were subtracted using a P/4 protocol. All experiments in which quinpirole was applied were carried out in small volume disposable glass chambers (300–500 μl volume) with a perfusion rate of 200–300 μl/s, which were used once only, excluding the possibility that tonic modulation was due to prior quinpirole exposure. Quinpirole was made up as a 10-mM stock solution, and aliquots were diluted as necessary and used once only.
Data were filtered at 2 kHz and digitized at 5–10 kHz. The holding potential was −100 mV, and pulses were delivered every 10 s. Test pulses were normally 40 ms in duration, and in the three pulses, protocol P1 and P2 were separated by at least 150 ms, and P2 was preceded by a 50-ms prepulse to +120 mV. Activation properties were determined from tail current measurements, as previously described [21]. Steady-state inactivation properties were measured by applying a 5- to 20-s pulse (depending on the inactivation properties of the currents) from −120 to +20 mV in 10-mV increments, followed by 11 ms repolarization to −100 mV before the 100-ms test pulse to +20 mV.
Data analysis and curve fitting
Current amplitude was measured 10 ms after the onset of the test pulse, and the average over a 2-ms period was calculated and used for subsequent analysis. The current density–voltage (I–V) relationships were fitted with a modified Boltzmann equation as follows: 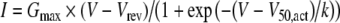 where I is the current density (in pA/pF), Gmax is the maximum conductance (in nS/pF), Vrev is the reversal potential, V50, act is the midpoint voltage for current activation, and k is the slope factor. Activation and steady-state inactivation data were fitted with a single Boltzmann equation of the form:
where I is the current density (in pA/pF), Gmax is the maximum conductance (in nS/pF), Vrev is the reversal potential, V50, act is the midpoint voltage for current activation, and k is the slope factor. Activation and steady-state inactivation data were fitted with a single Boltzmann equation of the form: 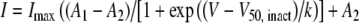 where Imax is the maximal current and V50, inact is the half-maximal voltage for current inactivation. For the steady-state inactivation, A1 and A2 represent the total and noninactivating current, respectively. Analysis was performed using Pclamp7 and Origin 7.
where Imax is the maximal current and V50, inact is the half-maximal voltage for current inactivation. For the steady-state inactivation, A1 and A2 represent the total and noninactivating current, respectively. Analysis was performed using Pclamp7 and Origin 7.
Data are expressed as the mean±SEM of the number of replicates, n. Error bars indicate the standard errors of multiple determinations. Statistical significance was analyzed using Student’s paired or unpaired t test or by ANOVA with Bonferroni’s post hoc test, if multiple comparisons were made.
Results
The isolated GK domains of CaVβ1b and β2a support voltage-dependent G protein modulation
In a previous study, we used a CaV2.2 construct with a mutation in the I–II loop (W391A), such that it did not show high-affinity interaction with CaVβ subunits, and observed a lack of voltage dependence of the quinpirole-mediated inhibition in the presence of β1b [21]. From that study, we concluded that the interaction of CaVβ with the I–II linker was necessary for voltage-dependent G protein modulation. However, it is possible that this interaction with the I–II linker is not sufficient in itself, but is required to bring another region of the CaVβ subunits into proximity with the channel. In particular, we found that for β2a, voltage-dependent G protein modulation was still present, despite the W391A mutation in the I–II linker. Therefore, there remained a question as to whether the palmitoylation of β2a resulted in a sufficiently high concentration of CaVβ subunit being present near to the I–II linker of the channel, such that there was high occupancy by β2a of the mutated I–II linker, despite a very low-affinity interaction or whether the residual interaction was with another domain of β2a on another part of the channel [21].
In order to examine which domain of CaVβ was necessary to promote the voltage dependence of G protein modulation, we compared the ability of full-length β1b or β2a and their isolated GK domains to support voltage-dependent G protein modulation. The constructs used are shown in Fig. 1. We coexpressed all the β subunit constructs with CaV2.2 and α2δ-2. For coexpression with full-length CaVβ1b, the peak IBa was −158.2 ± 25.7 pA/pF at +20 mV (n = 35). For comparison, in the absence of any CaVβ subunit, the peak IBa at +30 mV was −8.3 ± 1.0 pA/pF (n = 9), in experiments performed over the same time period. For CaV2.2/α2δ-2/β1b currents, application of the dopamine D2 receptor agonist quinpirole (100 nM) produced maximally 63.7 ± 6.6% inhibition at +10 mV (Fig. 2a,b). This inhibition showed a strong voltage dependence, as the P2/P1 ratio was 2.97 ± 0.23 at +10 mV (Fig. 2a,c). This is an example of complete voltage dependence, since full reversal of a 64% inhibition predicts a P2/P1 ratio of 2.8. In the absence of any coexpressed β subunit, quinpirole (100 nM) application still produced a substantial effect, resulting in 44 ± 13% inhibition at +10 mV (Fig. 2a,b). However, the voltage dependence of this inhibition was very low, the P2/P1 ratio being 1.3 ± 0.11 at +10 mV (Fig. 2a,c; P < 0.0001 compared to β1b), as we described previously for CaV2.2W391A, which did not interact with β1b [21].
Fig. 2.

GK domain of β1b restores voltage dependence to G protein modulation. aUpper panel example traces showing inhibition of CaV2.2 currents (Ctrl) by quinpirole (Quin, 100 nM) for CaV2.2/α2δ-2 coexpressed with β1b (left), without β (center), and with β1b-GK (179–426) (right). Traces are shown for 40 ms depolarizations to +10 mV before and after a depolarizing prepulse to +100 mV. Lower panel current–voltage relationships for the same conditions, prior to (filled squares) and during quinpirole application (open squares) (n = 10, 9, and 7, respectively). b Percentage inhibition by quinpirole between 0 and +30 mV for the three conditions depicted in a. Black bars +β1b (n = 10), white bars no β (n = 7), and gray bars +β1b-GK (179–426) (n = 7). c Facilitation (P2/P1) ratio between 0 and +30 mV in the presence of quinpirole for the same experiments as in b. The dotted line indicates a P2/P1 ratio of 1, i.e., no facilitation. d Basal facilitation (P2/P1) ratio between +10 and +30 mV for the same experiments as in b. The dotted line indicates a P2/P1 ratio of 1
We then utilized one of the truncated CaVβ1b subunit constructs described previously [32] to examine which domain(s) of CaVβ1b were required to promote the voltage dependence of G protein modulation. We found that a β1b-GK domain construct (β1b-HOOK+ɛSH3+GK (179–426)), containing both the HOOK region and the fifth β-strand, coexpressed with CaV2.2/α2δ-2, enhanced calcium channel currents to a smaller extent than full-length β1b subunit, the peak current at +20 mV being −79.8 ± 16.2 pA/pF (n = 7), as described previously [32]. However, quinpirole (100 nM) produced 67.5 ± 6.9% inhibition of IBa at +10 mV, and this inhibition could be relieved by a depolarizing prepulse to +120 mV (Fig. 2a,b). The P2/P1 ratio was greater than that in the absence of any β subunit at all potentials, being 2.06 ± 0.15 at +10 mV (Fig. 2a,c; P < 0.001 compared to no β subunit). It is important to note that for all β1b constructs, the basal facilitation prior to the application of quinpirole was not significantly different from unity (Fig. 2d). This result indicates that the interaction of the CaV2.2 I–II linker with the GK domain of the CaVβ1b subunit is sufficient to promote voltage dependence of G protein modulation, and the SH3 domain is not required.
For CaV2.2/α2δ-2 together with full-length β2a, the peak IBa was −145.9 pA/pF at +20 mV (Table 1). Quinpirole (100 nM) inhibited these currents to a smaller extent, producing maximally 38.7 ± 4.9% inhibition at +10 mV (Fig. 3a,b; P = 0.0042 compared to β1b). This inhibition showed a P2/P1 ratio of 1.88 ± 0.19 at +10 mV (Fig. 3a,c). The low P2/P1 ratio was to be expected, in view of the small inhibition by quinpirole.
Table 1.
The effect of various CaVβ subunit constructs on biophysical parameters of CaV2.2/α2δ-2 calcium channel currents expressed in tsA-201 cells
| β species | Peak IBa, pA/pF (n) | Voltage for peak IBa (mV) | Steady-state inactivation V50 inact, mV (n) |
|---|---|---|---|
| No β | −8.3 ± 1.0 (9) | +30 | −25.4 ± 7.5 (3) |
| β2a | −145.9 ± 34.9 (14)** | +20 | +0.47 ± 2.16 (6)** |
| C3,4S-β2a | −96.6 ± 12.0 (21)* | +20 | −47.6 ± 1.4 (3)** †† |
| β2-GK (226–442) | −79.9 ± 23.9 (17)* | +20 | −54.8 ± 3.0 (5)** †† |
| β2a-GK (136–442) | −63.2 ± 12.7 (18)† | +20 | −45.5 ± 3.0 (5)** †† |
| β2a-ΔvHOOK | −131.3 ± 26.8 (16)** | +20 | −14.2 ± 6.0 (6) |
| C3,4S-β2a-ΔvHOOK | −101.4 ± 15.4 (20)* | +20 | −46.0 ± 1.8 (5)** †† |
| β2-SH3 (1–135)+β2-GK (226–442) | −28.0 ± 11.1 (9)†† | +20 | −57.3 ± 1.4 (4)** †† |
Statistical significances (one-way analysis of variance and Bonferroni’s post hoc test) were determined for differences compared to CaV2.2 expressed without any β subunit or compared to CaV2.2 expressed with wild-type β2a subunit
*P < 0.05 and **P < 0.01, CaV2.2 expressed without any β subunit; †P < 0.05 and ††P < 0.01, CaV2.2 expressed with wild-type β2a subunit
Fig. 3.

GK domains of β2a support voltage-dependent G protein modulation. aUpper panel example traces showing inhibition of CaV2.2 currents (Ctrl) by quinpirole (Quin, 100 nM) for CaV2.2/α2δ-2 coexpressed with β2a (left), C3,4S-β2a (center), and with β2a-GK (226–442) (right). Traces are shown for 40 ms depolarizations to +10 mV before and immediately after a depolarizing prepulse to +120 mV. Lower panel current–voltage relationships for the same conditions prior to (filled squares) and during quinpirole application (open squares), (n = 6, 6, and 7, respectively). b Percentage inhibition by quinpirole between +10 and +30 mV for the three conditions depicted in a and the additional GK domain construct β2a-GK (136–442) (n = 9, 7, 11, and 11, respectively). c Facilitation (P2/P1) ratio between +10 and +30 mV in the presence of quinpirole for the three conditions depicted in a and β2a-GK (136–442) (n = 7, 8, 9, and 6, respectively). The dotted line indicates a P2/P1 ratio of 1. d Basal facilitation (P2/P1) ratio between +10 and +30 mV for the three conditions depicted in a and β2a-GK (136–442) (n = 7, 9, 10, and 9, respectively). The dotted line indicates a P2/P1 ratio of 1
Based on our previous findings [21], it is likely that there is increased interaction between the β2a subunit and CaV2.2, as a result of palmitoylation elevating its effective concentration at the plasma membrane and increasing its availability to interact with the channel. We found that removal of the palmitoylation motif on β2a (C3,4S-β2a) resulted in greater inhibition by 100 nM quinpirole (67.6 ± 7.5% inhibition at + 10 mV, Fig. 3a,b; P = 0.0047 compared to β2a), and correspondingly increased the P2/P1 ratio to 2.93 ± 0.67 at +10 mV (Fig. 3b,c).
Similar to our finding for the β1b-HOOK+ɛSH3+GK construct, we found that β2a-HOOK+ɛSH3+GK (136–442) supported voltage-dependent G protein modulation. This construct, coexpressed with CaV2.2/α2δ-2, enhanced calcium channel currents to a smaller extent than full-length β2a subunit, the peak IBa being −63.2 pA/pF at +20 mV (Table 1). However, the inhibition by 100 nM quinpirole was 56.0 ± 7.2% at +10 mV (Fig. 3b), and the P2/P1 ratio was 2.04 ± 0.48 at +10 mV (Fig. 3c).
Similar results were obtained for a minimal GK domain of β2a, β2-GK (226–442), coexpressed with CaV2.2/α2δ-2 where the peak current density at +20 mV was −79.9 pA/pF (Table 1). Although the interaction of this GK domain with the I–II linker of CaV2.2 was not confirmed in our yeast two-hybrid results (see Fig. 6), this may be a result of misfolding in the yeast system, as in tsA-201 cells, the peak IBa was significantly greater in the presence of β2a-GK (226–442) than in the absence of any β subunit (P < 0.05, Table 1). Furthermore, the steady-state inactivation was also significantly hyperpolarized compared to the absence of any β subunit (P < 0.01, Table 1), to a similar extent to full-length β1b [21] or C3,4S-β2a (Table 1) [32]. Both these results indicate that the calcium channel currents are influenced by β2a-GK (226–442) interacting with the I–II linker of CaV2.2. Moreover, quinpirole (100 nM) produced 62.0 ± 6.7% inhibition at +10 mV, and the inhibition could be relieved by a depolarizing prepulse (Fig. 3a,b). The P2/P1 ratio was 2.73 ± 0.37 at +10 mV (Fig. 3a,c). This result confirms that the interaction of the CaV2.2 I–II linker with the GK domain of any CaVβ subunit is sufficient to promote voltage dependence of G protein modulation. For all the CaVβ2 constructs depicted in Fig. 3c, their basal P2/P1 values were between 1 and 1.5, indicating little basal facilitation was present (Fig. 3d).
Fig. 6.

Protein interactions involving β2a domains. Protein interactions were demonstrated using β-galactosidase assays after cotransformation of plasmids into yeast. Blue colonies indicate a positive interaction. a Positive control showing CaV2.2 I–II loop (BI–II) pACT2 and β1b pAS2-1 (lane 1). β2a minus the C terminus (5–442), β2a-ɛSH3+GK (214–442), and β2a-HOOK+ɛSH3+GK (135–442) interacted with the I–II linker (lanes 2–4) whereas β2a-GK (225–442) showed no interaction (lane 5). b Positive control showing β2a minus the C terminus (5–442) interacting with BI–II (lane 1). No interactions were demonstrated between SH3 domains and BI–II (lanes 2–5). c The shortest GK construct (225–442) was not found to interact with β2a-SH3 (5–134) (lanes 1 and 2) whereas the longer GK constructs, β2a-ɛSH3+GK (214–442) and β2a-HOOK+ɛSH3+GK (135–442), showed a positive interaction (lanes 3 and 4)
Investigation of the roles of the CaVβ SH3 and HOOK domains in voltage-dependent G protein modulation
In order to examine whether the SH3 domain and the HOOK domain of β2a played a role in the reduced G protein modulation shown by β2a, we examined the effect of a β2a construct in which the SH3 domain and the GK domain are present, but which is missing amino acids 169–213, comprising the variable HOOK domain (β2a-δvHOOK) [32]. The basic properties of IBa resulting from the coexpression of CaV2.2/α2δ-2/β2a-ΔvHOOK are given in Table 1. For β2a-ΔvHOOK, the amount of inhibition by quinpirole was 39.9 ± 6.7% at +10 mV, not significantly different from β2a itself (Fig. 4a,b). Evidence from our previous study [32] indicated that this construct remains palmitoylatable, such that the concentration at the plasma membrane and thus the occupancy by this construct of the I–II linker is likely to remain high. However, unlike full-length β2a, the P2/P1 ratio was also high, being 2.85 ± 0.56 at +10 mV (Fig. 4c).
Fig. 4.

The determinants for voltage-dependent G protein modulation and facilitation in the presence of β2a constructs. aUpper panel example traces showing inhibition of CaV2.2 currents (Ctrl) by quinpirole (Quin, 100 nM) for CaV2.2/α2δ-2 coexpressed with β2a-ΔvHOOK (left) and C3,4S-β2a-ΔvHOOK (right). Traces are shown for 40 ms depolarizations to +10 mV before and after a depolarizing prepulse to +120 mV. Lower panel current–voltage relationships for the same conditions prior to (filled squares) and during quinpirole application (open squares) (n = 6 for both). b Percentage inhibition by quinpirole between +10 and +30 mV for the two conditions depicted in a (n = 10 and 8, respectively). Data for β2a from Fig. 3 is included for comparison (dashed bar). c Facilitation (P2/P1) ratio between +10 and +30 mV in the presence of quinpirole for the two conditions depicted in a (n = 9 and 8, respectively). Data for β2a from Fig. 3 is included for comparison (dashed bar). The dotted line indicates a P2/P1 ratio of 1
A potential reason for the discrepancy between the low percentage of inhibition and the high P2/P1 ratio for β2a-ΔvHOOK, compared to full-length β2a, is that currents in the presence of this construct were also observed to show strong basal facilitation. This is likely to account for the high P2/P1 ratio during quinpirole application, as the depolarizing prepulse also removes tonic facilitation. Prior to any agonist application, the P2/P1 ratio in the presence of β2a-ΔvHOOK was 2.34 ± 0.22 at +10 mV (Fig. 5a,b). For comparison, wild-type β2a showed only a small degree of basal facilitation, P2/P1 under control conditions being 1.43 ± 0.13 at +10 mV (Fig. 3d, P < 0.05 compared to β2a-ΔvHOOK), and C3,4S-β2a-GK also showed no tonic facilitation (Fig. 3d). Furthermore, β2-GK (226–442) showed no basal facilitation in the absence of agonist, the P2/P1 ratio being 1.2 ± 0.17 at +10 mV (Figs. 3d and 5a,b). For the proximally extended GK domain β2a-GK (136–442), there was also no significant basal facilitation, P2/P1 in the absence of agonist being 1.5 ± 0.17 at +10 mV (Fig. 3d).
Fig. 5.

The determinants for basal facilitation in the presence of β2a constructs. a Example traces showing basal facilitation of CaV2.2 currents for CaV2.2/α2δ-2 coexpressed with β2a-ΔvHOOK (left), lack of basal facilitation with β2a-GK (226–442) (center), and basal facilitation with β2a-SH3 (1–135)+GK (226–442). Traces are shown for 40 ms depolarizations to +10 mV before and after a depolarizing prepulse to +100 mV. b Basal facilitation (P2/P1) ratio between +10 and +30 mV for CaV2.2/α2δ-2 coexpressed with β2a-ΔvHOOK (n = 9, black bars), β2a-ΔvHOOK+transducin-α (n = 7, gray bars), C3,4S-β2a-ΔvHOOK (n = 11, hatched bars), β2a-SH3 (1–135)+GK (226–442) (n = 9, horizontal striped bars), and β2a-GK (226–442) (n = 10, dashed bars, repeated from Fig. 3d for comparison). The dotted line indicates a P2/P1 ratio of 1, i.e., no basal facilitation. c Time course of facilitation with increasing prepulse duration Δt for β2a (open squares, n = 10), β2a-ΔvHOOK (filled squares, n = 10), and β2a-SH3 (1–135) and GK (226–442) (open stars, n = 9). The lines are single exponential fits to the mean data. d Time course of reinhibition with increasing duration Δt between prepulse and P2 test pulse for β2a (open squares, n = 6) and β2a-ΔvHOOK (filled squares, n = 6). The lines are single exponential fits to the mean data. The inset shows an example set of traces showing the increasing inhibition of the P2 traces with increased Δt
This basal facilitation in the presence of β2a-ΔvHOOK was due to tonic G protein modulation since it could be removed by coexpression of transducin-α, which acts as a sink for free Gβγ subunits. The basal P2/P1 ratio in the presence of transducin-α was 1.14 ± 0.05 at +10 mV (Fig. 5b). Basal facilitation was also absent when guanosine 5′-O-(2-thiodiphosphate) (GDP-βS, 200 μM) was included in the patch pipette (data not shown). This concentration of GDP-βS also blocked quinpirole-mediated inhibition (data not shown).
In order to examine whether the presence of basal facilitation was favored by the palmitoylation of the β2a-ΔvHOOK, we also examined the properties of C3,4S-β2a-ΔvHOOK. The basic properties of currents in the presence of this construct are given in Table 1. Quinpirole-mediated inhibition was much higher than for the palmitoylatable construct, being 79.2 ± 3.1% at +10 mV, similar to that obtained for C3,4S-β2a (Fig. 4a,b). The P2/P1 ratio was correspondingly high, being 3.22 ± 0.74 at +10 mV (Fig. 4a,c). However, this construct showed no basal facilitation, the P2/P1 ratio prior to agonist application being 1.02 ± 0.05 at +10 mV (Fig. 5b).
In the presence of β2a-ΔvHOOK, the time constant for dissociation (τdissoc) of tonically bound Gβγ at +120 mV was 33.6 ± 6.5 ms (n = 10, Fig. 5c). In contrast, for the small amount of tonic facilitation shown by wild-type β2a, the τdissoc at +120 mV was 21.3 ± 4.5 ms (n = 10, Fig. 5c), indicating a higher dissociation rate of Gβγ from wild-type β2a at +120 mV of 46.9 s−1 compared to 29.7 s−1 for β2a-ΔvHOOK, although the difference did not reach statistical significance. The time constant for rebinding of Gβγ at −100 mV to wild-type β2a was 247.7 ± 25.9 ms, and for β2a-ΔvHOOK, it was 296.4 ± 24.2 ms (Fig. 5d, P > 0.05).
Assuming a basal Gβγ concentration of 50 nM, as estimated previously [34], we can utilize the tonic P2/P1 ratio and the rate of Gβγ rebinding after a depolarizing prepulse to determine the KD for Gβγ at −100 mV, as described previously [34]. For CaV2.2 coexpressed with wild-type β2a, the koff for Gβγ was calculated to be 2.83 s−1 and the kon was 24.2 μM−1 s−1, leading to a KD for Gβγ interaction with the channel of 116.9 nM. We estimated the in vitro affinity of the interaction of the isolated CaV2.2 I–II linker and Gβγ to be 62 nM [3]. This is likely to be modulated in the intact channel, particularly by the presence of the CaVβ subunit. In contrast, for CaV2.2 with β2a-ΔvHOOK, the koff was 1.44 s−1 and the calculated kon was 38.6 μM−1 s−1, leading to a much higher affinity for Gβγ, the KD being 37.3 nM. It is worth noting that, as expected, the koff was much higher at +120 mV than that estimated at −100 mV, nevertheless, the off-rate of Gβγ from the β2a-ΔvHOOK construct was slower than that for the wild-type β2a at both potentials.
To examine further whether the presence of the SH3 domain played any role in G protein modulation, we finally examined the effect of including the SH3 and GK domains as two separate constructs. We found previously that the β2a-SH3 (1–135) domain, which retains a palmitoylation site and can, therefore, accumulate in the vicinity of the plasma membrane, is able to interact with β2a-GK (136–442) and mimic full-length β2a in its biophysical properties [32]. In contrast, β2a-SH3 (1–135) did not show any interaction with the isolated β2a-GK (226–442), which is lacking the final (ɛ) β-strand of the SH3 domain that is required for the interaction, but did show interaction with β2a-GK (214–442), which is lacking only the variable HOOK region, but retains the ɛ-strand that completes the SH3 domain [32]. These results are confirmed by our yeast two-hybrid results (see Fig. 6).
The effect of including together the noninteracting pair of constructs β2a-SH3 (1–135) and β2a-GK (226–442) on the basic current properties was a reduction, rather than an increase, in the peak IBa compared to β2a-GK (226–442) alone (Table 1) and a depolarization of the voltage for 50% activation of the current to +24.9 ± 2.4 mV (n = 5) compared to +32.0 ± 3.6 (n = 7) for β2a-GK (226–442) alone. As a possible explanation and in confirmation of the results described above for β2a-ΔvHOOK, we found that the inclusion of β2a-SH3 (1–135) with β2a-GK (226–442) resulted in calcium channel currents that showed strong tonic modulation. In the absence of agonist, the peak IBa showed a basal facilitation of 2.61 ± 0. 72 at +10 mV (Fig. 5a,b), which is likely to be the reason that the peak IBa was smaller in the presence than in the absence of β2a-SH3 (1–135) (Table 1). This is in strong contrast to the lack of facilitation shown by either of the β2a-GK domains alone (Figs. 3d and 5b). For this pair of constructs, the time constant for the removal of tonic inhibition (τdissoc) at +120 mV was 29.1 ± 5.4 ms (Fig. 5c, P > 0.05 compared to β2a).
Yeast two-hybrid assays were performed in order to probe any direct interactions between the CaVβ subunit constructs and the I–II linker of CaV2.2 and whether the interactions between specific β subunit domains matched the results obtained in our electrophysiological experiments (Fig. 6). Our positive control was the interaction between the CaV2.2 I–II linker and full-length β1b (Fig. 6a, column 1), which has been demonstrated by many different techniques, including surface plasmon resonance [9, 11, 21]. We found a β2a construct missing the extreme N terminus and the C terminus (5–442) also interacted with the I–II linker (Fig. 6a, column 2). The truncated constructs β2a-ɛSH3+GK (214–442) and β2a-HOOK+ɛSH3+GK (135–442) also interacted with the I–II linker (Fig. 6a, columns 3 and 4), whereas the shortest construct β2a-GK domain (225–442) did not interact with the I–II linker in this assay (Fig. 6a, column 5). Furthermore, none of the β2a-SH3 domains tested interacted with the I–II of CaV2.2 (Fig. 6b). These included β2a-SH3+HOOK (5–224) (Fig. 6b, columns 2 and 3) and β2a-SH3 (5–134) (Fig. 6b, columns 4 and 5).
We also found that β2a-SH3 (5–134) did not interact with β2a-GK (225–442) (Fig. 6c, columns 1 and 2 with the constructs in either vector), whereas it showed strong interactions with β2a-ɛSH3+GK (214–442) (Fig. 6c, column 3) and with β2a-HOOK+ɛSH3+GK (135–442) (Fig. 6c, column 4).
Discussion
The core structure of all CaVβ subunits is characterized by a GK and an SH3 domain [13, 18, 27, 39]. The 18-amino-acid AID motif in the I–II loop of HVA CaV α1 subunits is crucial for binding to CaVβ subunits [4, 21, 29]. Recent structural data from three groups have provided detailed information about CaVβ subunits and their interaction with the AID sequence [13, 27, 39]. However, the structural studies provided no insight into the role of the disordered HOOK domain, which intervenes in the split SH3 domain, before its fifth β-strand. In this study, we have examined which CaVβ subunit domains are involved in promoting the voltage dependence of G protein modulation, which is lost in the absence of any CaVβ subunit [9, 21].
Requirement of CaVβ GK domains for plasma membrane expression of HVA calcium channels
One of the main effects of CaVβ subunits on HVA calcium channels is to increase current density. However, the mechanism for this increase remains controversial, either being attributed to increased trafficking [6], increased maximum open probability [26], or both. In agreement with the first hypothesis, we and others have shown biochemically that the proportion of HVA CaVα1 subunits in the plasma membrane is increased by CaVβ subunit coexpression [1, 9, 14, 21]. This finding was reinforced by the fact that fewer channels were present at the surface when the mutated CaV2.2W391A channels that did not interact with β subunits were cotransfected with a CaVβ [21]. However, it is clear that CaVβ subunits also increase the open probability for CaV2.2 as well as other HVA channels [20, 25].
The CaVβ GK domain is sufficient to restore voltage-dependent G protein modulation of CaV2.2 channels
We showed previously that abrogation of the interaction of CaV2.2 with a CaVβ subunit, by introduction of the W391A mutation in the AID motif, did not affect the ability of Gβγ to inhibit CaV2.2, but did prevent the removal of Gβγ by a depolarizing prepulse [21]. In this study, we have obtained similar results when CaV2.2 was expressed without any CaVβ subunit and we found that coexpression of GK domain constructs from either β2a or β1b is sufficient to restore the voltage dependence of G protein modulation to wild-type CaV2.2 channels.
It has been found in several studies [3, 15] that the Gβγ subunits, which are responsible for direct G protein modulation of calcium channels, bind to the AID region of the I–II linker, and it has been proposed that they may compete with CaVβ subunits [33]. In contrast, fluorescence resonance energy transfer (FRET) studies have shown that CaVβ and Gβγ are able to bind to calcium channels at the same time [19]. Also in disagreement with a simple competition between Gβγ and CaVβ subunits is the finding that the presence of CaVβ subunits does not reduce the amount of G protein modulation [10, 24]. However, we did find that the presence of CaVβ subunits promoted the voltage-dependent removal of Gβγ by depolarizing prepulses [10, 24]. A similar conclusion was reached by using CaV2.2 containing the W391A mutation in the I–II linker, such that it did not interact with CaVβ subunits [21].
Involvement of other β subunit domains in G protein modulation of calcium channels
The results described in this study and our previous study [32] indicate that the reduced G protein modulation of the palmitoylatable compared to the nonpalmitoylatable β2 constructs is likely to be related to the fact that palmitoylation maintains an elevated concentration of this CaVβ subunit associated with the inner leaflet of the plasma membrane and, therefore, in the vicinity of the channel. However, as we and others have discussed previously [24], there is unlikely to be a simple competition between Gβγ and CaVβ subunits for binding to the I–II linker. Furthermore, in this study, we found that there is little difference in the amount of G protein modulation in the absence compared to the presence of any of the free GK domains. These findings indicate that the reduced G protein modulation seen with β2a is unlikely to be due solely to the fact that its palmitoylation results in an increased occupancy by its GK domain of the I–II linker. Moreover, the results with the HOOK deletion constructs indicate that the proposed interaction of the HOOK domain of β2a with the channel is also not responsible alone for the reduced modulation observed with palmitoylated β2a. This suggests that the increased interaction of both the SH3 domain and the HOOK domain with the channel, resulting from palmitoylation of β2a, is responsible for this difference in extent of G protein modulation.
Determinants of tonic modulation of CaV2.2
The strong basal facilitation of the β2a construct lacking the variable part of the HOOK domain requires its palmitoylation and indicates that the absence of this HOOK domain of β2a promotes, either directly or indirectly, tonic Gβγ binding. It is also possible that removal of the variable HOOK domain constrains the structure of the CaVβ subunit and that other parts of the calcium channel are also involved in Gβγ binding. In support of this, we noted a trend to increased basal facilitation for GK (136–442), containing the HOOK domain, compared to GK (226–442) in which the HOOK domain was absent, although this did not reach statistical significance.
The results from the experiment utilizing the combination of β2a-SH3 (1–135) and β2-GK (226–442) lacking the HOOK domain confirm the results obtained with β2a-ΔvHOOK, since both show strong tonic modulation. We interpret these results as indicating that the presence of the palmitoylatable free SH3 domain, in the absence of the variable part of the HOOK domain of β2a, promotes basal facilitation of CaV2.2 channels and results in increased tonic Gβγ binding. Our previous evidence [32] indicates that β2a-SH3 (1–135) is palmitoylated, since when it is coexpressed with GK(136–442), it reconstitutes the properties of palmitoylated β2a, in terms of slow inactivation, an effect which has been attributed to palmitoylation [21, 30]. Our present results further indicate that an interaction between the GK domain and the SH3 domain of β2a is not necessary for the demonstration of tonic G protein modulation, since β2a-SH3 (1–135)+β2-GK (226–442) and β2a-ΔvHOOK showed quite similar properties with respect to expression of tonic facilitation.
Although our electrophysiological data indicate that additional interactions are likely to occur between the CaV2.2 channels and both the SH3 and the HOOK domains of CaVβ subunits, nevertheless, our yeast two-hybrid data do not indicate that there is an interaction between the β2a-SH3 domain and the I–II linker of CaV2.2, in contrast to a previous study using CaV2.1 [23]. This was also suggested previously from our binding results for β1b, since it showed the same binding affinity for the full-length CaV2.2 I–II linker as the I–II linker truncated just after the AID region, indicating that there are no additional binding sites for the β subunit distal to the AID motif [9, 21].
In the present study, we have not addressed the other regions of the CaV2.2 subunit involved in this interaction, but other studies have shown that Gβγ binds to the C terminus of CaV2.2 [22] and that the I–II linker itself interacts with other regions of CaV2 channels [36]. These results, among others, indicate that Gβγ is likely to bind to a complex state-dependent binding pocket, also including the N terminus of the channel [28].
Our electrophysiological data suggest that the presence of the HOOK domain is important for the voltage-dependent removal of Gβγ. In particular, we calculate that in the absence of the HOOK domain, the affinity of the CaV2.2/CaVβ2 complex for Gβγ is increased about threefold. The effect of this change in affinity is manifested particularly at resting Gβγ levels, previously estimated to be about 50 nM [34], when it leads to substantial tonic Gβγ modulation. It is also evidenced by a more rapid rate of rebinding of Gβγ, following its removal by a depolarizing prepulse. These findings are in agreement with the hypothesis proposed previously [9, 11, 21] that the greater the occupancy of the binding site for CaVβ subunit on the channel, the greater is the voltage dependence of G protein inhibition.
In our previous study [32], we provided evidence that the HOOK domain of β2a is involved in modulating voltage-dependent inactivation, since removal of the HOOK domain shifted the steady-state inactivation to more negative potentials and also increased the inactivation kinetics. The contribution of inactivation imposed by different CaVβ subunits on G protein regulation has been investigated previously [25, 41]. We found that the inactivation properties of expressed CaV2.2 channels depended on the CaVβ subunit species, but only to a minor extent on the presence or absence of Gβγ. Furthermore, the closed times and latency to first opening of the CaV2.2 channels were increased by Gβγ, but this effect was similar for both β1b and β2a subunits [25]. More recently, the effect of the inactivation on G protein modulation was studied, and an effect was observed on voltage-dependent recovery from G protein modulation because of the opposing effects of different CaVβ subunits on inactivation [41]. In this study, we show directly that removal of the β2a HOOK domain enhances Gβγ binding affinity. In the future, this may help to identify how Gβγ dimers modulate the CaV2 channels.
Conclusions
The present results indicate that the interaction of a CaVβ subunit GK domain alone with the CaV2.2 channel is sufficient to restore voltage dependence to the G protein modulation process. However, these results also suggest that the SH3 and HOOK domains of CaVβ subunits are likely to have a role in preventing tonic binding of Gβγ to the calcium channels.
Acknowledgements
This work was supported by The Wellcome Trust. We thank the following for the generous gifts of cDNAs: Dr. Y. Mori (Seriken, Okazaki, Japan) for the rabbit CaV2.2; Dr. E. Perez-Reyes (Loyola University, Chicago, IL, USA) for the rat β2a; Dr. R. T. Hughes (Yale, New Haven, CT, USA) for the mut-3 GFP; Genetics Institute (Cambridge, MA, USA) for the pMT2. We also thank K. Chaggar for the technical assistance and Selvan Bavan for performing some of the molecular biology.
Conflict of interests None.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Andriy V. Dresviannikov and Karen M. Page contributed equally to this work.
References
- 1.Altier C, Dubel SJ, Barrère C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E (2002) Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem 277:33598–33603 [DOI] [PubMed]
- 2.Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, PerezReyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC, Rees M (2001) Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci 21:6095–6104 [DOI] [PMC free article] [PubMed]
- 3.Bell DC, Butcher AJ, Berrow NS, Page KM, Brust PF, Nesterova A, Stauderman KA, Seabrook GR, Nurnberg B, Dolphin AC (2001) Biophysical properties, pharmacology and modulation of human, neuronal L-type (a1D, CaV1.3) voltage-dependent calcium currents. J Neurophysiol 85:816–828 [DOI] [PubMed]
- 4.Berrou L, Klein H, Bernatchez G, Parent L (2002) A specific tryptophan in the I–II linker is a key determinant of beta-subunit binding and modulation in Ca(V)2.3 calcium channels. Biophys J 83:1429–1442 [DOI] [PMC free article] [PubMed]
- 5.Bertaso F, Ward RJ, Viard P, Milligan G, Dolphin AC (2003) Mechanism of action of G(q) to inhibit gbetagamma modulation of Ca(V)2.2 calcium channels: probed by the use of receptor-galpha tandems. Mol Pharmacol 63:832–843 [DOI] [PubMed]
- 6.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M (2000) The I–II loop of the Ca2+ channel alpha(1) subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 25:177–190 [DOI] [PubMed]
- 7.Birnbaumer L, Qin N, Olcese R, Tarteilus E, Platano E, Costantin J, Stefani E (1998) Structures and functions of calcium channel b subunits. J Bioenerg Biomembranes 30:357–376 [DOI] [PubMed]
- 8.Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I, Dolphin AC (1997) Importance of the different b subunits in the membrane expression of the a1A and a2 calcium channel subunits: studies using a depolarisation-sensitive a1A antibody. Eur J Neurosci 9:749–759 [DOI] [PubMed]
- 9.Butcher AJ, Leroy J, Richards MW, Pratt WS, Dolphin AC (2006) The importance of occupancy rather than affinity of CaV{beta} subunits for the calcium channel I-II linker in relation to calcium channel function. J Physiol 574:387–398 [DOI] [PMC free article] [PubMed]
- 10.Canti C, Bogdanov Y, Dolphin AC (2000) Interaction between G proteins and accessory b subunits in the regulation of a1B calcium channels in Xenopus oocytes. J Physiol 527:419–432 [DOI] [PMC free article] [PubMed]
- 11.Canti C, Davies A, Berrow NS, Butcher AJ, Page KM, Dolphin AC (2001) Evidence for two concentration-dependent processes for b subunit effects on a1B calcium channels. Biophys J 81:1439–1451 [DOI] [PMC free article] [PubMed]
- 12.Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16:521–555 [DOI] [PubMed]
- 13.Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J (2004) Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 429:675–680 [DOI] [PubMed]
- 14.Cohen RM, Foell JD, Balijepalli RC, Shah V, Hell JW, Kamp TJ (2005) Unique modulation of L-type Ca2+ channels by short auxiliary beta1d subunit present in cardiac muscle. Am J Physiol Heart Circ Physiol 288:H2363–H2374 [DOI] [PubMed]
- 15.De Waard M, Liu HY, Walker D, Scott VES, Gurnett CA, Campbell KP (1997) Direct binding of G-protein bgamma complex to voltage-dependent calcium channels. Nature 385:446–450 [DOI] [PubMed]
- 16.Dolphin AC (2003) b subunits of voltage-gated calcium channels. J Bioenerg Biomembranes 35:599–620 [DOI] [PubMed]
- 17.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA (2000) Nomenclature of voltage-gated calcium channels. Neuron 25:533–535 [DOI] [PubMed]
- 18.Hanlon MR, Berrow NS, Dolphin AC, Wallace BA (1999) Modelling of a voltage-dependent Ca2+ channel b subunit as a basis for understanding its functional properties. FEBS Lett 445:366–370 [DOI] [PubMed]
- 19.Hummer A, Delzeith O, Gomez SR, Moreno RL, Mark MD, Herlitze S (2003) Competitive and synergistic interactions of G protein beta(2) and Ca(2+) channel beta(1b) subunits with Ca(v)2.1 channels, revealed by mammalian two-hybrid and fluorescence resonance energy transfer measurements. J Biol Chem 278:49386–49400 [DOI] [PubMed]
- 20.Jones LP, Wei SK, Yue DT (1998) Mechanism of auxiliary subunit modulation of neuronal a1E calcium channels. J Gen Physiol 112:125–143 [DOI] [PMC free article] [PubMed]
- 21.Leroy J, Richards MS, Butcher AJ, Nieto-Rostro M, Pratt WS, Davies A, Dolphin AC (2005) Interaction via a key tryptophan in the I–II linker of N-type calcium channels is required for beta1 but not for palmitoylated beta2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J Neurosci 25:6984–6996 [DOI] [PMC free article] [PubMed]
- 22.Li B, Zhong H, Scheuer T, Catterall WA (2004) Functional role of a C-terminal Gbetagamma-binding domain of Ca(v)2.2 channels. Mol Pharmacol 66:761–769 [DOI] [PubMed]
- 23.Maltez JM, Nunziato DA, Kim J, Pitt GS (2005) Essential Ca(v)beta modulatory properties are AID-independent. Nat Struct Mol Biol 12:372–377 [DOI] [PubMed]
- 24.Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC (2000) Calcium channel b subunit promotes voltage-dependent modulation of a1B by Gbg. Biophys J 79:731–746 [DOI] [PMC free article] [PubMed]
- 25.Meir A Dolphin AC (2002) Kinetics and Gbetagamma modulation of Ca(v)2.2 channels with different auxiliary beta subunits. Pflugers Arch 444:263–275 [DOI] [PubMed]
- 26.Neely A, Garcia-Olivares J, Voswinkel S, Horstkott H, Hidalgo P (2004) Folding of active calcium channel beta(1b)-subunit by size-exclusion chromatography and its role on channel function. J Biol Chem 279:21689–21694 [DOI] [PubMed]
- 27.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA (2004) Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha1 interaction domain. Neuron 42:387–399 [DOI] [PubMed]
- 28.Page KM, Canti C, Stephens GJ, Berrow NS, Dolphin AC (1998) Identification of the amino terminus of neuronal Ca2+ channel a1 subunits a1B and a1E as an essential determinant of G protein modulation. J Neurosci 18:4815–4824 [DOI] [PMC free article] [PubMed]
- 29.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP (1994) Calcium channel b-subunit binds to a conserved motif in the I-II cytoplasmic linker of the a1-subunit. Nature 368:67–70 [DOI] [PubMed]
- 30.Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L (1998) Unique regulatory properties of the type 2a Ca2+ channel b subunit caused by palmitoylation. Proc Natl Acad Sci U S A 95:4690–4695 [DOI] [PMC free article] [PubMed]
- 31.Richards MW, Butcher AJ, Dolphin AC (2004) Calcium channel beta-subunits: structural insights AID our understanding. Trends Pharmacol Sci 25:626–632 [DOI] [PubMed]
- 32.Richards MW, Leroy J, Pratt WS, Dolphin AC (2007) The HOOK-Domain Between the SH3 and the GK Domains of CaVbeta subunits contains key determinants controlling calcium channel inactivation. Channels 1:92–101 [DOI] [PubMed]
- 33.Sandoz G, Lopez-Gonzalez I, Grunwald D, Bichet D, Altafaj X, Weiss N, Ronjat M, Dupuis A, De Waard M (2004) Cavbeta-subunit displacement is a key step to induce the reluctant state of P/Q calcium channels by direct G protein regulation. Proc Natl Acad Sci U S A 101:6267–6272 [DOI] [PMC free article] [PubMed]
- 34.Stephens GJ, Brice NL, Berrow NS, Dolphin AC (1998) Facilitation of rabbit a1B calcium channels: involvement of endogenous Gbgamma subunits. J Physiol (Lond) 509:15–27 [DOI] [PMC free article] [PubMed]
- 35.Stephens GJ, Page KM, Bogdanov Y, Dolphin AC (2000) The a1B calcium channel amino terminus contributes determinants for b subunit mediated voltage-dependent inactivation properties. J Physiol (Lond) 525:377–390 [DOI] [PMC free article] [PubMed]
- 36.Stotz SC, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW (2000) Fast inactivation of voltage-dependent calcium channels—a hinged-lid mechanism? J Biol Chem 275:24575–24582 [DOI] [PubMed]
- 37.Takahashi SX, Miriyala J, Colecraft HM (2004) Membrane-associated guanylate kinase-like properties of beta-subunits required for modulation of voltage-dependent Ca2+ channels. Proc Natl Acad Sci U S A 101:7193–7198 [DOI] [PMC free article] [PubMed]
- 38.Takahashi SX, Miriyala J, Tay LH, Yue DT, Colecraft HM (2005) A CaVbeta SH3/guanylate kinase domain interaction regulates multiple properties of voltage-gated Ca2+ channels. J Gen Physiol 126:365–377 [DOI] [PMC free article] [PubMed]
- 39.Van Petegem F, Clark KA, Chatelain FC, Minor DL Jr (2004) Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429:671–675 [DOI] [PMC free article] [PubMed]
- 40.Walker D, Bichet D, Campbell KP, De Waard M (1998) A b4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel a1A subunit. J Biol Chem 273:2361–2367 [DOI] [PubMed]
- 41.Weiss N, Tadmouri A, Mikati M, Ronjat M, De Waard M (2007) Importance of voltage-dependent inactivation in N-type calcium channel regulation by G-proteins. Pflugers Arch 454:115–129 [DOI] [PMC free article] [PubMed]