

Abstract
The monocytic leukemia zinc finger (MOZ) gene encodes a large multidomain protein that contains, besides other domains, 2 coactivation domains for the transcription factor Runx1/acute myeloid leukemia 1 and a histone acetyl transferase (HAT) catalytic domain. Recent studies have demonstrated the critical requirement for the complete MOZ protein in hematopoietic stem cell development and maintenance. However, the specific function of the HAT activity of MOZ remains unknown, as it has been shown that MOZ HAT activity is not required either for its role as Runx1 coactivator or for the leukemic transformation induced by MOZ transcriptional intermediary factor 2 (TIF2). To assess the specific requirement for this HAT activity during hematopoietic development, we have generated embryonic stem cells and mouse lines carrying a point mutation that renders the protein catalytically inactive. We report in this study that mice exclusively lacking the HAT activity of MOZ exhibit significant defects in the number of hematopoietic stem cells and hematopoietic committed precursors as well as a defect in B-cell development. Furthermore, we demonstrate that the failure to maintain a normal number of hematopoietic precursors is caused by the inability of HAT−/− cells to expand. These results indicate a specific role of MOZ-driven acetylation in controlling a desirable balance between proliferation and differentiation during hematopoiesis.
Introduction
Hematopoiesis is orchestrated at the molecular level by the interplay between transcription factors and chromatin-modifying enzymes (reviewed by Rice et al1 and Kioussis and Georgopoulos2). The importance of the epigenetic regulation in hematopoiesis is highlighted by the findings that chromosomal translocations that alter the activity of chromatin-modifying enzymes are recurrently found associated with different forms of leukemia. One of these chromatin-modifying enzymes, the monocytic leukemia zinc finger (MOZ or recently renamed KAT6a3) protein, was first identified through positional cloning of t(8;16)(p11;p13) translocation with CREB binding protein (CBP) in acute myeloid leukemia (AML).4 Subsequently, MOZ was found translocated to P300,5 to transcriptional intermediary binding factor 2 (TIF2),6,7 and more recently to NcoA3.8 It has been proposed that aberrant acetylation by these fusion proteins might participate in the process of leukemogenesis.4
MOZ encodes a large multidomain protein (224 kDa) that contains, among others, 2 domains shown to interact with the transcription factor Runx1,9,10 a C4H3 domain also referred to as plant homeodomain, an atypical Cys(2)-His-Cys (C2HC) zinc finger domain with a putative nucleosome-binding activity and a region with homology to the active acetyl-coenzyme A (CoA) binding site of several histone acetyl transferase (HAT) proteins4 (Figure S1A, available on the Blood website; see the Supplemental Materials link at the top of the online article). MOZ is the founding member of the MOZ, Ybf2/Sas2, Tip60 (MYST) family of HATs, characterized by the presence of a conserved MYST domain that comprises both the C2HC nucleosome-binding domain and the putative HAT domain.4 MOZ has been shown to acetylate in vitro specific lysine residues of several proteins, including histones H2A, H3 and H4, Runx1, and MOZ itself.9,11 Although the HAT activity of MOZ is now well established, its biologic relevance is still unknown. Analysis of the MOZ-TIF2 leukemic fusion protein has shown that the C2HC nucleosome binding domain is necessary for leukemic transformation, but that, in contrast, the HAT activity seems dispensable.12 It has also been reported that MOZ can interact with the transcription factor Runx1, a critical protein in hematopoietic development13–15 and a gene frequently found translocated in leukemia.16,17 Interaction between MOZ and Runx1 leads to an increased activity of Runx1-responsive reporter genes.9,18 Although several domains of MOZ were shown to be required for the interaction and activation of Runx1-dependent transcription by MOZ, the HAT domain did not appear to play any role in that function.9
Two groups have recently assessed the physiologic role of the MOZ protein in vivo by generating mice with distinct targeted alleles of MOZ, both leading to the absence of the complete MOZ protein. Analyses of these mice indicate that MOZ is essential for the development19 and the maintenance20 of hematopoietic stem cells (HSCs), and that MOZ deletion results, respectively, in embryonic lethality by midgestation or death at birth. These phenotypes observed in absence of the whole MOZ protein are likely to reflect the cumulative consequences of the loss of both the Runx1 transcriptional coactivator function and the HAT activity. However, the specific importance of the HAT activity of MOZ in hematopoiesis has not been addressed by these previous studies.
To dissect the specific relevance of MOZ-driven acetylation in the regulation of hematopoiesis, we generated a mouse strain carrying a single amino acid change that inactivates the HAT activity of the MOZ protein. In this mouse strain, any phenotype is therefore directly resulting from the lack of HAT activity rather than from the inactivation of any other function that other domains of the protein might perform. In contrast to the complete MOZ knockouts, HAT−/− MOZ mice remain alive during all the gestation period, but approximately 40% of the homozygote mice subsequently die within the first 6 months of causes currently under investigation. Importantly, these mice exhibit significant defects in the number of HSCs and committed precursors, suggesting that reduction in HSC number previously reported for the MOZ complete knockout is likely to reflect, at least in part, a direct effect of the lack of MOZ HAT activity. Interestingly, we also observed in these mice a B-cell developmental defect, distinct to the defects observed in the total MOZ knockout. Using the in vitro differentiation of embryonic stem (ES) cells toward hematopoiesis, we demonstrate that the defect is established at the earliest stage of hematopoietic development. The defect in the numbers of hematopoietic precursors is consistently associated with the generation from these precursors of colonies containing fewer cells. Accordingly, culture of CD34+c-Kit+ purified hematopoietic progenitors revealed that HAT−/− MOZ hematopoietic precursors did not display defects in differentiation or increased levels of apoptosis, but had a profound deficiency in their proliferative potential. Altogether our study demonstrates a critical role of the HAT activity of MOZ in the proliferation and maintenance of hematopoietic precursors at all stages of development.
Methods
Construction of the MOZ HAT expression vector and purification of the mutated protein
The sequence containing amino acids 507-945 of MOZ (RCPSVIE to SEGVE), corresponding to the complete MYST domain plus some of the acidic domain, was cloned in the bacterial expression vector pQE-32 and fused to a His-tag for purification. The HAT mutation (G657E) was introduced using the QuickChange mutagenesis kit (Stratagene; Agilent Technologies, Cedar Creek, TX). The baculoviral MOZ protein was expressed in pFASTBAC. Purifications of the MOZ proteins were performed on nickel-nitrolotriacetic acid resin. The purified proteins were used in a standard HAT assay. Type IIA histones (Sigma-Aldrich, St Louis, MO; H-9250) were acetylated with tritiated acetyl enzyme CoA (Amersham, Princeton, NJ; TRK688). Assays were incubated for 1 hour at 30°C. Duplicate aliquots were run out on 15% to 20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels, and the gels were cut in half. Samples were run in duplicates. As loading control, one set of samples was stained with Coomassie blue, whereas the other half was fixed, dried, and exposed using a tritium screen on a phosphor imager.
Construction of the targeting vector
The regions of the targeting vector homologous to MOZ sequences were isolated from a 129/Ola strain bacterial artificial chromosome (BAC) library (Genome Systems, St Louis, MO). The polymerase chain reaction (PCR) conditions followed for the amplification of the homologous arms are detailed in Perez-Campo et al.21
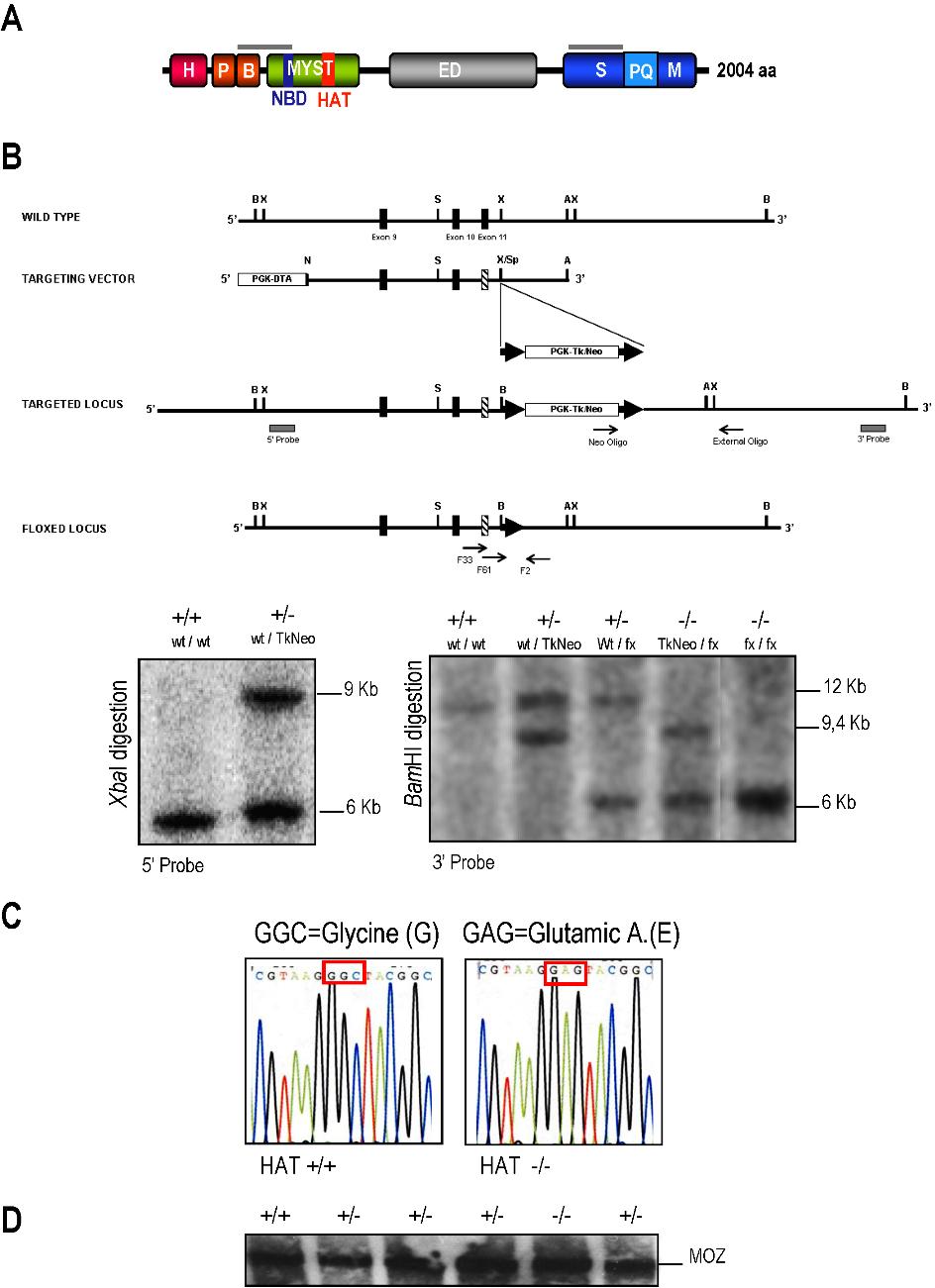
Generation of MOZ HAT−/− ES cells
Mouse ES cells (E14.1, 129/Ola) were electroporated with the ApaI-linearized targeting vector. Clones that had undergone a homologous recombination event were identified by PCR and confirmed by Southern blot analysis. Of 384 individually selected clones, 3 correctly targeted were identified. Two positive clones were transiently transfected with a Cre recombinase expression vector to excise the Tkneo cassette. TkNeo-deficient clones were identified by PCR. The intactness of the targeted locus and the absence of the TkNeo cassette in G418-sensitive clones after Cre-mediated excision were confirmed by Southern blot analysis. The second copy of the gene was targeted, and the TkNeo cassette was removed to generate MOZ HAT−/− ES cells.
ES cell growth and differentiation
ES cells were maintained and differentiated, as previously described.22 Differentiation of embryoid bodies (EBs) was carried out in 60-mm Petri-grade dishes in Iscove modified Dulbecco medium (IMDM) supplemented with 15% fetal calf serum (FCS), 2 mM l-glutamine (Invitrogen, Carlsbad, CA), transferrin (200 μg/mL; Roche, Basel, Switzerland), 0.5 mM ascorbic acid (Sigma-Aldrich), and 4.5 × 10−4 M monothioglycerol (MTG). Cultures were maintained in a humidified chamber in a 5% CO2 air mixture at 37°C. Serum-free conditions that sustain the growth of hematopoietic precursors in liquid culture were described by Mikula et al.23
Colony assays
Conditions for the generation and expansion of blast cell colonies (blast colony-forming cell [BL-CFC] assay) and for the growth of hematopoietic precursors have been previously described.15,24 For expansion of blast cell colonies, individual colonies were transferred to Matrigel-coated (Collaborative Research, San Jose, CA) microtiter wells containing IMDM with 10% FCS, 10% horse serum (Invitrogen), vascular endothelial growth factor (VEGF; 5 ng/mL), insulin-like growth factor 1 (IGF-1; 10 ng/mL), erythropoietin (2 U/mL), basic fibroblast growth factor (10 ng/mL), interleukin (IL)-11 (50 ng/mL), kit ligand (KL; 1% conditioned medium), IL-3 (1% conditioned medium), l-glutamine (1%), and 4.5 × 10−4 M MTG. After 4 days of growth, the presence of adherent (endothelial) and nonadherent (hematopoietic) cells was scored in each well. Pictures of colonies or cell cultures were taken with a Nikon Coolpix 995 camera on a DMIRB Leica microscope with objectives 10×/0.25 NA and 4×/0.1 NA, respectively. Cytospin slides were mounted with Pertex mounting media (Cell Path, Newtown, United Kingdom). Pictures of the cytospin slides were taken with a Roper Coolsnap HQ and a CRI Microcolour RGB tuneable filter on an Olympus BX51 Microscope with objective UPLFLN 40×/0.75 NA. Images were processed with Adobe Photoshop version 6.0 (Adobe, San Jose, CA).
Immunohistochemical staining
Aggregates or trypsinized colonies were plated on gelatin-coated coverslips and cultured in IMDM with 10% serum replacement (Invitrogen). Cells were fixed in 2% paraformaldehyde for 20 minutes, washed twice in PBS, permeabilized in 0.2% Triton X-100/PBS, and washed in 10% FCS/0.2% Tween 20/PBS. Cells were incubated for 1 hour with the corresponding antibody.
Animal work
All animal work was performed under regulation governed by the Home Office Legislation under the Animal Scientific Procedures Act 1986, with approval from the Paterson Institute for Cancer Research.
Embryo dissections
Heterozygote mice for the HAT point mutation were mated overnight, and noon of the day of the vaginal plug was defined as 0.5 days postconception (dpc). At 14.5 dpc, the uteri were removed from the peritoneum and washed with several changes of IMDM. Embryos were dissected, and fetal liver was harvested. Single-cell suspensions of tissues were obtained by mechanical disaggregation using a syringe.
Competitive repopulation assays
Experimental conditions for this assay were as published by Langer et al.25
Immunizations and enzyme-linked immunosorbent assay
Preimmune serum was collected 2 days before immunization with 100 μg of dinitrophenyl (DNP)–keyhole limpet hemocyanin (KLH; BioCat, Heidelberg, Germany) and Alum adjuvant (Pierce, Rockford, IL). Mice were bled at 2 weeks and boosted with 100 μg DNP-KLH at 3 weeks, and serum was collected at 4 weeks. Enzyme-linked immunosorbent assay plates were coated with DNP-bovine serum albumin (BioCat), and immunoglobulin (Ig) M and IgG responses against DNP were quantified.
Glucose-6-phosphate-isomerase assay
The contribution of ES cells to chimeric animals was determined by glucose-6-phosphate-isomerase (GPI) assay, performed as originally described,26 using Helena Laboratories (Beaumont, TX) reagents.
Flow cytometry
Single-cell suspensions were analyzed on a FACScan or a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) or sorted on a FACSVantage cell sorter (BD Biosciences). The antibodies used were as follows: CD3, Gr1, Mac1, B220, and Ter119 (biotinylated); Sca-1 (fluorescein isothiocyanate [FITC]), c-Kit (allophycocyanin [APC]), and IL7-R (phycoerythrin [PE]) were used for the HSC and common lymphoid progenitor (CLP) analysis; Sca-1, CD3, Gr1, Mac1, B220 and Ter119 (PE), c-Kit (APC), and CD34 (biotinylated) and CD16 (FITC) were used for the common myeloid progenitor (CMP), megakaryocyte-erythroid progenitor (MEP), and GMP analysis. Staining with mAb Flk1-bio, c-Kit-PE, CD34-bio, CD34-APC, and CD45-Bio was performed, as previously described.22 All the antibodies mentioned above were from BD Pharmingen (San Diego, CA). Annexin/7-aminoactinomycin D staining was performed using the annexin V-PE Apoptosis Detection Kit I (BD Pharmingen) following the manufacturer's instructions. For cell-cycle analysis, cells were stained with 1 μg/mL propidium iodide.
Results
Generation of the MOZ HAT+/− ES cells and mice
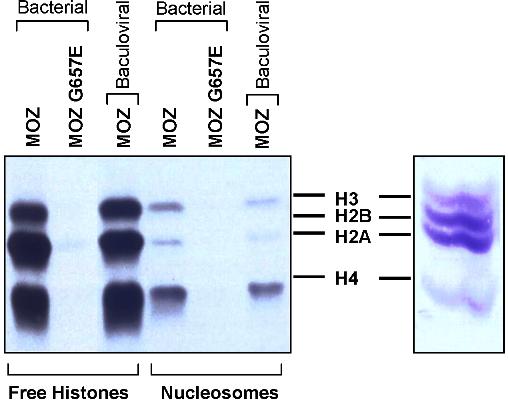
To investigate the importance of MOZ HAT activity in hematopoiesis, we introduced by gene targeting a single amino acid substitution (G657E) in the HAT catalytic domain of MOZ (Figure S1B,C). Previous works have shown that an analogous mutation abrogates the HAT activity of human MOZ12 and inactivates the HAT activity of the MOZ yeast and Drosophila homologues SAS3 (something about silencing protein 3)27 and MOF (males absent on the first),28 respectively. The same mutation also inactivates the ability of the mouse MOZ protein to acetylate free histones or nucleosomes, as demonstrated in Figure S2. Three independent heterozygous HAT+/− ES cell lines were established upon electroporation of the targeting vector in E14 ES cells. The same process was simultaneously conducted with a nonmutated version of the targeting vector. This control allowed us to assign the phenotype of the resulting mice solely to the presence of the point mutation. Injection of a HAT+/− ES cell line and the control ES cell line into blastocysts led to the generation of MOZ HAT+/− and MOZ control mice. For all studies, to minimize the background effects on the analysis of hematopoiesis, all mice used were age-matched littermates backcrossed onto the C57BL/6 background. Two independent homozygote HAT−/− cell lines were derived by targeting the second allele of MOZ in HAT+/− ES cell lines.
Crosses of HAT+/− heterozygotes produced viable HAT−/− offspring, but more than 40% of these homozygote mice died within the first 6 months of causes currently under investigation (Figure S3). The homozygous mice displayed in general low body weight and the size of the hematopoietic organs (thymus and spleen) were reproducibly reduced (Table S1). In contrast, no gross abnormalities were detected in HAT+/− mice (data not shown). The expected MOZ protein product of 224 kDa was detected at similar levels in cell extracts of wild-type (WT), heterozygous, and homozygous embryos by immunoblotting with a monoclonal antibody against MOZ (Figure S1D). The resulting HAT−/− protein is expected to assemble efficiently into higher order transcriptional complexes, preserving all domains as well as its ability to interact with Runx1 and to stimulate Runx1-mediated transcription.9,18
The mutation of the HAT domain affects B-lineage differentiation and hematopoietic stem/progenitor cell frequencies
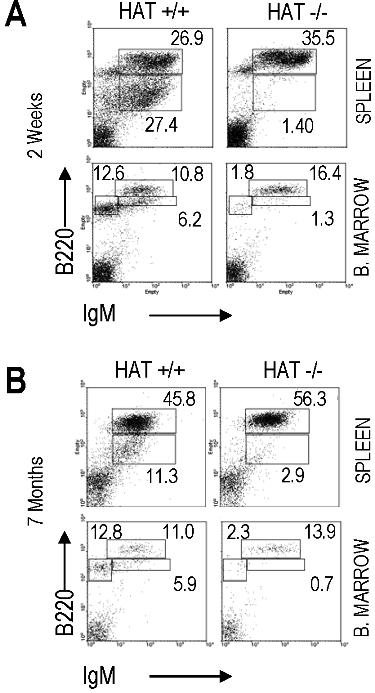
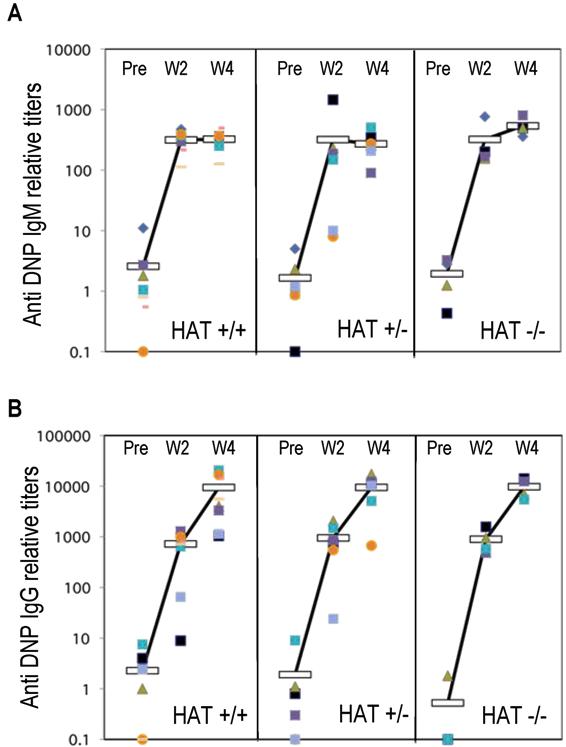
We analyzed the presence of lymphoid, myeloid, and erythroid cell populations in HAT+/+, HAT+/−, and HAT−/− mice by flow cytometry (Figure 1 and Table S2). The frequencies of both pro/pre-B cells (B220+/IgM−) and immature B cells (B220dim/high/IgMhigh) were reproducibly lower in the bone marrow of HAT−/− animals than in the bone marrow of WT and heterozygous mice. This lower frequency in immature B cells was also observed in the spleen of the HAT−/− animals (Figure 1) and was consistently encountered in all HAT−/− mice analyzed at different stages of development (Figure S4). The frequency of mature B cells (B220+/IgM+) was, in contrast, not affected. HAT−/− MOZ mice did not display any deficiencies in their abilities to raise, after immunization with DNP-KLH, IgM, and IgG antibody responses (Figure S5), indicating that these mice are indeed not immunocompromised.
Figure 1.

Analyses of hematopoietic cell populations in HAT+/+, HAT+/−, and HAT−/− mice. Flow cytometry analysis of T-cell, B-cell, erythroid, and macrophage populations in the bone marrow, thymus, and spleen of 3-month-old mice. FACS profiles shown in the figure correspond to 1 representative mouse of each phenotype.
In addition, we did not detect any defect in HAT−/− mice either in the frequency of thymic CD4−/CD8− T cells, as described by Thomas et al,19 or in the number of Ter119+/CD71+ erythroid cells either in bone marrow or E14.5 fetal liver, as described by Katsumo et al20 (Figure 1 and Table S2). The frequencies of mature myeloid CD45+/Mac1+ cells were equivalent in the bone marrow of HAT+/+, HAT+/−, and HAT−/− mice. Altogether these findings indicate that mature B, T, erythroid, and myeloid cells are represented at normal levels in HAT−/− mice and reveal a specific role for the HAT activity of MOZ in the generation of B-cell subpopulations.
We extended our FACS analyses to more immature hematopoietic compartments and quantified myeloid precursor, lymphoid precursor, and HSC populations in the fetal liver of HAT+/+, HAT+/−, and HAT−/− day 14.5 embryos. A substantial reduction in the frequency of the LSK (Lin−/Sca-1+/c-Kit+) cell population, which includes HSCs, was detected in the fetal liver of HAT−/− embryos (Figure 2 and Table S3). This decrease in the HSC population was equivalent to the ones observed in fetal liver of embryos lacking the full MOZ protein.19,20 Furthermore, the frequencies of CMPs (Lin−/Sca-1−/c-Kit+/CD34+/FcγRIII/IIlow), CLPs (Lin−/Sca-1low/IL-7-Rα+/c-Kitlow), granulocyte-macrophage progenitors (GMPs, Lin−/Sca-1−/c-Kit+/CD34+/FcγRIII/II+), and MEPs (Lin−/Sca-1−/c-Kit+/CD34−/FcγRIII/IIlow) were also reduced in HAT−/− compared with HAT+/+ (Figure 2A,B). We also observed a decrease in the number of cells with the KLSF (Lin−/Sca-1−/c-Kit+/CD34−/FcγRIII/II+) phenotype, whereas an increase in the frequency of cells in this subpopulation was detected in one of the complete MOZ knockouts.20 The HAT+/− animals displayed in most cases an intermediate phenotype. Altogether these data indicate that although all subpopulations of hematopoietic precursors are present in the homozygous HAT−/− mice, they are consistently represented at lower frequencies than in the WT and heterozygote animals.
Figure 2.

Analyses of HSC and precursor populations in HAT+/+, HAT+/−, and HAT−/− fetal liver. (A) Staining and (B) quantification of E14.5 fetal liver cells with the indicated antibodies to identify the HSC, CLP, GMP, CMP, MEP, and KLSF populations by flow cytometry. To analyze the HSC and CLP populations, 8 HAT+/+, 8 HAT+/−, and 8 HAT−/− mice were used. To analyze the GMP, KLSF, CMP, and MEP populations, 8 HAT+/+ 24 HAT+/−, and 8 HAT−/− mice were used. The values represent the percentage of cells in each cell population. Each sample is represented by a ▴, and the average of the samples is presented as a horizontal bar. *P < .5; **P < .01; ***P < .005.
The HAT activity of MOZ is essential for maintaining the functionality of HSCs
To directly assess the functional consequences of the inactivation of the HAT activity of MOZ on lineage-committed hematopoietic progenitors, we performed colony-forming cell assays with 14.5 dpc fetal liver cells. HAT−/− hematopoietic progenitors were able to give rise to all cell lineages; however, the number of committed progenitors was reduced by approximately half compared with their WT counterparts (Figure 3A). In the HAT+/− heterozygote, an intermediate phenotype was detected with reduction in numbers of both erythroid and mixed lineage precursors, whereas myeloid lineage progenitor levels were not affected. The lower numbers of HAT−/− colonies are likely to reflect the lower frequencies of precursor cell populations observed in HAT−/− fetal liver (Figure 2B). Interestingly, the size of all colonies generated by the HAT−/− hematopoietic progenitors was reproducibly reduced (Figure 3B).
Figure 3.

Hematopoietic colonies and long-term repopulation potential of HAT+/+, HAT+/−, and HAT−/− fetal liver cells. (A) Numbers of colonies generated by HAT+/+, HAT+/−, and HAT−/− fetal liver cells from littermate animals. A total of 30 × 103 cells/mL was replated in methylcellulose medium. Fetal liver cells were then genotyped, and myeloid, erythroid, and mixed lineage colonies were scored after 6 to 8 days. The average of the number of colonies generated is indicated by a horizontal bar. (B) Comparative size of representative HAT+/+ and HAT−/− colonies generated from day 14.5 fetal liver (magnification ×100). (C) Competitive long-term repopulation of irradiated mice. Data are expressed in log ratio between tested C57BL/6 fetal liver–derived (CD45.2+) and competitor PEP3 bone marrow–derived cells (CD45.1+). A total of 2 × 105 fetal liver cells from HAT+/+, HAT+/−, and HAT−/− embryos was transplanted into irradiated CD45.1+/CD45.2+ recipients with 4 × 105 competitor bone marrow cells from PEP3 mice (CD45.1+). A total of 2 × 106 cells from primary recipients was transplanted into secondary recipients 3 months after reconstitution. Bars represent standard error of the mean values. *P < .5; **P < .01; ***P < .005.
We next tested the functionality of HAT−/− HSCs by competitive repopulation assays. Day 14.5 fetal liver cells from C57BL/6 HAT+/+, HAT+/−, and HAT−/− embryos (expressing the allelic variant CD45.2) were transplanted into F1 C57BL/6 × PEP3 recipients (expressing both CD45.1 and CD45.2) together with competitor bone marrow cells from PEP3 mice (expressing CD45.1). We measured the ratio of the single-positive CD45.2+ cell population versus the single-positive CD45.1+ population in the peripheral blood of the recipients at 4-week intervals after transplantation. As shown in Figure 3C on the left panel, the repopulation capacity of HAT+/− and HAT−/− fetal liver cells was clearly inferior to that of WT cells. To eliminate the possibility that these results were just reflecting the initial lower frequency of HSCs in heterozygous and homozygous mice, we performed secondary transplantations by injection of bone marrow cells from primary recipients, 12 weeks after their reconstitution, into secondary PEP3 (CD45.1). The defect in the repopulation capacity of HAT−/− cells was worsened after the secondary transplantations (Figure 3C right panel). These results demonstrate the critical role of the HAT activity of MOZ in the maintenance of the HSC functionality and suggest that the defect in HSCs observed in both complete knockout of MOZ results from the absence of this activity.
The HAT mutation impairs the onset of development of the hematopoietic lineages
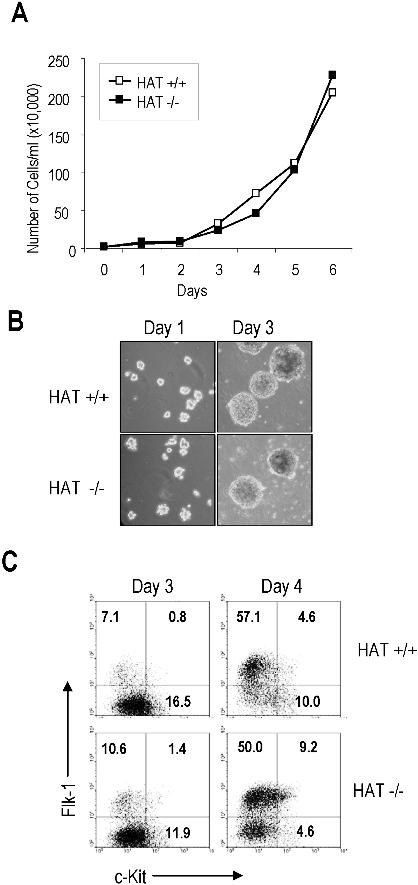
The in vitro differentiation of ES cells has been shown to recapitulate early events of hematopoietic development and, as such, to provide a good experimental system to characterize the function of genes during this process.29–31 The first blood precursor generated during ES cell differentiation is the blast colony-forming cell (BL-CFC), which gives rise to blast colonies containing progenitors of smooth muscle, endothelial, and hematopoietic lineages, and represents the in vitro equivalent of the hemangioblast.32,33 During in vitro differentiation, WT HAT+/+ and homozygous HAT−/− ES cell lines formed embryoid bodies (EBs) to the same extent, expanded at the same rate, and acquired the expression of Flk-1, the receptor 2 for VEGF34 with similar kinetics (Figure S6), indicating no gross impairment of HAT−/− cells. We next evaluated the potential of HAT+/+, HAT+/−, and HAT−/− ES cells to generate blast colonies. We observed, at all time points examined, that HAT−/− EB cells generated less than half the number of blast colonies compared with HAT+/+ and HAT+/− cell lines (Figure 4A). Two independent HAT−/− ES cell lines presented a similar defect, indicating that this phenotype was not restricted to a specific clone (data not shown). Strikingly, blast colonies generated by HAT−/− ES cells were reproducibly smaller than those generated by HAT+/+ and HAT+/− ES cells (Figure 4B). To evaluate the hematopoietic, smooth muscle, and endothelial potential of these blast colonies, 32 individual colonies from each genotype were picked and expanded in 96-well plates in conditions that support the proliferation of both hematopoietic and endothelial lineages. After 4 days, we scored each well for the presence of adherent smooth muscle/endothelial cells (E) and nonadherent hematopoietic cells (H). The majority of wells seeded with WT and heterozygous colonies contained both adherent vascular/smooth muscle cells and nonadherent hematopoietic cells (H+/E+; Figure 4C,D). In contrast, the majority of HAT−/− individual blast colonies generated only a layer of vascular/smooth muscle cells with the expansion of hematopoietic cells extremely limited or absent (H−/E+). These findings indicate a critical role of the HAT activity of MOZ in the expansion of early hematopoietic precursors from blast colonies.
Figure 4.

Blast colony potential and hematopoietic and smooth muscle/endothelial potential of blast colonies generated from HAT+/+, HAT+/−, and HAT−/− ES cell lines. (A) Number of blast colonies generated by HAT+/+, HAT+/−, and HAT−/− ES cell lines. Bars represent standard error of the mean number of colonies from at least 3 cultures. P values indicating that the differences between the number of colonies generated by the HAT+/+ and the HAT−/− are statistically significant are indicated. (B) Comparative size of HAT+/+ and HAT−/− individual blast colonies generated from day 3.5 EBs (magnification ×200). (C) Individual HAT+/+, HAT+/−, and HAT−/− blast colonies were analyzed for their ability to generate hematopoietic and endothelial cells. Thirty-two blast colonies from each cell line were grown in liquid culture. The ability of each individual blast colony to expand and generate or not hematopoietic (H+ or H−) and/or endothelial (E+ or E−) cells was scored 4 days later. (D) Generation by individual blast colonies of nonadherent hematopoietic (white arrowhead) and adherent endothelial cells (black arrowhead). Note the limited proliferation of hematopoietic cells from HAT−/− blast colonies.
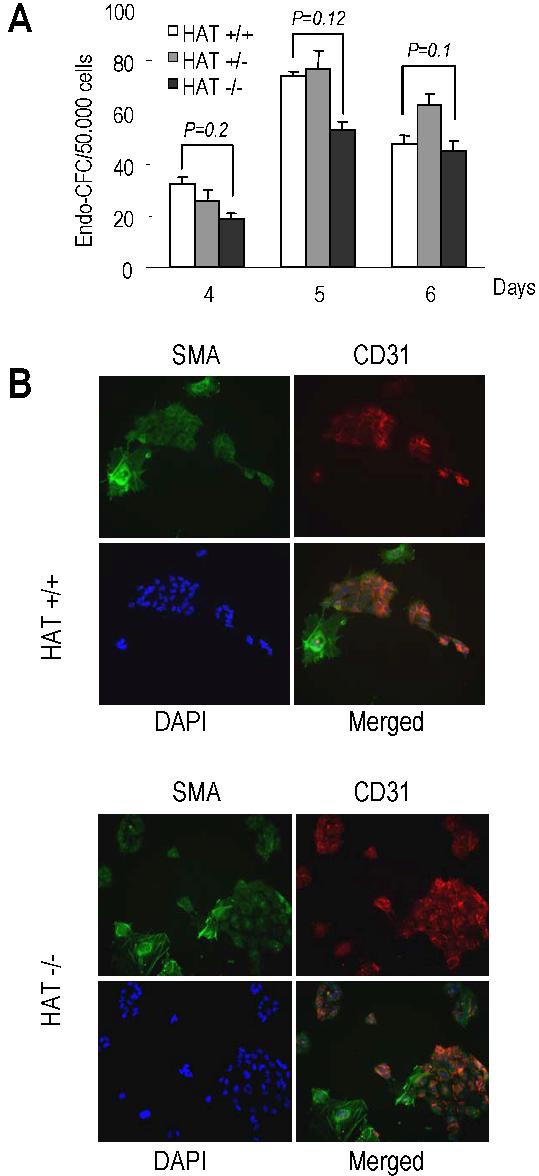
To further analyze the vascular endothelial/smooth muscle potential of the HAT−/− cells, we sorted precursors for these lineages on the basis of Flk-1 expression from day 4, 5, and 6 EBs. There were no statistically significant differences between the number of endothelial colonies generated by the HAT+/+ and HAT−/− MOZ Flk-1+ precursors (Figure S7A). When Flk-1+ cells were plated on gelatin-coated glass coverslips and allowed to grow for 10 days, similar frequencies of cells stained positively for the endothelial marker CD31 (platelet endothelial cell adhesion molecule) and the smooth muscle marker, smooth muscle actin (Figure S7B). Altogether these data indicate that the HAT activity of MOZ is critical for the normal expansion of early hematopoietic precursors, whereas, in contrast, generation and development of vascular and smooth muscle cells are largely unaffected.
We next quantitatively assessed hematopoietic development in more differentiated EBs (from day 5 to day 11). EB cells were replated daily in methylcellulose culture conditions that support the growth of myeloid and erythroid lineage precursors. As shown in Figure 5A, the HAT−/− cells generated dramatically fewer primitive erythroid (EryP), macrophage (Mac), definitive erythroid (EryD), and mast cell (Mast) colonies than HAT+/+ and heterozygous HAT+/− cells. The defect was strikingly more severe for hematopoietic lineages generated at later time points of differentiation, such as definitive erythrocytes or mast cells, suggesting a progressive defect in the generation of hematopoietic cells. In addition, the hematopoietic colonies generated by HAT−/− cells were consistently smaller than their HAT+/+ and heterozygous HAT+/− counterparts (Figure 5B). Taken together, these results indicate a critical function of the HAT activity of MOZ at the onset of hematopoietic development.
Figure 5.

Analysis of primitive and definitive hematopoietic potential of HAT+/+, HAT+/−, and HAT−/− ES cells. (A) EBs were harvested from day 4 to day 11 of differentiation, and single-cell suspensions were replated in conditions supporting the generation of both primitive and definitive hematopoietic colonies. Primitive erythroid (EryP), definitive erythroid (EryD), macrophage (Mac), and mast cell (Mast) colonies were scored. Days of differentiation are indicated. Bars represent standard error of the mean number of colonies from at least 3 cultures. (B) Comparative size of primitive erythroid and macrophage colonies generated from HAT+/+ and HAT−/− day 6 EBs (magnification ×200).
HAT−/− hematopoietic progenitors show a defect in proliferation
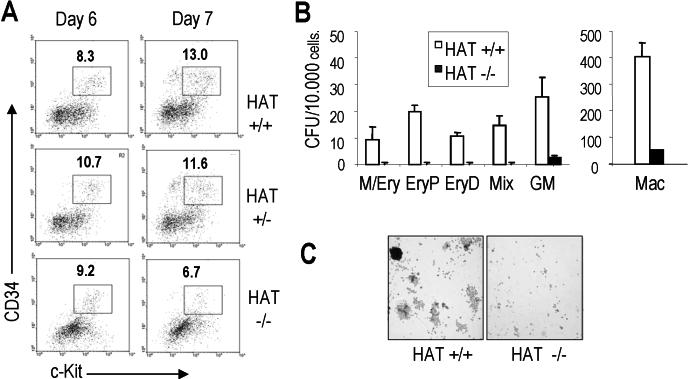
To assess whether the defect in generation of mature hematopoietic colonies from HAT−/− cells was reflecting a decrease in the generation of early hematopoietic progenitors in EBs, we analyzed EBs for the presence of CD34+c-Kit+ cells containing early hematopoietic precursors.35,36 At day 6 of differentiation, we observed similar frequencies of CD34+c-Kit+ cells in EBs generated from the 3 types of ES cells (Figure S8A). In contrast, by day 7, the percentage of CD34+/c-Kit+ cells was reduced by half in HAT−/− EBs relative to their WT and heterozygous counterparts. These results suggest a progressive defect in the generation (amplification) upon time of cell population of early hematopoietic precursors. We then sorted similar number of CD34+/c-Kit+ cells from day 6 HAT+/+ and HAT−/− EBs and replated them in methylcellulose cultures. HAT−/− cells generated dramatically fewer mature hematopoietic colonies than the equivalent cell population sorted from WT EBs (Figure S8B). The colonies generated from the mutant ES cells were again reproducibly smaller than their WT counterparts (Figure S8C). Altogether these findings indicate a major impairment in the ability of HAT−/− MOZ precursors to generate normal frequencies of colonies containing high number of mature hematopoietic cells.
The generation of lower number of colonies containing fewer cells could potentially result either from impaired differentiation, increased rate of cell death, or defects in proliferation. To examine these different possibilities, we sorted CD34+c-Kit+ hematopoietic precursors from WT and mutant EBs and cultured identical number of cells in a serum-free media with cytokines supporting the proliferation and differentiation of hematopoietic precursors. We observed mostly immature cells with a blastic morphology at the earliest time points and more differentiated cells with mostly a typical macrophage morphology at later time points (Figure 6A). We scored the morphology of 200 cells per culture at different time points and observed that cells with an immature morphology were persisting longer in the WT culture (Figure 6B). This observation was confirmed by flow cytometry analysis, indicating a prolonged persistence of early CD34+ hematopoietic precursors that had not yet acquired CD45 expression (CD45−) in the WT cultures compared with the HAT−/− cultures (Figure 6C). These data indicate that impaired differentiation is unlikely to account for the lower number of hematopoietic colonies generated by the homozygous HAT−/− cells. The cultured cells were also stained at days 2 and 7 for the apoptotic marker annexin V. In both HAT+/+ and HAT−/− MOZ cultures, a low frequency of apoptotic cells was detected, but this level was not significantly higher in HAT−/− cell cultures (Figure 6D), excluding the possibility of an increased rate of cell death as the cause of the hematopoietic defect observed with the HAT−/− cells. Finally, when the proliferation of the sorted progenitors was examined by scoring the cell numbers at different time points, we observed that cells in the HAT+/+ culture expanded steadily, whereas the number of mutant cells barely increased (Figure 6E). A similar defect in proliferation was observed in cultures initiated from nonsorted day 14.5 HAT−/− fetal liver cells (Figure 6E). The difference was, however, less marked in fetal liver cultures, potentially reflecting the presence of a more mixed population of progenitors, and therefore, allowing some compensation. These findings demonstrate that the HAT activity of MOZ is critical for the expansion and proliferation of early hematopoietic precursors. In absence of this HAT activity, immature precursors are able to differentiate and give rise to all lineages, but with a limited amplification, leading to smaller pools of mature hematopoietic cells. In agreement with this conclusion, we observed a higher frequency of cells in G1 phase in the HAT−/− cultures, indicative of cell cycle exit (Figure 6F).
Figure 6.

Comparative analysis of the proliferation and differentiation potential of HAT+/+ and HAT−/− CD34+/c-kit+ hematopoietic progenitors. (A) Cell morphology. Sorted CD34+/c-Kit+ hematopoietic populations from HAT+/+ and HAT−/− ES cell lines were resuspended in serum-free proliferation media at a concentration of 2 × 105 cells/mL, harvested at indicated time points, and stained by May-Grünwald-Giemsa for morphologic analysis. (B) Relative frequency of undifferentiated/blast cells (□) and macrophages (■) at different stages of maturation in the HAT+/+ and HAT−/− cultures. A total of 200 cells was scored for each sample. (C) Analysis of CD34+/CD45− cell population in HAT+/+ and HAT−/− cell cultures. Days of differentiation are indicated. Numbers represent the percentage of total population. (D) Percentage of apoptotic cells in HAT+/+ and HAT−/− asynchronous cultures. Percentage of annexin V–positive cells is indicated for each time point. (E) Expansion of CD34+/c-Kit+ cells and fetal liver cells. Total number of cells (×104) generated during the culture. CD34+/c-Kit+ cells isolated from day 6 EBs or total fetal liver cells from embryos were seeded at a density of 2 × 106 or 106 cells/mL, respectively. (F) Cell cycle status of HAT+/+ and HAT−/− CD34+/c-Kit+ hematopoietic progenitors in serum-free proliferation media. Asynchronous cultures of CD34+/c-Kit+ hematopoietic progenitors were stained with propidium iodide and analyzed by flow cytometry. Percentages of cells in G1 and S/G2 in HAT+/+ and HAT−/− at day 3 of culture are indicated.
Discussion
Although the critical role of the complete MOZ protein in hematopoietic development was recently established,19,20 the specific biologic function of the HAT activity of this protein has to date remained elusive. To address this question, we have generated ES cells and mouse lines mutated specifically in the HAT catalytic domain of MOZ. Our study reveals that mice carrying a mutated version of the MOZ protein that is normally expressed, but lacks HAT activity, exhibit important defects in the number of HSCs and committed precursors. The control mouse line containing the LoxP site, but no HAT point mutation, was found to be, in contrast, completely normal (data not shown). The HSCs from these HAT−/− MOZ mice also displayed a significant defect in their long-term repopulation potential upon serial transplantation. In addition, we observed a depletion of immature B cells in the HAT−/− mice. A mechanism involving changes in acetylation and chromatin decondensation has been recently shown to regulate the expression of 2 genes involved in B-cell differentiation (Crlz1 and IgJ).37 In a similar way, the defect observed in the MOZ HAT−/− could reflect a role of MOZ HAT activity in regulating the specific expression of other genes also implicated in B-cell development by modifying their chromatin accessibility.
We used the in vitro differentiation of ES cells toward hematopoiesis to determine whether the hematopoietic defect was established at the earliest stage of development. We observed with this model an even more dramatic reduction in number of mature hematopoietic cells generated by HAT−/− MOZ ES cells than in the embryos or young and adult animals. We interpret the greater amplitude of the defect in hematopoietic cells as a consequence of the absence of homeostasis in this in vitro model system. We took further advantage of this experimental system to access large number of precursors of the correct genotype and to investigate the mechanisms leading to generation of fewer mature hematopoietic cells. Our results indicate that HAT−/− MOZ hematopoietic precursors display no substantial defect in differentiation, or increased rate of apoptosis, but a profound deficiency in their proliferative potential. Altogether these findings demonstrate that the HAT activity of MOZ is critical for the expansion and proliferation of early hematopoietic precursors. In its absence, the pools of immature precursors are substantially reduced and these progenitors differentiate without noticeable amplification, leading to the generation of fewer mature hematopoietic cells.
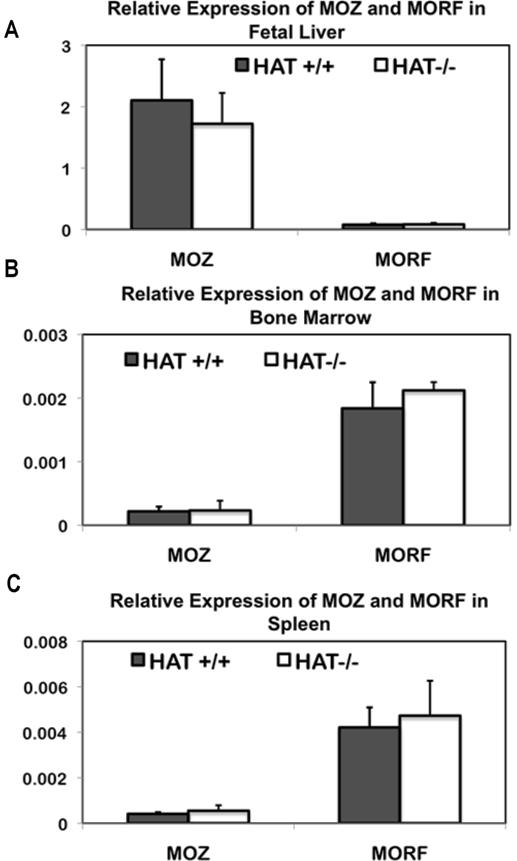
Crosses of HAT+/− heterozygotes produced viable HAT−/− offspring, but more than 40% of these homozygote mice died within the first 6 months. These observations indicate that the consequences of inactivation of the HAT activity of MOZ and total knockout of MOZ are distinct because complete knockouts lead to early embryonic or around birth lethality.19,20 The absence of a full penetrance of the phenotype in the HAT−/− mice could potentially be explained by some compensatory mechanisms by the closely related MYST protein, monocytic leukemia zinc finger protein-related factor (MORF). The MORF (also known as Querkopf or KAT6b3) protein has a high level of sequence similarity with MOZ38 and the same histone acetylation specificity in vitro.38–40 However, we did not detect any increased levels of MORF transcription in HAT−/− mice compared with HAT+/+ mice in either fetal liver or spleen and bone marrow of 3-month mice (Figure S9). An increased expression of MORF is therefore unlikely to compensate for the absence of the HAT activity of MOZ.
Although our results unambiguously establish hematopoietic defects in the HAT−/− MOZ homozygote mice, the extent of the defect is unlikely to directly lead to death. One alternative cause to explain the death of these mice could be strong requirements for the HAT activity of MOZ in other tissues/organs of the mouse. In support of this hypothesis, the contribution of HAT−/− ES cells to different organs of chimeric mice measured by GPI assay was consistently lower compared with WT ES cells in hematopoietic tissues such as thymus, spleen, and blood, as expected, but also more surprisingly in brain and gut (Figure S10). The contribution of the HAT−/− ES cells to heart, kidney, smooth muscle, and total bone marrow was in contrast similar to the ones observed with HAT+/+ ES cells. These findings strongly suggest a potential role of the HAT activity of MOZ in specific tissues other than the hematopoietic system, in which, by analogy, the HAT activity of MOZ might be required for the proliferation of stem cells or precursors in these tissues. Concurring with this hypothesis, the closely related MORF protein has been implicated in the self-renewal and differentiation of neural progenitor cells.41–43 Furthermore, it has been demonstrated that MOZ expression is under the direct control of Nanog, Oct4, and Sox2 in ES cells.44 These observations suggest that MOZ might play important roles in stem cell compartments other than the HSC compartment.
Our study establishes for the first time a critical and specific role of the HAT activity of MOZ in hematopoiesis. Abrogation of the HAT activity of MOZ leads to a drastic decrease in the proliferation potential of most blood lineages from the onset of hematopoietic development. With the exception of B-lymphocyte maturation, MOZ HAT mutation does not appear to affect the differentiation process as all blood lineages are generated, but to impair more specifically the proliferation required for the generation of normal pools of hematopoietic precursors and mature cells. The precise molecular mechanism by which the HAT activity of MOZ regulates the proliferation of HSC and hematopoietic precursors is the subject of current investigations.
Supplementary Material
Acknowledgments
We are grateful to the members of the FACS laboratory for help in cell analysis and cell sorting, to members of the Molecular Biology Core Facility for help in sequencing and genotyping, and to members of the Biological Resource Unit for help in animal maintenance and breeding. We thank K. Labib, A. Sanchez-Diaz, and members of the laboratory for critical reading of the manuscript, and S. Pearson for help with hematopoietic cell morphology analysis.
This work was supported by Cancer Research UK grant C147/A6058.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: F.M.P.-C. and G.L. designed and performed research, analyzed data, and wrote the paper; J.B. designed and performed research; and V.K. designed research and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dr Georges Lacaud, Paterson Institute for Cancer Research, University of Manchester, Wilmslow Rd, M20 4BX, United Kingdom; e-mail: glacaud@picr.man.ac.uk.
References
- 1.Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26:6697–6714. doi: 10.1038/sj.onc.1210755. [DOI] [PubMed] [Google Scholar]
- 2.Kioussis D, Georgopoulos K. Epigenetic flexibility underlying lineage choices in the adaptive immune system. Science. 2007;317:620–622. doi: 10.1126/science.1143777. [DOI] [PubMed] [Google Scholar]
- 3.Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 4.Borrow J, Stanton VP, Jr, Andresen JM, et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 5.Kitabayashi I, Aikawa Y, Yokoyama A, et al. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia. 2001;15:89–94. doi: 10.1038/sj.leu.2401983. [DOI] [PubMed] [Google Scholar]
- 6.Carapeti M, Aguiar RC, Goldman JM, Cross NC. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood. 1998;91:3127–3133. [PubMed] [Google Scholar]
- 7.Carapeti M, Aguiar RC, Watmore AE, Goldman JM, Cross NC. Consistent fusion of MOZ and TIF2 in AML with inv(8)(p11q13). Cancer Genet Cytogenet. 1999;113:70–72. doi: 10.1016/s0165-4608(99)00007-2. [DOI] [PubMed] [Google Scholar]
- 8.Esteyries S, Perot C, Adelaide J, et al. NCOA3, a new fusion partner for MOZ/MYST3 in M5 acute myeloid leukemia. Leukemia. 2008;22:663–665. doi: 10.1038/sj.leu.2404930. [DOI] [PubMed] [Google Scholar]
- 9.Kitabayashi I, Aikawa Y, Nguyen LA, Yokoyama A, Ohki M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 2001;20:7184–7196. doi: 10.1093/emboj/20.24.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelletier N, Champagne N, Stifani S, Yang XJ. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene. 2002;21:2729–2740. doi: 10.1038/sj.onc.1205367. [DOI] [PubMed] [Google Scholar]
- 11.Champagne N, Pelletier N, Yang XJ. The monocytic leukemia zinc finger protein MOZ is a histone acetyltransferase. Oncogene. 2001;20:404–409. doi: 10.1038/sj.onc.1204114. [DOI] [PubMed] [Google Scholar]
- 12.Deguchi K, Ayton PM, Carapeti M, et al. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cells. 2003;3:259–271. doi: 10.1016/s1535-6108(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 15.Lacaud G, Gore L, Kennedy M, et al. Runx1 is essential for hematopoietic commitment at the hemangioblast stage of development in vitro. Blood. 2002;100:458–466. doi: 10.1182/blood-2001-12-0321. [DOI] [PubMed] [Google Scholar]
- 16.Miyoshi H, Kozu T, Shimizu K, et al. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993;12:2715–2721. doi: 10.1002/j.1460-2075.1993.tb05933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 18.Bristow CA, Shore P. Transcriptional regulation of the human MIP-1α promoter by RUNX1 and MOZ. Nucleic Acids Res. 2003;31:2735–2744. doi: 10.1093/nar/gkg401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas T, Corcoran LM, Gugasyan R, et al. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006;20:1175–1186. doi: 10.1101/gad.1382606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsumoto T, Aikawa Y, Iwama A, et al. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006;20:1321–1330. doi: 10.1101/gad.1393106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez-Campo FM, Spencer HL, Elder RH, Stern PL, Ward CM. Novel vectors for homologous recombination strategies in mouse embryonic stem cells: an ES cell line expressing EGFP under control of the 5T4 promoter. Exp Cell Res. 2007;313:3604–3615. doi: 10.1016/j.yexcr.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Fehling HJ, Lacaud G, Kubo A, et al. Tracking mesoderm induction and its specification to the hemangioblast during embryonic stem cell differentiation. Development. 2003;130:4217–4227. doi: 10.1242/dev.00589. [DOI] [PubMed] [Google Scholar]
- 23.Mikula M, Schreiber M, Husak Z, et al. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 2001;20:1952–1962. doi: 10.1093/emboj/20.8.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacaud G, Kouskoff V, Trumble A, Schwantz S, Keller G. Haploinsufficiency of Runx1 results in the acceleration of mesodermal development and hemangioblast specification upon in vitro differentiation of ES cells. Blood. 2004;103:886–889. doi: 10.1182/blood-2003-06-2149. [DOI] [PubMed] [Google Scholar]
- 25.Langer JC, Henckaerts E, Orenstein J, Snoeck HW. Quantitative trait analysis reveals transforming growth factor-β2 as a positive regulator of early hematopoietic progenitor and stem cell function. J Exp Med. 2004;199:5–14. doi: 10.1084/jem.20030980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaren A, Buehr M. GPI expression in female germ cells of the mouse. Genet Res. 1981;37:303–309. doi: 10.1017/s0016672300020309. [DOI] [PubMed] [Google Scholar]
- 27.Takechi S, Nakayama T. Sas3 is a histone acetyltransferase and requires a zinc finger motif. Biochem Biophys Res Commun. 1999;266:405–410. doi: 10.1006/bbrc.1999.1836. [DOI] [PubMed] [Google Scholar]
- 28.Akhtar A, Becker PB. The histone H4 acetyltransferase MOF uses a C2HC zinc finger for substrate recognition. EMBO Rep. 2001;2:113–118. doi: 10.1093/embo-reports/kve022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keller GM. In vitro differentiation of embryonic stem cells. Curr Opin Cell Biol. 1995;7:862–869. doi: 10.1016/0955-0674(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 30.Lacaud G, Keller G, Kouskoff V. Tracking mesoderm formation and specification to the hemangioblast in vitro. Trends Cardiovasc Med. 2004;14:314–317. doi: 10.1016/j.tcm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Lacaud G, Robertson S, Palis J, Kennedy M, Keller G. Regulation of hemangioblast development. Ann N Y Acad Sci. 2001;938:96–107. doi: 10.1111/j.1749-6632.2001.tb03578.x. discussion 108. [DOI] [PubMed] [Google Scholar]
- 32.Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- 33.Huber TL, Kouskoff V, Fehling HJ, Palis J, Keller G. Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature. 2004;432:625–630. doi: 10.1038/nature03122. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita J, Itoh H, Hirashima M, et al. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408:92–96. doi: 10.1038/35040568. [DOI] [PubMed] [Google Scholar]
- 35.Pearson S, Sroczynska P, Lacaud G, Kouskoff V. The stepwise specification of embryonic stem cells to hematopoietic fate is driven by sequential exposure to Bmp4, activin A, bFGF and VEGF. Development. 2008;135:1525–1535. doi: 10.1242/dev.011767. [DOI] [PubMed] [Google Scholar]
- 36.Williamson AJ, Smith DL, Blinco D, et al. Quantitative proteomics analysis demonstrates post-transcriptional regulation of embryonic stem cell differentiation to hematopoiesis. Mol Cell Proteomics. 2008;7:459–472. doi: 10.1074/mcp.M700370-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Lim JH, Cho SJ, Park SK, et al. Stage-specific expression of two neighboring Crlz1 and IgJ genes during B cell development is regulated by their chromatin accessibility and histone acetylation. J Immunol. 2006;177:5420–5429. doi: 10.4049/jimmunol.177.8.5420. [DOI] [PubMed] [Google Scholar]
- 38.Champagne N, Bertos NR, Pelletier N, et al. Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J Biol Chem. 1999;274:28528–28536. doi: 10.1074/jbc.274.40.28528. [DOI] [PubMed] [Google Scholar]
- 39.Pelletier N, Champagne N, Lim H, Yang XJ. Expression, purification, and analysis of MOZ and MORF histone acetyltransferases. Methods. 2003;31:24–32. doi: 10.1016/s1046-2023(03)00084-7. [DOI] [PubMed] [Google Scholar]
- 40.Doyon Y, Cayrou C, Ullah M, et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Thomas T, Voss AK, Chowdhury K, Gruss P. Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development. 2000;127:2537–2548. doi: 10.1242/dev.127.12.2537. [DOI] [PubMed] [Google Scholar]
- 42.Thomas T, Voss AK. Querkopf, a histone acetyltransferase, is essential for embryonic neurogenesis. Front Biosci. 2004;9:24–31. doi: 10.2741/1208. [DOI] [PubMed] [Google Scholar]
- 43.Merson TD, Dixon MP, Collin C, et al. The transcriptional coactivator Querkopf controls adult neurogenesis. J Neurosci. 2006;26:11359–11370. doi: 10.1523/JNEUROSCI.2247-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}