

Abstract
Neutropenia and related infections are the most important dose-limiting toxicities in anticancer chemotherapy and radiotherapy. In this study, we explored a new strategy for augmenting host defense in neutropenia-related pneumonia. Phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) signaling in neutrophils was elevated by depleting PTEN, a phosphatidylinositol 3′-phosphatase that hydrolyzes PtdIns(3,4,5)P3. In myeloid-specific PTEN knockout mice, significantly more neutrophils were recruited to the inflamed lungs during neutropenia-associated pneumonia. Using an adoptive transfer technique, we demonstrated that this enhancement could be caused directly by PTEN depletion in neutrophils. In addition, disruption of PTEN increased the recruitment of macrophages and elevated proinflammatory cytokines/chemokine levels in the inflamed lungs, which could also be responsible for the enhanced neutrophil recruitment. Depleting PTEN also significantly delayed apoptosis and enhanced the bacteria-killing capability of the recruited neutrophils. Finally, we provide direct evidence that enhancement of neutrophil function by elevating PtdIns(3,4,5)P3 signaling can alleviate pneumonia-associated lung damage and decrease pneumonia-elicited mortality. Collectively, these results not only provide insight into the mechanism of action of PTEN and PtdIns(3,4,5)P3 signaling pathway in modulating neutrophil function during lung infection and inflammation, but they also establish PTEN and related pathways as potential therapeutic targets for treating neutropenia-associated pneumonia.
Introduction
Pneumonia is usually triggered when a person's defense system is weakened.1–4 It represents a major cause of infectious complication in cancer patients with treatment-related neutropenia.5–7 Neutropenia-related lung infections have been treated with broad-spectrum antibiotic therapy and granulocyte colony-stimulating factor (G-CSF) therapy. Here, we explored an alternative strategy for augmenting host defense in neutropenia-related pneumonia by enhancing neutrophil functions (ie, recruitment, survival, and bacteria killing) in neutropenic patients. We achieved this by augmenting the intracellular phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) signaling pathway, which has been implicated in a variety of neutrophil functions, such as survival, polarization, chemotaxis, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation.8–11 We recently demonstrated that augmenting PtdIns(3,4,5)P3 signal by depleting PTEN, a phosphatidylinositol 3′-phosphatase that negatively regulates PtdIns(3,4,5)P3 signaling, prevents neutrophil death.12 In addition, PTEN-null neutrophils had enhanced sensitivity to chemoattractant stimulation. A larger fraction of these neutrophils displayed membrane ruffles in response to chemoattractant stimulation. Chemoattractant-induced transwell migration and superoxide production were also augmented.13 In the present study, we further investigated the consequences of PTEN disruption in neutropenia-related pneumonias. Our data revealed that PTEN-null neutrophils possess an enhanced bacteria-killing capability in bacterial pneumonia, and their recruitment to the inflamed lungs was also augmented. Moreover, we provided direct evidence that enhancement of neutrophil function by elevating PtdIns(3,4,5)P3 signaling can alleviate pneumonia-associated lung damage and decrease the related mortality rate. Collectively, these results not only provide insight into the mechanism of action of the PtdIns(3,4,5)P3 pathway in elevating neutrophil function in lung infection and inflammation, but they also establish PTEN and its related pathways as potential therapeutic targets for treating neutropenia-related pneumonia.
Methods
Mice
The conditional PTEN knockout (KO) mice (PTENloxP/loxP) and the myeloid-specific Cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The experimental myeloid-specific PTEN KO mice were generated as previously described.12,13 Six- to 12-week-old mice were used in all experiments. Mouse bone marrow neutrophils were prepared as described by Zhu et al.12 All procedures involving mice were approved and monitored by the Children's Hospital Animal Care and Use Committee at Harvard Medical School.
Escherichia coli or LPS-induced acute pneumonia
After anesthesia with ketamine hydrochloride (100 mg/kg intraperitoneally) and xylazine (10 mg/kg intraperitoneally), mouse trachea was surgically exposed, and a total volume of 50 μL saline or a dose of 106 colony-forming units (CFU) of E coli (strain 19138; ATCC, Manassas, VA) or 5 mg/kg lipopolysaccharide (LPS; E coli o55:B5, Sigma-Aldrich, St Louis, MO) per mouse was instilled intratracheally to the left bronchus. Colloidal carbon (1%) was included in the instillate to indicate deposition. At the end of the experiments, mice were killed by CO2.
Cyclophosphamide and irradiation-induced mouse neutropenia model
Cyclophosphamide powder (Cytoxan; Bristol-Myers Squibb, Princeton, NJ) was dissolved in distilled water for injection at a final concentration of 20 mg/mL. Cyclophosphamide was injected intraperitoneally at a total dose of 250 mg/kg (two 0.5-mL injections on day 1 [150 mg/kg] and day 4 [100 mg/kg]). Blood samples (∼ 30 μL) were taken from the retroorbital sinuses of anesthetized uninfected mice using heparinized capillary tubes (Modulohm, Herlev, Denmark) on days 1, 4, 5, 6, and 7. Total and differential white blood cell counts (neutrophils, lymphocytes, and monocytes) were performed using a Hemavet 850 hematology system (Drew Scientific, Ramsey, MN), which is a multiparameter, automated hematology analyzer designed for in vitro diagnostic use. For irradiation-induced neutropenia, mice were exposed to a single dose of 600 cGy in a Gammacell 40 137Cs Irradiator (MDS Nordion, Ottawa, ON). Total and differential white blood cell counts were monitored on days 3, 4, 5, and 6 as described above (data not shown).
Adoptive transfer of neutrophils
Bone marrow neutrophils were isolated from wild-type (WT) and myeloid-specific PTEN-deficient mice as previously described14 and were labeled with 5- (and 6-)carboxyfluorescein diacetate succinimidyl ester (CFSE; final concentration 5 μM) or seminaphthorhodafluor-1 (SNARF-1) acetate (final concentration 5 μM) at 37°C for 10 minutes. Labeled cells were mixed (1:1) as indicated and then injected intravenously (via tail vein) into WT neutropenic mice that had been challenged with 105 CFU of E coli for 2.5 hours. Bronchoalveolar lavage fluid (BALF) was harvested 2.5 hours after the injection of the cell mixture. The amount of adoptively transferred neutrophils recruited to the bronchoalveolar air space was analyzed using a FACSCanto II flow cytometer and FACSDiva software (BD Biosciences, San Jose, CA). Relative recruitment of WT and PTEN-null neutrophils was calculated as the ratio of indicated populations in the BALF (Figure 5).
Figure 5.

Disruption of PTEN directly increases the efficiency of neutrophil recruitment to the inflamed lungs. (A) Recruitment of adoptively transferred neutrophils to the lungs in the E coli–induced neutropenia-related pneumonia model. Neutrophils isolated from WT and myeloid-specific PTEN-deficient mice were labeled with CFSE or SNARF-1. Labeled cells were mixed (1:1) as indicated and then injected intravenously (via tail vein) into WT neutropenic mice that had been challenged with 105 CFU of E coli for 2.5 hours. BALF was harvested 2.5 hours after the injection of the cell mixture. (B) The amount of adoptively transferred neutrophils recruited to the lungs was analyzed using a FACSCanto II flow cytometer and FACSDiva software. (C) Relative recruitment of neutrophils was calculated as the ratio of indicated populations in the BALF. Data shown are mean (± SD) of 3 mice. **P < .01 versus WT neutrophils.
Assays for neutrophil/macrophage functions
The related assays, including neutrophil/macrophage recruitment, morphometric analyses of histologic lung sections, BALF collection, BALF cytokine and chemokine levels and total protein levels, in vitro cytokine and chemokine release by alveolar macrophages, in situ detection of apoptosis, neutrophil apoptosis in the inflamed peritoneal cavity, bacterial burden, in vitro bactericidal assay, pulmonary mechanics measurement, gentamicin protection assay, phagocytosis assay, superoxide production during phagocytosis, and myeloperoxidase activity assay, are described in detail in Document S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Statistical analysis
The Kaplan-Meier and log-rank methods were used to analyze survival rates. Other values were compared using the Student t test. Differences were considered significant when P was less than .05.
Results
Accumulation of PTEN-null neutrophils in inflamed lungs is enhanced in bacterial pneumonia
We recently reported that the responsiveness of neutrophils to chemoattractant stimulation is enhanced in PTEN KO mice in which PtdIns(3,4,5)P3 signaling is hyperactivated. Using a mouse peritonitis model, we showed that the recruitment of neutrophils to inflamed peritoneal cavity was highly elevated in these mice.13 To investigate whether elevation of PtdIns(3,4,5)P3 signaling can also lead to increased neutrophil recruitment to inflamed lungs, we used a mouse bacterial pneumonia model. In this model, lung inflammation was induced by intratracheal instillation of the Gram-negative bacterium E coli, which is one of the most common pathogens in neutropenia-related pneumonia.5–7 Because of the early embryonic lethality of conventional Pten−/− mice,15 we used a conditional PTEN KO mouse, in which 2 loxP sequences were inserted on either side of exon 5 of the PTEN gene encoding the phosphatase domain. We crossed this mouse with a myeloid-specific Cre line, in which the Cre recombinase gene was under the control of a lysozyme M promoter. Thus, disruption of PTEN occurred only in mice with the myeloid linage, including monocytes, mature macrophages, and neutrophils.12 Mice that are homozygous for this allele are viable, fertile, normal in size, and do not display any gross physical or behavioral abnormalities.13
Neutrophil accumulation in inflamed lungs was assessed 8 and 24 hours after bacteria instillation using 2 independent methods: bronchoalveolar lavage (BAL; Figure 1A) and morphometric analyses of histologic lung sections (Figures 1B,C and S1). As expected, very few neutrophils were detected in the lungs of unchallenged mice. Similar to previously published data from other laboratories using the same model,16–19 the number of neutrophils in the WT BALF reached nearly 106 cells/lung 8 hours after bacteria instillation and nearly 2 × 106 cells/lung 24 hours after bacteria instillation. Myeloid-specific PTEN−/− mice showed a dramatic increase in bacteria-induced neutrophil recruitment. Nearly 2 × 106 neutrophils were recruited to the lungs 8 hours after induction, and 3.5 × 106 were recruited 24 hours after induction (Figure 1A). We detected a similar effect when we quantified the number of emigrated neutrophils in alveolar air spaces by morphometric analyses of histologic lung sections (Figures 1B,C and S1). Because PTEN disruption did not affect peripheral blood neutrophil count before or after bacteria instillation (Figure S2), the enhanced neutrophil accumulation is most likely due to elevated neutrophil recruitment and/or delayed neutrophil death in the inflamed lungs. In these experiments, specific increases in PtdIns(3,4,5)P3 signaling in recruited neutrophils was confirmed by measuring phosphorylation of Akt, a well-known downstream factor in the PtdIns(3,4,5)P3 signaling pathway (Figure 1D).
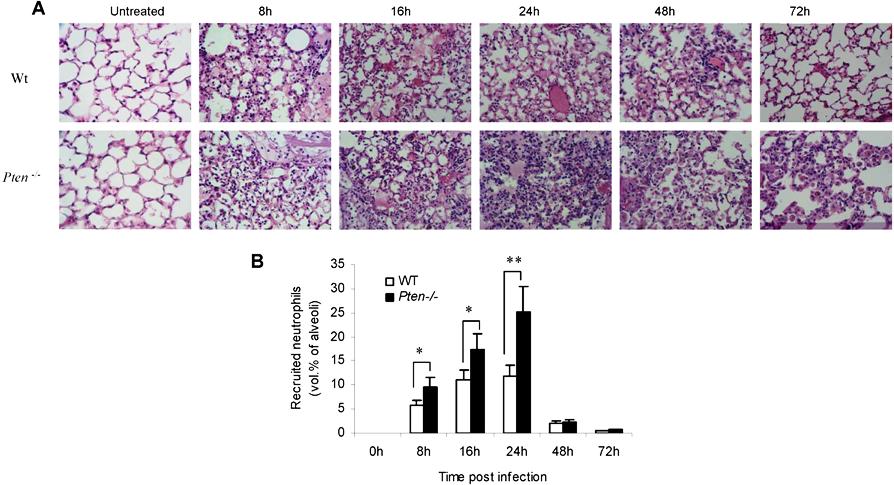
Figure 1.

Accumulation of PTEN-null neutrophils in inflamed lungs was enhanced during E coli pneumonia. Mice were intratracheally instilled with 106 CFU of E coli and killed at each indicated time point. (A) Neutrophils in BALF. The total number of cells in the lungs was counted by hemocytometer. Differential cell counts were conducted on cytospin preparations stained with a modified Wright-Giemsa stain (Volu-Sol, Salt Lake City, UT). Neutrophils were recognized by their lobular or segmented nuclei. The percentage of pulmonary neutrophils (PMNs) in the whole population was determined accordingly. Total number of PMNs recruited was calculated as follows: number of PMNs = cell density × volume × % PMN. All data are presented as mean (± SD); n ≥ 4 mice in each group. *P < .05, **P < .01 versus wild type. (B) Staining of histologic lung sections shows emigrated neutrophils and polymerized fibrin in the pulmonary parenchyma. Lungs were fixed with Bouin solution at 23 cm H2O pressure. Tissues were embedded in paraffin, and 6-μm–thick sections were stained with hematoxylin and eosin (H&E). (C) Emigrated neutrophils in alveolar air spaces were quantified as volume fraction of the alveolar air space using standard point-counting morphometric techniques.63 The relative volumes of the parenchymal regions occupied by emigrated neutrophils were calculated by investigators blinded to the identities of the mice and were expressed as the percentage of the total parenchymal region volume (including both tissue and air space). (D) PtdIns(3,4,5)P3 signaling is specifically elevated in PTEN-null neutrophils. Neutrophils, primary spleen cells, and brain cells collected from WT and myeloid-specific PTEN KO mice were stimulated with 25 ng/mL MIP-2 and lysed at 0 and 10 minutes. Phosphorylated and total Akt were detected by Western blotting using anti–phospho-Akt (Ser473; 1:1000) and anti-Akt (1:1000) antibodies (Cell Signaling Technology, Beverly, MA), respectively. Densitometry of the blots was performed using the ImageJ software, Gel Analyzer plug-in. Phospho-Akt levels were normalized based on loading (total Akt level). (E) Pulmonary edema formation was quantified as the percentage of edema area in the total parenchymal region using IPlab imaging software. (F) BALF total protein. Protein accumulated in the inflamed lung was measured using a Bio-Rad (Hercules, CA) protein assay kit. The standard curve was constructed using bovine serum albumin (BSA). (G) Rate of mortality due to E coli–induced pneumonia in WT and myeloid-specific PTEN KO mice. Age- and sex-matched WT and PTEN KO mice were intratracheally challenged with 106 live E coli and monitored for 7 days. The Kaplan-Meier and log-rank methods were used to analyze survival rates. *P < .01 versus WT.
Dramatically increased lung neutrophil number often leads to aggravated lung damage. We consistently detected augmented pulmonary edema formation (Figure 1E) and increased protein accumulation (Figure 1F) in the lungs of the PTEN−/− mice. In addition, a much increased pneumonia-associated death rate was observed in the PTEN−/− mice. More than 80% of WT experimental mice survived the challenge. In contrast, only 50% of PTEN KO mice survived (Figure 1G). Collectively, these results demonstrated that elevating PtdIns(3,4,5)P3 signaling by PTEN disruption can result in enhanced neutrophil accumulation and more severe lung inflammation in bacterial pneumonia in nonneutropenic mice. We also examined neutrophil accumulation and associated lung inflammation in pneumonia induced by sterile ligands LPS. Essentially the same results were observed: PTEN disruption led to elevated neutrophil recruitment and aggravated lung inflammation (Figure S3).
Augmenting the PtdIns(3,4,5)P3 signal by disrupting PTEN delays neutrophil death in bacterial pneumonia
One mechanism that can lead to enhanced pulmonary neutrophil accumulation is increasing the lifespan of recruited neutrophils. Neutrophils are terminally differentiated and usually have a short lifespan (1-4 days in tissue). They die via programmed cell death (apoptosis). We have shown that augmenting the PtdIns(3,4,5)P3 signal by depleting PTEN dramatically delays the spontaneous death of cultured neutrophils.13 In this study, we explored whether neutrophil death in inflamed lungs is also delayed in PTEN KO mice. We first measured the apoptosis of neutrophils collected from BALF. Few apoptotic neutrophils were detected in WT or PTEN-null mice (Figure S4). In the inflamed lung, the apoptotic neutrophils might have already been cleared by macrophages before they could emigrate into the alveolar space. Thus, we checked the apoptosis of neutrophils in the lung tissues. The viability of neutrophils in histologic lung sections was determined by a terminal deoxynucleotidyltransferase–mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay, which detects fragmented DNA in apoptotic cells20,21 (Figure 2A,B). In WT mice, approximately 10% of recruited pulmonary neutrophils were TUNEL-positive 24 hours after the bacteria instillation. This number dropped to 7% in PTEN KO mice, suggesting reduced neutrophil death in these mice.
Figure 2.

Depletion of PTEN delays neutrophil death in bacterial pneumonia. (A) Apoptotic neutrophils in the inflamed lungs. Mice were infected by intratracheal instillation of 106 CFU of E coli and killed 24 hours later. The viability of accumulated neutrophils was determined by TUNEL assay. Green arrows indicate apoptotic neutrophils. (B) Quantification of apoptotic neutrophils. Data shown are mean (± SD; n = 4). *P < .05 versus WT. (C-E) Examination of in vivo death of adoptively transferred neutrophils. Neutropenia in receipt mice was induced by a widely used chemotherapeutic drug, Cy (Figure S5). The mature WT or PTEN-null neutrophils were purified from the inflamed peritoneal cavity and labeled with intracellular fluorescent dye CFSE or 5-(and -6)-chloromethyl SNARF-1 acetate (SNARF-1). Labeled cells were mixed (1:1) and intraperitoneally injected to WT mice challenged with 3% thioglycollate (TG). Peritoneal lavage was collected 15 hours after cell injection, and the ratios of adoptively transplanted WT and PTEN-null neutrophils were analyzed using a FACSCanto II flow cytometer and FACSDiva software (D). Relative death rates were quantified and expressed as the ratio of adoptively transplanted PTEN-null neutrophils to WT neutrophils in the inflamed peritoneal cavity (E). Data shown are mean (± SD; n = 3 mice in each group). *P < .05 versus WT.
To further investigate whether the reduction of in vivo neutrophil death was due to alterations of the intrinsic apoptotic/survival pathway in the PTEN-null neutrophils or due to an altered lung inflammatory environment in the myeloid-specific PTEN KO mice, we conducted an adoptive transfer experiment using a mouse peritonitis model (Figure 2C). We labeled purified PTEN-null neutrophils with intracellular fluorescent CFSE (green) and WT neutrophils with another dye (5-(and 6-)chloromethyl SNARF-1 acetate [red]), or vice versa. The mixed (1:1) population was injected into the peritoneal cavity of the same WT mice. The relative rate of apoptosis was calculated by measuring the ratio between PTEN-null and WT neutrophils at the inflammation site. By doing this, we skipped the step of neutrophil recruitment. Thus, the number of neutrophils in the peritoneal cavity will directly reflect the death rate of each population. In addition, because the WT and PTEN-null neutrophils were in exactly the same environment, variability caused by the difference of each individual recipient mouse was eliminated. PTEN-null and WT neutrophils were identified by their unique fluorescent labels using FACS analysis. Supporting our hypothesis that PTEN-null neutrophils have a prolonged lifespan, we detected greatly delayed clearance of transplanted PTEN-null neutrophils compared with WT neutrophils (Figure 2D,E).
Augmenting PtdIns(3,4,5)P3 signal by depleting PTEN enhances the bacteria-killing capability of neutrophils
In the mouse bacterial pneumonia model, we detected elevated neutrophil recruitment to inflamed lungs in PTEN-null mice (Figure 1). We subsequently explored the survival rate of intratracheally instilled live E coli cells (Figure 3A). Due to cell proliferation, the number of bacteria gradually increased after initial instillation. When a significant number of neutrophils accumulated in the lungs (8-24 hours after the instillation), the number of bacteria stopped increasing, reflecting the bacteria-killing capability of neutrophils. We detected fewer bacteria in inflamed myeloid-specific PTEN-null mice, suggesting that these mice have enhanced bacteria-killing capability (Figure 3A). This finding could be the result of elevated neutrophil recruitment detected in PTEN-null mice. To test whether the intrinsic bacteria-killing capability of neutrophils is also enhanced by PTEN disruption, we conducted an in vitro assay using the same number of WT and PTEN-null neutrophils (Figure 3B,C). The bacteria-killing capability of PTEN-null neutrophils was increased by 40% at 30 minutes and by 60% at 2 hours compared with WT neutrophils. Augmented phagocytosis could be responsible for enhanced bacteria killing. To test this, we quantified the number of bioparticles engulfed by each neutrophil using an in vitro phagocytosis assay (Figure 3D,E). After 1 hour of incubation at 37°C, an average of 60 mouse serum–opsonized fluorescein-conjugated zymosan particles were engulfed by 100 WT neutrophils (phagocytic index). PTEN−/− neutrophils had a dramatically increased phagocytic index: nearly 100 bacteria were engulfed by 100 neutrophils (Figure 3D). A similar effect was detected when the in vitro phagocytosis assay was conducted using purified mouse neutrophils and serum-opsonized fluorescein-conjugated bacteria bioparticles (Figure 3E). These results are consistent with a previous report indicating an increased phagocytosis in PTEN-null macrophages.22 The augmented phagocytosis was likely a result of enhanced engulfment, because there was essentially no difference in the initial bacteria/zymosan-binding capability between WT and PTEN-null neutrophils (Figure 3D,E). We also monitored phagosome-lysosome fusion using a LysoTracker fluorescent dye. No obvious alteration was detected in the PTEN-null neutrophils, suggesting that elevation of the PtdIns(3,4,5)P3 signal does not affect phagosome maturation (Figure 3F).
Figure 3.

Disrupting PTEN enhances the bacteria-killing capability of neutrophils. (A) Bacterial killing in inflamed lungs. Mice were intratracheally instilled with 106 CFU of E coli and killed at the indicated times. Whole lungs were homogenized and serially diluted with sterile cold water. Aliquots were spread on Luria-Bertani agar plates and incubated overnight at 37°C. Live bacteria were quantified as CFU/lung. (B) In vitro bacterial killing assay. Purified bone marrow WT or PTEN-null neutrophils were incubated with E coli for 2 hours. Diluted aliquots were spread on agar plates and incubated overnight at 37°C. (C) In vitro bacterial killing capabilities were reflected by the decrease of CFU after indicated incubation periods. (D,E) In vitro phagocytosis assay. FITC-labeled zymosans (D) or E coli bioparticles (E) were opsonized with mouse serum and incubated with neutrophils at 4°C (control) or 37°C for 1 hour. Extracellular fluorescence was quenched by trypan blue. Phagocytosis index (PI) was expressed as the number of bioparticles engulfed by 100 neutrophils. Binding index was expressed as the number of bioparticles bound to 100 neutrophils. More than 200 neutrophils were counted in each group. (F) Phagosome maturation. WT or PTEN-null neutrophils were incubated with mouse serum-opsonized latex beads for 60 minutes. Phagosomes were stained with LysoTracker and visualized under fluorescent microscopy. (G) Intracellular reactive oxygen species (ROS) production during phagocytosis. Bone marrow–derived neutrophils from WT and PTEN KO mice were incubated with (or without) mouse serum–opsonized E coli or zymosan bioparticles in the presence of superoxide dismutase (SOD; 50 U/mL) and catalase (2000 U/mL). ROS production was monitored in a luminometer at 37°C. Chemiluminiscence (arbitrary light units) was recorded every 150 seconds for 1 hour. (H) Intracellular killing assay. Bone marrow–derived neutrophils from WT and PTEN KO mice were incubated with mouse serum-opsonized live E coli for 1 hour and then with 100 μg/mL gentamicin for an additional hour. Viable intracellular bacteria were quantified by subsequent plating the lysed samples on Luria-Bertani agar. The results were normalized by phagocytosis indexes. All data are presented as mean (± SD; n = 3-4 mice in each group). *P < .05, **P < .01, and ***P < .001 versus WT.
Elevated bacteria killing capability could also be a result of enhanced superoxide production in phagosomes. Accordingly, we measured phagocytosis-associated superoxide production in both PTEN KO and WT neutrophils. A significant enhancement was observed in the PTEN-null neutrophils for both E coli and zymosan-induced superoxide production (Figure 3G). Recently, Anderson et al reported that superoxide production elicited by serum-opsonized bioparticles is mainly mediated by complement and CD18. Interestingly, this process requires class III phosphatidylinositol 3-kinase (PI3K) and its product PtdIns(3)P, and it is independent of class I PI3K and its product PtdIns(3,4,5)P3.23 PTEN acts as a lipid phosphatase, removing the phosphate in the D3 position of the inositol ring from PtdIns(3,4,5)P3, PtdIns(3,4)P2, and PtdIns(3)P. Thus, its effect on phagocytosis-associated superoxide production is most likely mediated by its lipid phosphatase activity on PtdIns(3)P. Finally, we measured neutrophil intracellular bactericidal activity using a gentamicin protection assay. Consistent with the elevated superoxide production, the capability of the PTEN-null neutrophils to kill engulfed bacteria was much increased compared with the WT neutrophils (Figure 3H). Taken together, these findings further demonstrate the enhanced bacteria-killing capability of neutrophils in which the PtdIns(3,4,5)P3 signal is augmented.
Disruption of PTEN enhances pulmonary neutrophil accumulation and reduces bacterial burden in neutropenia-associated pneumonia
Excessive neutrophil accumulation or hyperresponsiveness of neutrophils can damage surrounding tissues and cause unwanted and exaggerated tissue inflammation. However, neutrophils are the major cell type in innate immunity, and they protect their host by engulfing, killing, and digesting invading bacterial and fungal pathogens. Thus, elevated neutrophil function might be beneficial in certain pathologic conditions. Accordingly, we investigated whether enhancing neutrophil function by PTEN disruption can be used as a therapeutic strategy to augment host defense in neutropenia-related pneumonia. We first examined neutrophil recruitment and its bacteria-killing capability under neutropenic conditions. We induced neutropenia using a widely used chemotherapeutic drug, cyclophosphamide (Cy; Figure S5), which is used primarily to treat cancer. The mechanism of action is thought to involve cross-linking and strand breakage of tumor cell DNA.24,25 The induction of neutropenia by Cy has been well documented.26–28 Two intraperitoneal injections for a total dose of 250 mg/kg (150 mg/kg at day 1 and 100 mg/kg at day 4) were sufficient to induce severe neutropenia. On day 5, Cy-treated mice had approximately 90% fewer circulating neutrophils than di the untreated group. The profound neutropenia persisted through days 6 and 7 (Figure S5 and Figure 4A). Due to the severe neutropenia, some WT and PTEN KO mice cannot survive bacterial challenge at a dose of 106 CFU/mouse (data not shown). Thus, we used 105 CFU/mouse in all experiments involving neutropenic mice. After bacterial instillation, the number of neutrophils accumulated in the lungs was significantly lower in both WT and myeloid-specific PTEN-null neutropenic mice compared with untreated mice. However, PTEN-null mice still had approximately 3-fold more bacteria-induced pulmonary neutrophil accumulation compared with WT mice (Figure 4B,C). Consistently, the activity of myeloperoxidase (MPO), a peroxidase enzyme most abundantly present in phagocytes, was substantially elevated in the lungs of PTEN-null mice (Figure 4D). Moreover, the enhanced neutrophil accumulation in the PTEN-null mice led to better clearance of instilled bacteria, as revealed by the reduced bacteria number in the inflamed lungs of these mice (Figure 4E-G). As a result, the resolution of bacteria-induced lung inflammation was accelerated. The inflammation-associated lung damage, which was evaluated by pulmonary edema formation, was alleviated in PTEN-null mice (Figure 4H). We also examined neutrophil recruitment, bacteria-killing capability, and lung inflammation in irradiation-induced neutropenic mice, and essentially the same results were obtained, demonstrating that the protective effects detected in the PTEN-null mice were not model-dependent (Figure S6).
Figure 4.

Disruption of PTEN enhances pulmonary neutrophil accumulation and reduces bacterial burden in neutropenia-related pneumonia. (A) PTEN depletion does not affect the amount of circulating neutrophils. The complete blood count (CBC) and white blood cell count of each sample were analyzed using a Hemavet 850 hematology system. (B) Histologic analysis of lungs reveals bacterial colonies and polymerized fibrin in the pulmonary parenchyma. Lungs were fixed with Bouin solution, and 6-μm sections were stained with H&E. (C) Total numbers of neutrophils in BALF were quantified as described in Figure 1A. (D) MPO activity, an indicator of tissue neutrophil accumulation, was determined using an EnzChek MPO activity assay kit (Invitrogen, Carlsbad, CA). (E) Bacterial colonies in histologic lung sections were quantified and expressed as colony numbers in each ×400 field. (F,G) Bacterial killing in inflamed lungs. Live bacteria in lung homogenates were assessed with the colony assay as described in Figure 3B,C. (H) Pulmonary edema formation was quantified as the percentage of edema in the total parenchymal region using IPlab imaging software. All data are presented as mean (± SD; n ≥ 4 mice in each group). **P < .01, ***P < .001 versus WT.
Disruption of PTEN directly increases the efficiency of neutrophil recruitment to the inflamed lungs
We have shown that the accumulation of neutrophils in both inflamed lungs (Figures 1, 4) and inflamed peritoneal cavity13 was enhanced in myeloid-specific PTEN-null mice. We further demonstrated that prolonged neutrophil survival is at least partially responsible for this elevated accumulation (Figure 2). However, whether the efficiency of neutrophil recruitment to the sites of inflammation is also increased in the myeloid-specific PTEN-null mice has not been directly examined in vivo. Thus, we next investigated neutrophil recruitment in neutropenic mice using an adoptive transfer assay (Figure 5). As described in Figure 2C, we labeled in vitro–purified PTEN-null neutrophils with CFSE and WT neutrophils with SNARF-1 acetate, or vice versa. The mixed (1:1) population was intravenously injected into a WT neutropenic recipient mouse 2.5 hours after the intratracheal instillation of E coli (Figure 5A). By doing this, we were able to compare the recruitment of these 2 types of neutrophils in exactly the same environment. Independent of the dye used to stain the neutrophils, we consistently detected enhanced (> 2-fold) pulmonary recruitment of PTEN-null neutrophils compared with WT neutrophils (Figures 5B,C and S7). This result suggests that the observed elevation of neutrophil accumulation in PTEN-null mice is a combination of enhanced neutrophil recruitment and delayed neutrophil death.
Disruption of PTEN increases the recruitment of macrophages in the inflamed lungs
Besides neutrophils, alveolar macrophages are also considered major effector cells in host defense against respiratory tract infections by virtue of their potent phagocytic properties.29–33 They are also important in initiating the inflammatory responses. In response to danger, alveolar macrophages produce various proinflammatory mediators to orchestrate the inflammatory response, leading to the recruitment of other types of cells, including neutrophils, to the lungs.34,35 In the myeloid-specific PTEN KO mice used in this study, PTEN expression is also ablated in macrophages.13 Thus, it is possible that elevation of PtdIns(3,4,5)P3 signaling can also alter the recruitment and function of macrophages in neutropenia-related pneumonia. To test this, we first measured the number of alveolar macrophages in resting and inflamed lungs. Two populations of macrophages have been identified in the mouse: resident macrophages and inflammatory macrophages.36–40 The extravasation of resident macrophages can occur in uninflamed lungs and is independent of neutrophils, while the recruitment of inflammatory macrophages to inflamed lungs occurs after infection and is a neutrophil-dependent process.41 Because both macrophage populations play a crucial role in host defense and bacterial killing, we measured the number of alveolar macrophages both before and after bacteria challenge.
The numbers of resident alveolar macrophages in the BALF of unchallenged mice were accessed by Wright-Giemsa staining in which alveolar macrophages were identified by their large size, large cytoplasmic region, and single, round nucleus (Figure 6A) and by FACS analysis in which resident alveolar macrophages were identified as CD11b−F4/80+ cells (Figures 6B and 8).42 This number was increased by more than 60% in the PTEN KO mice in both assays. During lung inflammation, recruitment of inflammatory macrophages occurs after neutrophil recruitment and usually peaks at 24 to 48 hours after bacterial infection. Accordingly, we measured the amount of inflammatory macrophages in inflamed lungs at 24 hours. We used FACS analysis to more precisely characterize the population of macrophages. Briefly, BALF cells were gated according to their forward light scatter (FSC) versus side light scatter (SSC) characteristics, thereby excluding contaminating red blood cells and cell debris. This was followed by hierarchical subgating according to their CD11b versus F4/80 antigen expression. Resident alveolar macrophages were then identified as F4/80+ and CD11b− cells. Inflammatory-induced exudate macrophages were identified as F4/80+ and CD11bhi cells (Figure S8). As previously reported,42 the total number of alveolar macrophages decreased after bacterial challenge. However, similar to neutrophil recruitment during lung inflammation, the recruitment of inflammatory macrophages was dramatically elevated in the PTEN KO mice (Figure 6B).
Figure 6.

Disruption of PTEN enhances alveolar macrophage accumulation and elevates proinflamatory cytokine/chemokine production in the inflamed lungs. Mice were intratracheally instilled with 106 CFU of E coli and killed at indicated time points. BALF were collected using ice-cold PBS/15 mM EDTA (ethylenediaminetetraacetic acid; 1 mL ×10). (A) Differential cell counts were conducted on cytospin preparations stained with a modified Wright-Giemsa stain (Volu-Sol). Macrophages were identified morphologically. BALF total cell numbers were counted using a hemocytometer. Macrophage population was calculated according to its percentage. (B) Total numbers of leukocytes, neutrophils, resident macrophages, and inflammatory macrophages in BALF were determined by FACS analysis. (C) BALF chemokine and cytokine levels were determined using enzyme-linked immunosorbent assay (ELISA) kits. (D) LPS-induced chemokine and cytokine release from purified primary alveolar macrophages. Alveolar macrophages were collected from unchallenged WT and PTEN KO mice and stimulated with LPS (1 μg/mL) for up to 24 hours. Supernatants were harvested and the chemokine/cytokine levels were determined using ELISA kits. Data are presented as mean (± SD; n ≥ 3 mice in each group). *P < .05, **P < .01 versus WT.
Cytokine and chemokine production was increased in the PTEN KO mice
Inflammation is always associated with a large amount of cytokine and chemokine release from the site of infection. These cytokines and chemokines can subsequently induce accumulation and activation of neutrophils, monocytes/macrophages, eosinophils, and lymphocytes. They also directly facilitate the clearance of pathogens by immune cells. In PTEN KO mice, the enhanced recruitment of neutrophils to the inflamed lungs could also be a result of elevated proinflammatory cytokine/chemokine levels in the lungs. Neutrophil recruitment to the inflamed lungs is mainly mediated by CXCR2 receptor.43–46 Keratinocyte-derived cytokine (KC) and macrophage-inflammatory protein-2 (MIP-2) are 2 of the CXCR2 receptor ligands in mice, and their levels in the lungs increase dramatically during the course of pneumonia.47–50 PTEN disruption may directly increase the production of these chemokines in the inflamed lungs. Alternatively, PTEN disruption may enhance neutrophil recruitment via affecting some other early cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin 6 (IL-6), and IL-1, which are rapidly induced by bacteria and promote neutrophil recruitment indirectly. It was reported that neutrophil recruitment was significantly decreased by combined deficiency of TNF-α and IL-1 signaling.51–54 Accordingly, we measured the level of these chemokines/cytokines in the inflamed lungs of both WT and PTEN KO mice. In BALF collected at 24 hours after E coli instillation, the concentrations of all 5 cytokines/chemokines were significantly increased (Figure 6C) in the PTEN KO mice, demonstrating that the elevated neutrophil recruitment in these mice is also partially contributed by the elevated cytokine/chemokine levels.
Among the known CXCR2 ligands, macrophages are thought to be important sources for MIP-2 and KC.47–50 Activated macrophages are also a major source of some early cytokines such as TNF-α and IL-6 in inflamed lungs. Thus, we directly examined whether PTEN disruption in macrophages can affect the LPS-induced production of MIP-2, KC, TNF-α, and IL-6 using an in vitro assay. Resident alveolar macrophages were prepared from WT and PTEN KO mice and stimulated with LPS for 24 hours. Our results showed that PTEN disruption did not increase the production of cytokines/chemokines by macrophages. Interestingly, PTEN depletion even led to a very small but statistically significant decrease of TNF-α and IL-6 production (Figure 6D). Collectively, these results suggest that the elevated cytokine/chemokine levels in BALF is mainly caused by the increased numbers of resident alveolar macrophages, and not by their enhanced capability of producing cytokines or chemokines.
Disruption of PTEN alleviates the severity of, and decreases the mortality associated with, neutropenia-related pneumonia
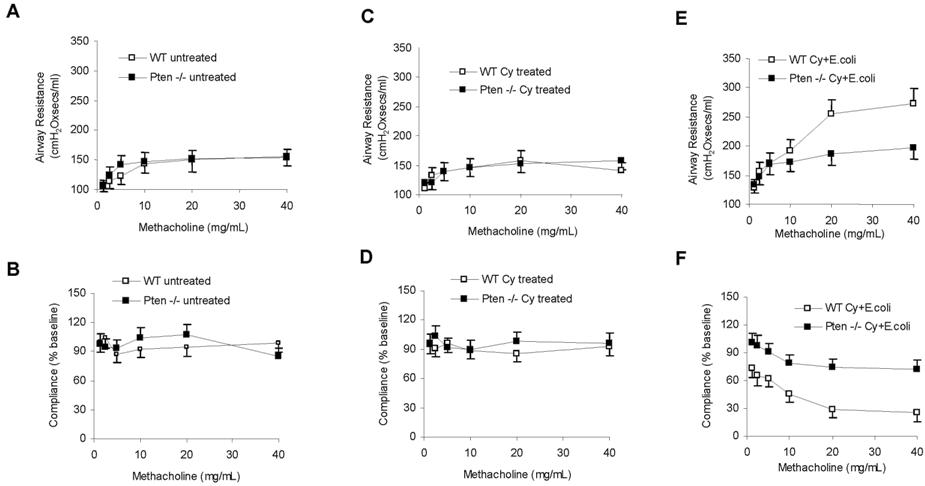
We demonstrated that augmentation of the PtdIns(3,4,5)P3 signal by depleting PTEN enhances neutrophil/macrophage recruitment to the inflamed lungs and increases the bacteria-killing capability of the recruited neutrophils in both normal and neutropenic mice. As a result, the bacterial burden in the infected myeloid-specific PTEN KO mice was reduced. Because the direct cause of neutropenia-related pneumonia is the lack of neutrophils in the infected lungs to clear the invading bacteria, the enhanced phagocyte accumulation and the augmented bacteria-killing capability in PTEN KO mice should lead to a quicker resolution of lung inflammation under neutropenic conditions. This was supported by the reduced formation of pulmonary edema in bacteria-challenged neutropenic PTEN-null mice (Figure 4H). To provide more direct evidence, we conducted an X-ray radiographic analysis to assess the severity of lung inflammation. In WT mice, bacterial instillation induced severe inflammation in 24 hours, as shown by pulmonary infiltrates and abnormal diffuse radiographic opacities throughout the lungs. In contrast, the inflammation in the PTEN-null mice was less severe and usually lasted for less than 24 hours (Figure 7A). Severe pneumonia is often accompanied with vascular leakage. BAL protein level increase has been used as an indicator of vascular leakage and a key parameter of inflammatory lung injury. Consistent with the enhanced bacteria killing capability and the alleviated lung inflammation in the PTEN KO mice, we detected much decreased total protein level in BALF in the inflamed lungs (Figure 7B). Pneumonia is also accompanied by compromised lung mechanics52,55; thus, we further explored the integrity of lung function by measuring pulmonary compliance and resistance using an invasive monitoring method (Figure 7C). Lung compliance, which is measured as the pulmonary volume change per unit of pressure change, reflects the comparative stiffness or elasticity of the lung: the stiffer the lung, the lower the compliance. Compliance is often reduced by edema in the alveolar spaces during lung inflammation. Lung resistance, which is the amount of pressure required to cause a unit change of gas flow, reflects both narrowing of the conducting airways and parenchymal viscosity. In the absence of pneumonia, there were no significant differences in pulmonary compliance and resistance between WT and myeloid-specific PTEN-null mice (Figure S9). As expected, lung compliance decreased and pulmonary resistance increased in mice with pneumonia. However, such alterations in lung mechanics were not as pronounced in neutropenic PTEN-null mice compared with their WT littermates at 24 hours after E coli instillation, supporting the idea that disruption of PTEN alleviates the severity of lung inflammation in neutropenic mice (Figure 7C).
Figure 7.

PTEN disruption alleviates the severity of and decreases the mortality associated with neutropenia-related pneumonia. (A) X-ray images were taken using an MX-20 Radiography System (Faxitron X-ray) at the Kresge Imaging Center (Rodent X-ray Unit) at Children's Hospital Boston. Representative X-ray images from untreated (left panel), Cy-treated (middle panel), and E coli–challenged, Cy-treated (right panel) WT or myeloid-specific PTEN KO mice are shown. Mice were anesthetized and images were taken in the postanterior view. The “cloudy areas” in the lungs are indicated. At least 4 mice were examined for each group and essentially the same results were observed. (B) BALF total protein level. Protein accumulated in the inflamed lung was measured using a Bio-Rad protein assay kit. The standard curve was constructed using BSA. (C) Airway resistance (left panel) and dynamic compliance (right panel) were measured in untreated, Cy-treated, and E coli–challenged, Cy-treated mice. For the E coli–challenged, 105 CFU of E coli were instilled intratracheally. Lung mechanics were measured 24 hours later. All data are presented as mean (± SD; n ≥ 4 mice in each group). *P < .05 versus WT. (D,E) Higher survival rates in PTEN KO mice with neutropenia-related pneumonia. Age- and sex-matched WT and PTEN KO mice were pretreated with Cy (250 mg/kg; panel D) or exposed to irradiation (600 cGy) as described in “Methods” and then challenged with 105 live E coli. The survival rates were analyzed using the Kaplan-Meier and log-rank methods. The differences in survival were statistically significant (*P < .01 by log-rank test).
Lastly, severe neutropenia-related pneumonia can lead to death. Thus, we investigated whether augmentation of the PtdIns(3,4,5)P3 signal by depleting PTEN can reduce such lethality. Consistent with the X-ray and lung function data, the increased bacteria clearance capability and less severe inflammation in the myeloid-specific PTEN-null mice resulted in a higher survival rate of bacteria-challenged neutropenic mice. More than 50% of PTEN experimental mice survived the challenge. In contrast, only 7% of WT mice survived (Figure 7D). In the above experiments, the mouse neutropenia was induced with the chemotherapeutic drug Cy. We also examined pneumonia-associated lethality of irradiation-induced neutropenic mice. Essentially the same results were obtained: PTEN disruption led to a higher survival rate of neutropenic mice, demonstrating that the protective effect observed in the PTEN-null mice was not a model-dependent phenomenon (Figure 7E). Taken together, these observations provide direct evidence that augmenting PtdIns(3,4,5)P3 by disrupting PTEN can alleviate the severity of neutropenia-related pneumonia. This effect is mainly mediated by the elevated bacterial killing capability, which is largely due to the augmented recruitment and enhanced function of neutrophils. Supporting this idea, when the neutropenia-related pneumonia was induced by sterile ligands LPS, we could not detect statistically significant difference in the survival rate between the WT and PTEN KO neutropenic mice. The elevated neutrophil recruitment was still observed in the PTEN KO mice. Nevertheless, this led to even more severe lung damage instead of alleviation of lung inflammation as observed in the pneumonia induced by live E coli (Figure S10).
Discussion
In this study, we explored the possibility of treating and preventing neutropenia-related pneumonia by enhancing various neutrophil functions. We did so by augmenting the intracellular PtdIns(3,4,5)P3 signaling pathway. Augmentation of this signal was achieved by disrupting PTEN, a phosphatidylinositol 3′-phosphatase that negatively regulates PtdIns(3,4,5)P3 signaling. We demonstrated that PTEN-null neutrophils have enhanced bacteria-killing capability, and their recruitment to inflamed lungs was augmented under both normal and neutropenic conditions. Most recently, Heit et al reported that under a certain situation, PTEN is required for prioritizing and integrating responses to multiple chemotactic cues. In agreement with our findings, they detected enhanced recruitment of neutrophils to the peritoneum in response to formyl-methionyl-leucyl-phenylalanine (fMLP) in the PTEN KO mice. However, disruption of PTEN led to “distraction” in migrating neutrophils in an in vivo model of inflammatory arthritis.56 Thus, the exact role of PTEN in neutrophils might rely on the diversity of chemoattractants involved, as well as on the relative doses and route used to induce the neutrophil inflammatory reactions. Whether PTEN functions to prevent distraction during lung inflammation is still largely unknown.
In response to inflammatory stimuli, neutrophils migrate from the blood to infected tissues, where they protect their host by engulfing, killing, and digesting invading bacterial and fungal pathogens. Conversely, excessive neutrophil accumulation or hyperresponsiveness of neutrophils can be detrimental to the system. Hence, the response of neutrophils to inflammatory stimuli needs to be well controlled. Augmenting neutrophil function by elevating the PtdIns(3,4,5)P3 signaling might increase the chance of aggravated inflammation and tissue damage. In the PTEN KO mice, more severe pulmonary edema and increased mortality rate were observed when bacterial pneumonia was induced in nonneutropenic mice (Figure 1E). This is most likely due to the excessive neutrophil accumulation and hyperactivation of the recruited PTEN-null neutrophils. Nevertheless, this should be less of a concern in neutropenic patients in whom the number of neutrophils is dramatically reduced and the release of noxious compounds, such as oxidants, proteinases, and DNA, by neutrophils is also minimal. The direct cause of neutropenia-related pneumonia is the lack of neutrophils in the infected lungs to clear the invading bacteria. When the dose of E coli is in a range that can be handled by the host innate immune system, the enhanced neutrophil accumulation and the augmented bacteria-killing capability in the PTEN KO mice will lead to faster resolution/alleviation of lung inflammation under neutropenic conditions. Consequently, the increased bacteria clearance capability and less severe inflammation in the PTEN-null mice will result in a much increased survival rate of bacteria-challenged neutropenic mice. In support of this idea, less severe pulmonary edema and increased survival rate were observed in infected neutropenic PTEN-null mice. However, if the amount of bacteria instilled into the lungs is too high, and thus even the PTEN-null neutrophils cannot clear all of the pathogens, PTEN KO mice might even display more aggravated lung inflammation due to the enhance neutrophil accumulation and superoxide production.
Note that although we focused on neutrophils and used myeloid-specific PTEN KO mice in this study, augmentation of PtdIns(3,4,5)P3 signaling might also enhance host defenses during neutropenia-associated pneumonia by affecting other immune cell types. For example, it is well documented that enhancing the PtdIns(3,4,5)P3 signal can strengthen T-cell and B-cell functions.57–61 It was also reported that disruption of PTEN in macrophages significantly increases their phagocytic capability,22 and that overexpression of PTEN inhibits macrophage invasion and proinflammatory cytokine expression.62 In our myeloid-specific PTEN KO mice, PTEN expression was also ablated in monocytes, macrophages, and some dendritic cells, which also play critical roles in host defense in bacterial pneumonia. Although the competitive adoptive transfer and in vitro bacteria killing/phagocytosis assays definitively demonstrated that PTEN disruption in neutrophils is at least partially responsible for the enhanced bacteria clearance capability in KO mice, the involvement of other cell types cannot be ruled out. Indeed, we demonstrated that disruption of PTEN also enhanced the accumulation of alveolar resident and inflammatory macrophages. As a result, the levels of proinflammatory cytokines/chemokines, such as MIP-2, KC, TNF-α, IL-1, and IL-6, in the BALF were elevated.
In present study, we demonstrated that neutrophil function can be enhanced by elevating PtdIns(3,4,5)P3 signaling in neutropenia-related pneumonia. In addition, we provided direct evidence that enhancement of neutrophil function by elevating PtdIns(3,4,5)P3 signaling can alleviate pneumonia-associated lung damage and decrease pneumonia-elicited mortality. These results provide insight into the mechanism of action of PTEN and PtdIns(3,4,5)P3 signaling pathway in modulating neutrophil function during lung infection and inflammation. As physiologic regulators of neutrophil function, the PTEN and related pathways could be promising therapeutic targets for modulating neutrophil performance in neutropenia-related pneumonia.
Supplementary Material
Acknowledgments
We thank Leslie Silberstein, Kulandayan Subramanian, Hidenori Hattori, Hakryul Jo, John Manis, and Li Chai for helpful discussions. We are also grateful to Roderick Terry Bronson and Li Zhang for their assistance with histology analyses.
B.S. is supported by National Institutes of Health (National Institutes of Health, Bethesda, MD) training grant HL066987. H.R.L. is supported by National Institutes of Health grants HL085100 and GM076084 and by a Research Scholar Grant from the American Cancer Society (Atlanta, GA).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.L. designed and carried out experiments, analyzed data, and prepared the manuscript; Y.J. contributed to Figure 2C and D; M.P. contributed to Figure 7C; F.L., B.S., and A.K. helped with planning and analyzing data; J.Y. bred mice; B.E.R. helped with setting up the pneumonia model; D.T.U., J.P.M., and K.Y. helped with analyzing data and evaluating the manuscript; and H.R.L. designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hongbo R. Luo, Karp Family Research Bldg, Rm 10214, 1 Blackfan Circle, Boston, MA 02115; e-mail: hongbo.luo@childrens.harvard.edu.
References
- 1.Hoare Z, Lim WS. Pneumonia: update on diagnosis and management. Br Med J. 2006;332:1077–1079. doi: 10.1136/bmj.332.7549.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loeb M. Community acquired pneumonia. Clin Evid. 2006;15:2015–2024. [PubMed] [Google Scholar]
- 3.Ostendorf U, Ewig S, Torres A. Nosocomial pneumonia. Curr Opin Infect Dis. 2006;19:327–338. doi: 10.1097/01.qco.0000235158.40184.28. [DOI] [PubMed] [Google Scholar]
- 4.Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358:716–727. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joos L, Tamm M. Breakdown of pulmonary host defense in the immunocompromised host: cancer chemotherapy. Proc Am Thorac Soc. 2005;2:445–448. doi: 10.1513/pats.200508-097JS. [DOI] [PubMed] [Google Scholar]
- 6.Viscoli C, Varnier O, Machetti M. Infections in patients with febrile neutropenia: epidemiology, microbiology, and risk stratification. Clin Infect Dis. 2005;40(suppl 4):S240–245. doi: 10.1086/427329. [DOI] [PubMed] [Google Scholar]
- 7.Leung AN, Gosselin MV, Napper CH, et al. Pulmonary infections after bone marrow transplantation: clinical and radiographic findings. Radiology. 1999;210:699–710. doi: 10.1148/radiology.210.3.r99mr39699. [DOI] [PubMed] [Google Scholar]
- 8.Iijima M, Huang YE, Devreotes P. Temporal and spatial regulation of chemotaxis. Dev Cell. 2002;3:469–478. doi: 10.1016/s1534-5807(02)00292-7. [DOI] [PubMed] [Google Scholar]
- 9.Stephens L, Ellson C, Hawkins P. Roles of PI3Ks in leukocyte chemotaxis and phagocytosis. Curr Opin Cell Biol. 2002;14:203–213. doi: 10.1016/s0955-0674(02)00311-3. [DOI] [PubMed] [Google Scholar]
- 10.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 11.Bourne HR, Weiner O. A chemical compass. Nature. 2002;419:21. doi: 10.1038/419021a. [DOI] [PubMed] [Google Scholar]
- 12.Zhu D, Hattori H, Jo H, et al. Deactivation of phosphatidylinositol 3,4,5-trisphosphate/Akt signaling mediates neutrophil spontaneous death. Proc Natl Acad Sci U S A. 2006;103:14836–14841. doi: 10.1073/pnas.0605722103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanian KK, Jia Y, Zhu D, et al. Tumor suppressor PTEN is a physiologic suppressor of chemoattractant-mediated neutrophil functions. Blood. 2007;109:4028–4037. doi: 10.1182/blood-2006-10-055319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia Y, Subrahmanyam KK, Erneux C, et al. Inositol 1,3,4,5-tetrakisphosphate negatively regulates PtdIns(3,4,5)P3 signaling in neutrophils. Immunity. 2007;27:453–467. doi: 10.1016/j.immuni.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groszer M, Erickson R, Scripture-Adams DD, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 16.D'Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26:204–213. doi: 10.1183/09031936.05.00095204. [DOI] [PubMed] [Google Scholar]
- 17.Dallaire F, Ouellet N, Simard M, Bergeron Y, Bergeron MG. Efficacy of recombinant human granulocyte colony-stimulating factor in a murine model of pneumococcal pneumonia: effects of lung inflammation and timing of treatment. J Infect Dis. 2001;183:70–77. doi: 10.1086/317652. [DOI] [PubMed] [Google Scholar]
- 18.Allen L, Dockrell DH, Pattery T, et al. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J Immunol. 2005;174:3643–3649. doi: 10.4049/jimmunol.174.6.3643. [DOI] [PubMed] [Google Scholar]
- 19.Brazil TJ, Dagleish MP, McGorum BC, Dixon PM, Haslett C, Chilvers ER. Kinetics of pulmonary neutrophil recruitment and clearance in a natural and spontaneously resolving model of airway inflammation. Clin Exp Allergy. 2005;35:854–865. doi: 10.1111/j.1365-2222.2005.02231.x. [DOI] [PubMed] [Google Scholar]
- 20.Walker JA, Quirke P. Viewing apoptosis through a ‘TUNEL’. J Pathol. 2001;195:275–276. doi: 10.1002/path.979. [DOI] [PubMed] [Google Scholar]
- 21.Lawry J. Detection of apoptosis by the TUNEL assay. Methods Mol Med. 2004;88:183–190. doi: 10.1385/1-59259-406-9:183. [DOI] [PubMed] [Google Scholar]
- 22.Cao X, Wei G, Fang H, et al. The inositol 3-phosphatase PTEN negatively regulates Fcγ receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172:4851–4857. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 23.Anderson KE, Boyle KB, Davidson K, et al. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. 2008;112:5202–5211. doi: 10.1182/blood-2008-04-149450. [DOI] [PubMed] [Google Scholar]
- 24.Grunberg SM. Cyclophosphamide and etoposide for non-small cell and small cell lung cancer. Drugs. 1999;58(suppl 3):11–15. doi: 10.2165/00003495-199958003-00002. [DOI] [PubMed] [Google Scholar]
- 25.Decker DG, Mussey E, Malkasian GD, Jr, Johnson CE. Cyclophosphamide in the treatment of ovarian cancer. Clin Obstet Gynecol. 1968;11:382–400. doi: 10.1097/00003081-196806000-00001. [DOI] [PubMed] [Google Scholar]
- 26.van't Wout JW, Linde I, Leijh PC, van Furth R. Effect of irradiation, cyclophosphamide, and etoposide (VP-16) on number of peripheral blood and peritoneal leukocytes in mice under normal conditions and during acute inflammatory reaction. Inflammation. 1989;13:1–14. doi: 10.1007/BF00918959. [DOI] [PubMed] [Google Scholar]
- 27.Zuluaga AF, Salazar BE, Rodriguez CA, Zapata AX, Agudelo M, Vesga O. Neutropenia induced in outbred mice by a simplified low-dose cyclophosphamide regimen: characterization and applicability to diverse experimental models of infectious diseases. BMC Infect Dis. 2006;6:55. doi: 10.1186/1471-2334-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cote CK, Van Rooijen N, Welkos SL. Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect Immun. 2006;74:469–480. doi: 10.1128/IAI.74.1.469-480.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358:716–727. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeyaseelan S, Young SK, Fessler MB, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia. J Immunol. 2007;178:3153–3160. doi: 10.4049/jimmunol.178.5.3153. [DOI] [PubMed] [Google Scholar]
- 31.Laskin DL, Weinberger B, Laskin JD. Functional heterogeneity in liver and lung macrophages. J Leukocyte Biol. 2001;70:163–170. [PubMed] [Google Scholar]
- 32.Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C. Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of Gram-negative bacteria. J Immunol. 2005;175:5596–5600. doi: 10.4049/jimmunol.175.9.5596. [DOI] [PubMed] [Google Scholar]
- 33.Knapp S, Leemans JC, Florquin S, et al. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med. 2003;167:171–179. doi: 10.1164/rccm.200207-698OC. [DOI] [PubMed] [Google Scholar]
- 34.Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. 2000;173:39–51. doi: 10.1034/j.1600-065x.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- 35.Twigg HL., 3rd Pulmonary host defenses. J Thorac Imaging. 1998;13:221–233. doi: 10.1097/00005382-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 37.van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods. 2004;288:111–121. doi: 10.1016/j.jim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 38.von Garnier C, Filgueira L, Wikstrom M, et al. Anatomical location determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J Immunol. 2005;175:1609–1618. doi: 10.4049/jimmunol.175.3.1609. [DOI] [PubMed] [Google Scholar]
- 39.Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry. 2004;61:170–177. doi: 10.1002/cyto.a.20064. [DOI] [PubMed] [Google Scholar]
- 40.GeurtsvanKessel CH, Willart MA, van Rijt LS, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J Exp Med. 2008;205:1621–1634. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soehnlein O, Zernecke A, Eriksson EE, et al. Neutrophil secretion products pave the way for inflammatory monocytes. Blood. 2008;112:1461–1471. doi: 10.1182/blood-2008-02-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maus UA, Backi M, Winter C, et al. Importance of phosphoinositide 3-kinase gamma in the host defense against pneumococcal infection. Am J Respir Crit Care Med. 2007;175:958–966. doi: 10.1164/rccm.200610-1533OC. [DOI] [PubMed] [Google Scholar]
- 43.McColl SR, Clark-Lewis I. Inhibition of murine neutrophil recruitment in vivo by CXC chemokine receptor antagonists. J Immunol. 1999;163:2829–2835. [PubMed] [Google Scholar]
- 44.Mehrad B, Strieter RM, Moore TA, Tsai WC, Lira SA, Standiford TJ. CXC chemokine receptor-2 ligands are necessary components of neutrophil-mediated host defense in invasive pulmonary aspergillosis. J Immunol. 1999;163:6086–6094. [PubMed] [Google Scholar]
- 45.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun. 2000;68:4289–4296. doi: 10.1128/iai.68.7.4289-4296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tateda K, Moore TA, Newstead MW, et al. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect Immun. 2001;69:2017–2024. doi: 10.1128/IAI.69.4.2017-2024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Introna M, Bast RC, Jr, Tannenbaum CS, Hamilton TA, Adams DO. The effect of LPS on expression of the early “competence” genes JE and KC in murine peritoneal macrophages. J Immunol. 1987;138:3891–3896. [PubMed] [Google Scholar]
- 48.Rossi DL, Hurst SD, Xu Y, et al. Lungkine, a novel CXC chemokine, specifically expressed by lung bronchoepithelial cells. J Immunol. 1999;162:5490–5497. [PubMed] [Google Scholar]
- 49.Smith JB, Herschman HR. Glucocorticoid-attenuated response genes encode intercellular mediators, including a new C-X-C chemokine. J Biol Chem. 1995;270:16756–16765. doi: 10.1074/jbc.270.28.16756. [DOI] [PubMed] [Google Scholar]
- 50.Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci U S A. 1989;86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Lung NF-κB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol. 2005;175:7530–7535. doi: 10.4049/jimmunol.175.11.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mizgerd JP, Lupa MM, Hjoberg J, et al. Roles for early response cytokines during Escherichia coli pneumonia revealed by mice with combined deficiencies of all signaling receptors for TNF and IL-1. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1302–1310. doi: 10.1152/ajplung.00353.2003. [DOI] [PubMed] [Google Scholar]
- 53.Mizgerd JP, Spieker MR, Doerschuk CM. Early response cytokines and innate immunity: essential roles for TNF receptor 1 and type I IL-1 receptor during Escherichia coli pneumonia in mice. J Immunol. 2001;166:4042–4048. doi: 10.4049/jimmunol.166.6.4042. [DOI] [PubMed] [Google Scholar]
- 54.Mehrad B, Standiford TJ. Role of cytokines in pulmonary antimicrobial host defense. Immunol Res. 1999;20:15–27. doi: 10.1007/BF02786504. [DOI] [PubMed] [Google Scholar]
- 55.Nuckton TJ, Alonso JA, Kallet RH, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med. 2002;346:1281–1286. doi: 10.1056/NEJMoa012835. [DOI] [PubMed] [Google Scholar]
- 56.Heit B, Robbins SM, Downey CM, et al. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol. 2008;9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- 57.Leslie NR, Yang X, Downes CP, Weijer CJ. The regulation of cell migration by PTEN. Biochem Soc Trans. 2005;33:1507–1508. doi: 10.1042/BST0331507. [DOI] [PubMed] [Google Scholar]
- 58.Donahue AC, Fruman DA. PI3K signaling controls cell fate at many points in B lymphocyte development and activation. Semin Cell Dev Biol. 2004;15:183–197. doi: 10.1016/j.semcdb.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 59.Fruman DA. Phosphoinositide 3-kinase and its targets in B-cell and T-cell signaling. Curr Opin Immunol. 2004;16:314–320. doi: 10.1016/j.coi.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 60.Okkenhaug K, Bilancio A, Emery JL, Vanhaesebroeck B. Phosphoinositide 3-kinase in T cell activation and survival. Biochem Soc Trans. 2004;32:332–335. doi: 10.1042/bst0320332. [DOI] [PubMed] [Google Scholar]
- 61.Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- 62.Koide S, Okazaki M, Tamura M, et al. PTEN reduces cuff-induced neointima formation and proinflammatory cytokines. Am J Physiol Heart Circ Physiol. 2007;292:H2824–2831. doi: 10.1152/ajpheart.01221.2006. [DOI] [PubMed] [Google Scholar]
- 63.Mizgerd JP, Peschon JJ, Doerschuk CM. Roles of tumor necrosis factor receptor signaling during murine Escherichia coli pneumonia. Am J Respir Cell Mol Biol. 2000;22:85–91. doi: 10.1165/ajrcmb.22.1.3733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}