

Abstract
Background
The entomopathogenic nematode Heterorhabditis bacteriophora and its symbiotic bacterium, Photorhabdus luminescens, are important biological control agents of insect pests. This nematode-bacterium-insect association represents an emerging tripartite model for research on mutualistic and parasitic symbioses. Elucidation of mechanisms underlying these biological processes may serve as a foundation for improving the biological control potential of the nematode-bacterium complex. This large-scale expressed sequence tag (EST) analysis effort enables gene discovery and development of microsatellite markers. These ESTs will also aid in the annotation of the upcoming complete genome sequence of H. bacteriophora.
Results
A total of 31,485 high quality ESTs were generated from cDNA libraries of the adult H. bacteriophora TTO1 strain. Cluster analysis revealed the presence of 3,051 contigs and 7,835 singletons, representing 10,886 distinct EST sequences. About 72% of the distinct EST sequences had significant matches (E value < 1e-5) to proteins in GenBank's non-redundant (nr) and Wormpep190 databases. We have identified 12 ESTs corresponding to 8 genes potentially involved in RNA interference, 22 ESTs corresponding to 14 genes potentially involved in dauer-related processes, and 51 ESTs corresponding to 27 genes potentially involved in defense and stress responses. Comparison to ESTs and proteins of free-living nematodes led to the identification of 554 parasitic nematode-specific ESTs in H. bacteriophora, among which are those encoding F-box-like/WD-repeat protein theromacin, Bax inhibitor-1-like protein, and PAZ domain containing protein. Gene Ontology terms were assigned to 6,685 of the 10,886 ESTs. A total of 168 microsatellite loci were identified with primers designable for 141 loci.
Conclusion
A total of 10,886 distinct EST sequences were identified from adult H. bacteriophora cDNA libraries. BLAST searches revealed ESTs potentially involved in parasitism, RNA interference, defense responses, stress responses, and dauer-related processes. The putative microsatellite markers identified in H. bacteriophora ESTs will enable genetic mapping and population genetic studies. These genomic resources provide the material base necessary for genome annotation, microarray development, and in-depth gene functional analysis.
Background
The entomopathogenic nematode, Heterorhabditis bacteriophora, and its mutualistic bacterium, Photorhabdus luminescens, are important biological control agents of insect pests [1] and represent an emerging model for research on mutualistic and parasitic symbiosis [2,3]. The use of H. bacteriophora as a biological control agent is hampered by its susceptibility to environmental extremes including temperature, desiccation, and UV radiation, differences in virulence towards different insect pests, and short shelf life. Elucidation of molecular mechanisms underlying these biological processes may serve as a foundation for improving the biological control potential of the nematode-bacterium complex.
The entomopathogenic nematode H. bacteriophora has a distinct life style. The infective juveniles (IJs) or dauer juveniles (DJs) persist in soil in search of a suitable insect host [4]. Following entry into the insect host through natural body openings and cuticle, the IJs regurgitate the symbiotic bacteria into the insect hemocoel [5]. The bacteria kill the insect host via septicemia, usually within 24–48 h [5]. The nematodes feed on the multiplying bacteria and disintegrated host tissues and produce 1 to 3 generations within the cadaver. When the food source depletes and nematode density reaches a threshold, next-generation IJs are formed which exit the cadaver in search of a new host [3].
In contrast to the closely related genetic model Caenorhabditis elegans, few genomic resources are available for H. bacteriophora. However, some progress has been made over the past few years with the generation and analysis of ~1,000 ESTs from H. bacteriophora GPS11 strain [3,6], the start of an H. bacteriophora complete genome sequence project (supported by the National Human Genome Research Institute), and the development of a reverse genetics tool using RNA interference [7]. Release of the genomes of C. elegans [8], C. briggsae [9], Brugia malayi [10], bacterium Photorhabdus luminescens subsp. laumondii TTO1 (obligate endosymbiont) [11] and over 1 million ESTs from various nematode species deposited in GenBank offers unprecedented opportunities for the genetics of entomopathogenic nematodes. Here, we report on the construction of cDNA libraries from H. bacteriophora TTO1 adult hermaphrodites and the generation and analysis of 31,485 ESTs. The TTO1 strain is different from GPS11 strain in insect toxicity and their symbiotic bacteria (Grewal et al., unpublished). ESTs are valuable for gene discovery and can also be used in the identification of microsatellite markers [12]. Therefore, we also identified microsatellite loci in H. bacteriophora ESTs for use in population genetic studies. In addition, ESTs will also aid the prediction of protein-coding genes in the annotation of complete genomes. However, domain identification, secretome prediction, phylogenetic and evolution analysis on the ESTs that are of short-length and low coverage are not very informative, and therefore were not performed.
Results
Generation of ESTs and assembly
A total of 31,586 ESTs from adult H. bacteriophora TTO1-M13e strain were generated from cDNA libraries and were deposited in GenBank under the accession numbers [GenBank:EG025323] – [GenBank:EG025806], [GenBank:ES408468] – [GenBank:ES414355], [GenBank:ES738967] – [GenBank:ES744677], [GenBank:EX006911] – [GenBank:EX015306], [GenBank:EX910019] – [GenBank:EX916843], and [GenBank:FF678120] – [GenBank:FF681586]. The removal of vector and short length (20 bp or less) sequences resulted in 31,485 high-quality ESTs with an average length of 531 bp. The cumulative length of all high-quality EST sequences was 16,713,919 bases.
The ESTs were subjected to cluster analysis using Vector NTI Advance™ 10 (Invitrogen), with the final assembly generated by Vector NTI Advance™ 10. The final assembly contained 3,051 contigs generated from 23,650 ESTs and 7,835 singletons (Table 1). The contigs consisted of 2 to 283 ESTs (See additional file 1: Components of H. bacteriophora assembled contigs) with lengths ranging from 60 to 2,856 bp and a mean of 743 bp (Table 1). The average length of singletons was 461 bp (Figure 1). In total, we identified 10,886 distinct EST sequences.
Table 1.
Summary statistics of ESTs generated from H. bacteriophora
| Feature | Value |
| Number of high quality ESTs | 31,485 |
| Average length of high quality ESTs (bp) | 531 |
| Number of contigs | 3,051 |
| Number of ESTs in contigs | 23,650 |
| Number of singletons | 7,835 |
| Number of distinct sequences | 10,886 |
| Average length of contigs (bp) | 743 |
| Average length of singletons (bp) | 461 |
Figure 1.

Length distribution of H. bacteriophora ESTs before and after assembly.
Putative functional identifications of the ESTs
In order to assess the putative identities, all distinct ESTs were subjected to BLASTx sequence similarity searches against GenBank's nr database and Wormpep190 database consisting of extensively curated C. elegans proteins from Wormbase [13]. Of the 10,886 distinct ESTs, 7,828 (71.9%) had significant matches (E value cutoff 1e-5) to proteins in GenBank's nr database. As expected, most of the best matches (95.9%) were to nematode proteins (Figure 2). A small proportion (0.3%) of the best matches was to prokaryotic proteins with localized sequence similarity ranging from 28% to 98% and a median of 81% (Table 2) and the remaining 3.8% of the best matches was to other eukaryotes including humans, fungi, plants, and insects. Of the remaining 3,058 H. bacteriophora distinct ESTs, 119 had significant matches to nucleotide sequences in GenBank nt database, including 31 that matched to mitochondrial genes.
Figure 2.

Categories of organisms with significant protein matches of H. bacteriophora distinct ESTs. The percentage was calculated considering the total number of H. bacteriophora distinct ESTs having significant matches (E value < 1e-5) as 100%.
Table 2.
Summary of H. bacteriophora distinctive ESTs matching to prokaryotic proteins
| Best BLASTx match | ||||
| H. bacteriophora distinctive ESTs | Description | Organism | E value | Identitiesa |
| Contig131 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 2e-44 | 308/507, 86% |
| ES410513 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 6e-11 | 125/227, 86% |
| ES410515 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 7e-18 | 227/231, 67% |
| ES410516 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 2e-16 | 161/171, 81% |
| ES410519 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 8e-17 | 158/251, 87% |
| ES410521 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 2e-25 | 188/288, 95% |
| ES410525 | MTA/SAH nucleosidase | Enterococcus faecalis V583 | 9e-15 | 149/249, 84% |
| Contig145 | DpnD protein | Streptococcus pneumoniae TIGR4 | 7e-21 | 152/375, 98% |
| Contig672 | Adenylate cyclase | Burkholderia oklahomensis EO147 | 2e-16 | 500/962, 33% |
| Contig1258 | GTP-binding protein HSR1-related | Thauera sp. MZ1T | 4e-21 | 596/649, 31% |
| Contig1517 | hypothetical protein Bmal10_03004924 | Burkholderia mallei 10399 | 2e-10 | 230/809, 47% |
| Contig2004 | binding-protein-dependent transport systems inner membrane component | Pyrobaculum calidifontis JCM 11548 | 2e-15 | 290/325, 39% |
| Contig2104 | conserved hypothetical protein | uncultured beta proteobacterium CBNPD1 BAC clone 578 | 2e-25 | 185/390, 85% |
| EG025466 | Hypothetical protein COLAER_01671 | Collinsella aerofaciens ATCC 25986 | 2e-41 | 404/436, 64% |
| ES410527 | post-segregation antitoxin CcdA | Escherichia coli E24377A | 2e-21 | 137/325, 91% |
| ES410742 | hypothetical protein MS53_0454 | Mycoplasma synoviae 53 | 9e-06 | 539/686, 28% |
| ES410920 | Sodium/pantothenate symporter | Rickettsia akari str. Hartford | 0 | 44/278, 60% |
| ES739471 | conserved hypothetical protein | Burkholderia ambifaria MC40-6 | 0 | 44/628, 60% |
| EX911257 | transposase, IS4 family protein | Escherichia coli O157:H7 str. EC869 | 4e-52 | 311/657, 98% |
| FF681770 | rod shape-determining protein | Xanthomonas oryzae pv. oryzae KACC10331 | 2e-26 | 272/274, 76% |
| FF681812 | ATP-dependent Clp protease proteolytic subunit | Rhodococcus sp. RHA1 | 3e-12 | 116/168, 82% |
| FF681935 | Ribonucleotide reductase, alpha subunit | Leuconostoc citreum KM20 | 8e-11 | 155/175, 66% |
| FF681952 | rod shape-determining protein | Xanthomonas oryzae pv. oryzae KACC10331 | 4e-12 | 119/191, 85% |
| FF681963 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | Tenacibaculum sp. MED152 | 3e-15 | 179/183, 65% |
| FF682197 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | Algoriphagus sp. PR1 | 3e-17 | 179/221, 70% |
| FF682207 | rod shape-determining protein | Xylella fastidiosa 9a5c | 1e-26 | 248/269, 78% |
a. "Identities" was presented as "length of aligned H. bacteriophora EST"/"full length of H. bacteriophora EST", percentage of identities over the aligned sequences.
The similarity search against C. elegans-specific database Wormpep190 returned essentially similar results (Figure 3). Of the 10,886 H. bacteriophora distinct ESTs, 7,699 (70.7%) had significant matches (E value cutoff 1e-5) to 4,460 C. elegans proteins in Wormpep190 database. Based on sequence similarity results, 12 H. bacteriophora ESTs were identified to be involved in RNA interference (RNAi) pathway (Table 3). The currently identified H. bacteriophora RNAi genes are a small portion of those identified in C. elegans and B. malayi (Figure 4). We also identified 22 ESTs corresponding to 14 genes potentially involved in dauer-related processes (Table 4) and 51 ESTs corresponding to 27 genes involved in defense and stress responses (see Additional File 2: H. bacteriophora distinct ESTs similar to C. elegans genes involved in defense and stress responses).
Figure 3.

Distribution of bit scores from the best matches of BLASTx searches of H. bacteriophora distinct ESTs. BLASTx searches with bit scores less than 20 were not significant, roughly corresponding to E value > 1e-5.
Table 3.
H. bacteriophora distinct ESTs similar to C. elegans genes involved in RNA interference
| H. bacteriophora distinct EST | C. elegans homolog | Putative function | E value | Similaritya |
| FF679415 | WBGene00009163 (drsh-1) | Predicted RNase III-type ribonuclease that is orthologous to Drosophila and human Drosha | 5e-12 | 555/628, 43% |
| ES739421 | WBGene00001585 (gfl-1) | Ortholog of human GAS41. | 1e-73 | 591/599, 77% |
| ES741260 | WBGene00004326 (rde-4) | dsRNA binding protein (dsRBP) | 7e-08 | 369/505, 50% |
| EX913423 | WBGene00004880 (smg-2) | RNA helicase nonsense mRNA reducing factor | 2e-47 | 509/576, 66% |
| Contig658 | WBGene00004883 (smg-5) | Component of nonsense-mediated mRNA decay (NMD) pathway | 5e-22 | 498/672, 61% |
| EX014403 | WBGene00006626 (TSN-1) | Component of the RNA-induced silencing complex (RISC) | 2e-64 | 651/652, 71% |
| Contig211 | WBGene00006924 (vig-1) | Predicted RNA-binding protein orthologous to Drosophila VIG (Vasa Intronic Gene) | 2e-26 | 594/1573, 45% |
| EX915778 | WBGene00006975 (zfp-1) | A leucine zipper, zinc finger, and PHD/LAP domain-containing protein | 1e-10 | 144/567, 83% |
| EX912737 | WBGene00006975 (zfp-1) | A leucine zipper, zinc finger, and PHD/LAP domain-containing protein | 5e-09 | 594/607, 38% |
a. "Similarity" was presented as "length of aligned H. bacteriophora EST"/"full length of H. bacteriophora EST", percentage of positives over the aligned sequences.
Figure 4.

RNA interference related genes identified in H. bacteriophora distinct ESTs. Genes in red circle were identified in C. elegans. Genes in green circle were identified in B. malayi. Genes in blue circle were identified in H. bacteriophora.
Table 4.
H. bacteriophora distinct ESTs similar to C. elegans genes involved in dauer larval development, dauer entry, and dauer exit
| H. bacteriophora distinct EST | Matching C. elegans gene | Description | BLASTx E value | Alignmenta | % similarity |
| Dauer larval development | |||||
| Contig58 | WBGene00018294 | atgr-18 WD40 repeat-containing protein | 6e-70 | (55–624)/625; (16–208)/394 | 87% |
| Contig349 | WBGene00010882 | atgr-7 E1 ubiquitin-activating-like enzyme | 4e-85 | (3–743)/743; (373–609)/647 | 77% |
| EX010274 | WBGene00010882 | atgr-7 E1 ubiquitin-activating-like enzyme | 4e-44 | (31–597)/601; (2–197)/647 | 67% |
| Contig1114 | WBGene00000247 | bec-1 beclin (human autophagy) homolog | 7e-33 | (214-747)/748; (2–181)/375 | 62% |
| EX916779 | WBGene00000247 | bec-1 beclin (human autophagy) homolog | 6e-10 | (215–442)/444; (2–79)/375 | 59% |
| Contig1259 | WBGene00004860 | sma-6 Activin receptor | 5e-15 | (72–518)/693; (505–634)/636 | 54% |
| Contig1603 | WBGene00006786 | unc-51 serine/threonine protein kinase | 1e-122 | (222–932)/1459; (39–275)/856 | 81% |
| ES740745 | WBGene00006786 | unc-51 serine/threonine protein kinase | 2e-35 | (101–460)/552; (1–120)/856 | 68% |
| FF681332 | WBGene00006786 | unc-51 serine/threonine protein kinase | 4e-63 | (41–502)/503; (671–833)/856 | 83% |
| Contig2206 | WBGene00004736 | sca-1 E1–E2 ATPases | 3e-77 | (2–460)/608; (844–996)/1004 | 95% |
| EX012478 | WBGene00004736 | sca-1 E1–E2 ATPases | 5e-89 | (37–597)/599; (1–187)/1004 | 94% |
| FF679293 | WBGene00004736 | sca-1 E1–E2 ATPases | 1e-50 | (1–399)/430; (780–912)/1004 | 81% |
| ES739311 | WBGene00006600 | tph-1 tryptophan hydroxylase | 2e-63 | (12–587)/587; (23–217)/532 | 81% |
| ES741003 | WBGene00000090 | age-1 phosphatidylinositol 3-kinase | 3e-09 | (30–128)/497; (1113–1145)/1146 | 85% |
| EX909983 | WBGene00000903 | daf-7 transforming growth factor beta proteins | 1e-39 | (9–491)/643; (190–350)/350 | 65% |
| FF678376 | WBGene00006527 | tax-6 calcineurin A | 3e-56 | (2–334)/348; (358–468)/597 | 95% |
| FF681873 | WBGene00003965 | pdk-1 3-phosphoinositide-dependent kinase 1 | 5e-22 | (9–221)/244; (517–587)/636 | 79% |
| Dauer entry | |||||
| Contig710 | WBGene00004789 | sgk-1 serine/threonine protein kinase | 1e-103 | (1–720)/1010; (220–459)/463 | 84% |
| EX913794 | WBGene00004789 | sgk-1 serine/threonine protein kinase | 6e-68 | (20–664)/664; (17–231)/463 | 74% |
| ES739212 | WBGene00000102 | akt-1 serine/threonine kinase | 5e-60 | (128–526)/630; (1–132)/546 | 89% |
| Dauer exit | |||||
| Contig933 | WBGene00003831 | nuo-1 NADH-ubiquinone oxidoreductase | 1e-108 | (9–659)/676; (1–219)/479 | 90% |
| EX007842 | WBGene00003831 | nuo-1 NADH-ubiquinone oxidoreductase | 7e-87 | (3–488)/645; (317–478)/479 | 97% |
a. The alignment is presented in the following format: (H. bacteriophora EST start coordinate – H. bacteriophora EST end coordinate)/H. bacteriophora length; (C. elegans start coordinate – C. elegans end coordinate)/C. elegans length.
Identification of parasitic nematode-specific ESTs
In order to identify parasitic nematode-specific ESTs, a comparison of H. bacteriophora ESTs to all nematode EST sequences from GenBank (see additional file 3: Nematode species that ESTs used in the analysis came from) was performed. The nematode taxa having ESTs were divided into animal- and human-parasitic nematodes (AHPNs), plant-parasitic nematodes (PPNs), and free-living nematodes (FLNs) to enable a more informative comparison using tBLASTx algorithm with an E value cutoff of 1e-5 (Figure 5A). Of the 10,886 H. bacteriophora ESTs, 2,523 had no matches to nematode ESTs (Figure 5B) of which 2,371 had no matches to proteins but 152 had matches to proteins of other organisms. There were 351 H. bacteriophora ESTs matching to ESTs of FLNs only, which encoded proteins potentially involved in processes shared by FLNs and entomopathogenic nematodes, such as dauer formation and response to environmental stresses. There were 540 H. bacteriophora ESTs matching only to ESTs of AHPNs, 43 matching only to ESTs of PPNs, and 105 matching only to ESTs of AHPNs and PPNs. Therefore, there were collectively 688 H. bacteriophora ESTs not matching to any of the ESTs of FLNs. When these 688 H. bacteriophora ESTs were searched against GenBank's nr database using BLASTx algorithm we found that 554 had no matches to proteins of FLNs. These 554 H. bacteriophora ESTs were designated as parasitic nematode-specific ESTs and listed in the additional file 4: Summary of BLASTx identification of parasitic nematode-specific H. bacteriophora ESTs. Among these 554 ESTs, 476 (86%) matched to ESTs from clade V parasitic nematodes and the remaining 78 ESTs (14%) matched to ESTs from parasitic nematodes in other clades. Among these parasitic nematode-specific ESTs, 142 had matches to proteins of other organisms, enabling putative function assignment. Among these ESTs are those encoding F-box-like/WD-repeat protein, theromacin, Bax inhibitor-1-like protein, and PAZ domain containing protein, which represent interesting targets for in-depth functional analysis. The remaining 412 had no matches to any protein in the current databases, and are thus considered novel sequences.
Figure 5.
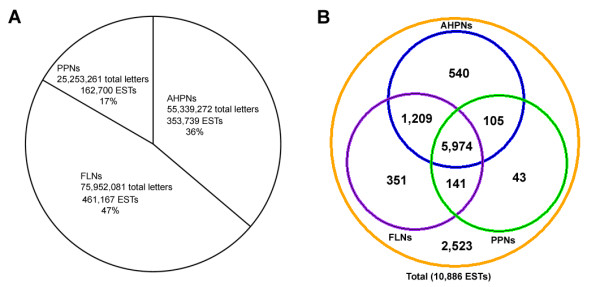
Summary of comparison between H. bacteriophora distinct ESTs and other ESTs from other nematode groups. (A) Summary of EST databases build from publicly-available animal- and human-parasitic nematodes (AHPNs), plant-parasitic nematodes (PPNs), and free-living nematodes (FLNs). (B) Results of EST comparison with numbers of matches to the ESTs of different categories. Numbers in brown circle are the total numbers of H. bacteriophora distinct ESTs. Numbers in green circle are the numbers of H. bacteriophora distinct ESTs matching to ESTs from PPNs. Numbers in purple circle are the numbers of H. bacteriophora ESTs matching to ESTs from FLNs. Numbers in blue circle are the numbers of H. bacteriophora ESTs matching to ESTs from AHPNs. All matches were tBLASTx hits better than E value of 1e-5.
Gene ontology annotation
Gene Ontology (GO) terms were assigned to 6,685 distinct ESTs with BLASTx search against the April 2008 release of GO database (see additional file 5: Summary of GO assignment of H. bacteriophora distinct ESTs). The GO assignment included 1,117 Biological Process terms assigned 13,438 times to 4,653 distinct ESTs, 244 Cellular Component terms assigned 2,454 times to 1,778 distinct ESTs, and 669 Molecular Function terms assigned 4,035 times to 3,190 distinct ESTs. "Embryonic development ending in birth or egg hatching" (40.9%) was the most dominant term out of the 4,653 distinct ESTs assigned to Biological Process GO category, followed by "nematode larval development" (28.3%), "positive regulation of growth rate" (27.6%), "reproduction" (24.7%), and "growth" (23,6%). Biological Process term associations of H. bacteriophora distinct ESTs that may present potential interests included: (i) 164 with "determination of adult life span"; (ii) 22 with "defense response"; (iii) 22 with dauer-related biological processes, including "dauer larval development", "dauer entry", and "dauer exit"; and (iv) 37 with stress-related biological processes. Protein binding (53.8%) was the most dominant term among the 3,190 H. bacteriophora ESTs annotated to the Molecular Function category, followed by identical protein binding (2.6%) and cytochrome-c oxidase activity (2.5%). Among the 1,778 H. bacteriophora ESTs annotated to Cellular Component category, 221 were nuclear and 218 were in cytoplasm. The GO assignment for each H. bacteriophora EST is given in additional file 4: Summary of GO assignment of H. bacteriophora distinct ESTs.
Microsatellite-containing ESTs
In total, we identified 168 microsatellite loci from 157 H. bacteriophora distinct EST sequences. Among these 157 H. bacteriophora ESTs, 77 had no matches to proteins in GenBank's nr database. The identified microsatellites were di-nucleotide (39.4%), tri-nucleotide (46.3%), tetra-nucleotide (2.7%), penta-nucleotide (0.5%), and hexa-nucleotide (0.5%) (Table 5). Among the 168 microsatellite loci, 141 had good flanking sequences for primer design while the remaining 27 had either short flanking sequences or the flanking sequences had too low GC contents for primer design (see additional file 6: Summary of microsatellite loci identified in H. bacteriophora distinct ESTs). The primers designed for the 141 microsatellite loci are potentially useful for genetic linkage mapping and population genetic studies.
Table 5.
Summary of microsatellite loci identified from H. bacteriophora distinct ESTs
| Number of repeatsa | Di-nt unit | Tri-nt unit | Tetra-nt unit | Penta-nt unit | Hexa-nt unit |
| 5 | 0 | 55 | 4 | 1 | 0 |
| 6 | 29 | 11 | 1 | 0 | 0 |
| 7 | 16 | 11 | 0 | 0 | 0 |
| 8 | 10 | 6 | 0 | 0 | 0 |
| 9 | 7 | 1 | 0 | 0 | 1 |
| 10 | 2 | 2 | 0 | 0 | 0 |
| 11 | 6 | 0 | 0 | 0 | 0 |
| 12 | 1 | 0 | 0 | 0 | 0 |
| 14 | 1 | 0 | 0 | 0 | 0 |
| 15 | 0 | 1 | 0 | 0 | 0 |
| 16 | 1 | 0 | 0 | 0 | 0 |
| : : |
|||||
| 31 | 1 | 0 | 0 | 0 | 0 |
| Sub-total | 74 | 87 | 5 | 1 | 1 |
| Percentage | 39.4% | 46.3% | 2.7% | 0.5% | 0.5% |
a. The numbers omitted from this column indicate that there were no microsatellite loci containing such repeats.
Discussion
This work produced a total of 31,485 high quality ESTs representing 10,886 distinct sequences. Sequence similarity searches of H. bacteriophora distinct ESTs showed 71.9% (7,828) matches to proteins from GenBank's nr database. The remaining 28.1% H. bacteriophora distinct ESTs represented novel genes yet to be assigned a function, demonstrating enormous novel gene discovery potential of this EST study. Among H. bacteriophora distinct ESTs having matches to proteins of other organisms in GenBank's nr database, a vast majority (95.9%) matched nematode proteins. About 71% (7,699) H. bacteriophora distinct ESTs match to 4,460 proteins from Wormpep190 that contains 23,771 extensively curated C. elegans proteins. H. bacteriophora homologs in C. elegans represent 18.8% of proteins in C. elegans. This finding suggests that H. bacteriophora and C. elegans have vastly different in proteomes, which may be explained in part by free-living versus parasitic life styles.
Interestingly, 26 distinct ESTs (0.3%) matched to proteins from various prokaryotic organisms (Table 2), all of which had less than 100% local sequence identities to prokaryotic sequences. These transcripts could result from horizontal gene transfer from bacteria encountered by H. bacteriophora during its life cycle. None of these ESTs matched to genes or proteins of P. luminescens subsp. laumondii TTO1, the natural symbiont of H. bacteriophora TTO1 [11]. Given the fact that poly(A) RNA was used in EST sequencing and the prokaryotic sequences were less than 100% identical to known prokaryotes, the possibility that these sequences are contaminants from other bacteria is low, although the possibility cannot be ruled out completely. The identification of sequences of putative prokaryotic origin in H. bacteriophora ESTs are consistent with our previous observations [6] and those observed in plant parasitic nematodes [14]. The putative prokaryotic origin of these sequences could be tested more rigorously once the complete genome sequence becomes available.
Sequence similarity searches of H. bacteriophora distinct ESTs against ESTs of other nematodes and proteins revealed the presence of 554 parasitic nematode-specific ESTs (Additional file 3). Eighty-six percent of these ESTs matched ESTs from parasitic nematodes in clade V. Taken into consideration the fact that most FLNs included in this study were also from clade V, we are confident that these ESTs reflect the differences between parasitic and free-living nematodes and are not the result of phylogenetic constraint. These 554 H. bacteriophora ESTs had sequence similarities with ESTs from other parasitic nematodes, suggesting that these genes may participate in parasitism-related activities. Although the 2,523 ESTs without matches to any nematode ESTs could be H. bacteriophora specific, we hesitate to consider them as parasitic nematode specific at this time because they lack sequence similarity to ESTs of parasitic nematodes. Among these, the 2,371 ESTs without matches to any proteins in the current GenBank nr database represent potentially novel H. bacteriophora genes, and 81 of these were identified in the EST dataset of H. bacteriophora GPS11 strain [3,6]. These findings suggest the enormous potential of discovering new genes and gene functions, genetic networks, and metabolic pathways specific to H. bacteriophora and other entomopathogenic nematodes. The identification of H. bacteriophora ESTs shared with other parasitic nematodes through our EST comparison opens the door for conducting in-depth research on gene functions that will ultimately elucidate the parasitic nematode-specific biological processes.
Among the 554 parasitic nematode-specific ESTs are those encoding F-box-like/WD-repeat protein, theromacin, Bax inhibitor-1-like protein, and PAZ domain containing protein. EST FF678238 encodes a homolog of the F-box-like/ED-repeat protein in Brugia malayi [10]. The WD-repeat is commonly associated with F-box domain that mediates protein-protein interactions in a variety of contexts such as polyubiquitination [15]. EST FF678397 encodes a protein similar to the PAZ domain containing protein in Brugia malayi [10]. The PAZ (Piwi, Argonaut and Zwille) domain has nucleic acid-binding capability and is potentially involved in post-transcriptional gene silencing [16]. Further investigation is needed to elucidate the functions of these two ESTs with common domains and whether they are related to parasitic nematode-specific processes.
Two other parasitic nematode-specific ESTs are Contig2528 and Contig1066. Contig2528 encodes a homolog of theromacin in the segmented worm Theromyzon tessulatum [17]. Theromacin is a novel antimicrobial peptide acting against Gram-positive bacteria but without any similarities to other known antimicrobial peptides [17]. H. bacteriophora TTO1 is obligately symbiotic with Photorhabdus luminescens subsp. laumondii TTO1 in natural environments. The production of an antimicrobial peptide could help establish the symbiotic relationship by selectively eliminating competing microbes. It is also possible that the antimicrobial peptide is a defense mechanism against potentially harmful microbes in the environment. Contig1066 encodes a protein similar to the uncharacterized protein family UPF0005 containing protein in Brugia malayi (GenBank accession number XP_001896958 http://www.ncbi.nlm.nih.gov/protein/170584338) and BAX inhibitor-1-like protein in wasp Nasonia vitripennis (GenBank accession number XP_001605379 http://www.ncbi.nlm.nih.gov/protein/156542785). BAX inhibitor-1 is a member of Bcl-2 family that suppresses programmed cell death [18]. Transmembrane BAX inhibitor motif protein (TMBI) homologs have been identified in C. elegans, C. briggsae, C. japonica, C. remanei, and Pristioncus pacifiucs (Wormbase). However, these genes have very low sequence similarities to H. bacteriophora Contig1066. BAX inhibitor-1 is involved in preventing endoplasmic reticulum stress-related programmed cell death in Arabidopsis [19] and humans [20].
GO assignments based on sequence similarity searches aid identification of H. bacteriophora distinct ESTs involved in different biological processes. Here we discuss the genes involved in some biological processes of interest in detail. A number of ESTs related to defense responses and stress responses were identified in these H. bacteriophora distinct ESTs (Additional File 2) based on GO assignments. Among H. bacteriophora distinct ESTs involved in defense response are 3 ESTs encoding a homolog of C. elegans SMEK (Dictyostelium suppressor of MEK null) homolog that is essential for DAF-16-mediated defense response to pathogenic bacteria and increased resistance to oxidative and UV-induced damage [21]. EST ES410098 encodes a heat shock protein HSP16-1 that is induced solely in response to heat shock or other environmental stresses [22]. Another 13 ESTs encoding 5 different proteins whose C. elegans homologs exhibit a "pathogen susceptibility increased" phenotype when silenced by RNAi [23]. However, the molecular functions of these proteins have yet to be elucidated. The defense response transcripts may be involved in the protection of entomopathogenic nematode IJs from bacterial or fungal pathogens and the insect innate immune system.
Five H. bacteriophora distinct ESTs involved in stress response encode a homolog to C. elegans catalase CTL-2 that likely is involved in protecting cells from reactive oxygen species as an antioxidant enzyme [24]. Another EST (ES742296) encodes a protein whose C. elegans homolog showed an "organism stress response abnormal" phenotype when silenced with RNAi [25]. Functions of other ESTs involved in stress response are yet to be clearly characterized. These transcripts related to stress responses provide workable targets for the improvement of ultraviolet, desiccation, and heat stress tolerance, traits desperately sought for improving the biological pest control potential of H. bacteriophora. Once the functions of these genes are determined, they can be potentially used for genetic manipulations of entomopathogenic nematodes. ESTs involved in dauer larval development, dauer entry, and dauer exit were also identified (Table 4) according to GO assignments. The infective juvenile stage of entomopathogenic nematodes is developmentally similar to the dauer stage in many bacteria feeding nematodes, including C. elegans and C. briggsae. The dauer is a developmentally arrested stage triggered by food deprivation, high population density, and other harsh environmental conditions [26]. Elucidation of this process is of specific interest in the case of entomopathogenic nematodes because the dauer juvenile is the only life stage capable of infecting insects [4].
RNA interference represents a powerful technique for analysis of gene function. An RNAi system relying on soaking in double-stranded RNA solution has been established in H. bacteriophora [7]. Interestingly, we were able to identify only a small number of known RNAi related genes in H. bacteriophora (Table 3) compared to C. elegans and B. malayi [10].
We have identified genes encoding RNAi induced silencing complex (RISC) components. One EST (EX014403) encodes a homolog of C. elegans TSN-1 (71% similarity at the amino acid level) and another EST (Contig211) encodes a homolog of C. elegans VIG-1 (45% similarity at the amino acid level). TSN-1 (Tudor staphylococcal nuclease) containing 5 staphylococcal/micrococcal nuclease domains and a tudor domain is a RISC component in C. elegans, Drosophila and mammals [27]. The purified TSN-1 from C. elegans was shown to have nuclease activity and therefore thought to contribute to RNA degradation in RNAi [27]. The product of the vig-1 gene was also shown to be a component of RISC [28]. We did not identify a member of Argonaute family in this EST set based on sequence similarity. However, we identified an EST (FF678397) encoding a protein similar to a PAZ domain containing protein from Brugia malayi (57% similarity at the amino acid level). Another EST (FF679415) encodes a putative homolog to Drosophila and human Drosha [29] rather than Dicer in C. elegans. These findings suggest that H. bacteriophora may have structurally different RNAi pathway components than its relative, C. elegans.
Other RNAi related genes we were able to identify are those encoding homologs of SMG-2, SMG-5, RDE-4, GFL-1, and ZFP-1. SMG-2 and SMG-5 are involved in nonsense-medicated mRNA decay (NMD) where eukaryotic mRNAs with premature stop codons are selectively and rapidly degraded [30,31]. The other three genes, rde-4 [32], gfl-1 and zfp-1 [33] were shown to be involved in RNAi via RNAi evidence. We currently are not able to identify a gene encoding a SID-1 homolog in H. bacteriophora TTO1 that was shown to be necessary for systemic RNAi [34]. However, a sid-1 gene has been found in H. bacteriophora GPS11 [3]. It is possible that more known genes may be identified when the complete genome of H. bacteriophora TTO1 is sequenced.
This EST project also enabled the development of genetic markers. We have identified 168 microsatellite loci from H. bacteriophora distinct ESTs, of which we were able to design primers for 141 based on the flanking sequences. These microsatellite markers may be useful for genetic mapping, linkage analysis, and population genetic studies. In a separate effort, microsatellite loci with 2- or 3-bp repeat units were selected for microsatellite marker development, along with the microsatellite loci enriched from genomic DNA of H. bacteriophora [35]. Eight polymorphic microsatellite loci were demonstrated within a Northeast Ohio population.
Conclusion
We have generated 31,485 high quality H. bacteriophora ESTs representing 10,886 distinct sequences. Among these, 7,828 (71.9%) ESTs matched to proteins in GenBank's nr database. The vast majority (95.9%) of the best matches was to nematode proteins, a small portion (0.3%) to prokaryotic proteins and the remaining 3.8% to other eukaryotic proteins. GO terms were assigned to 6,685 H. bacteriophora distinct ESTs. "Embryonic development ending in birth or egg hatching" and "protein binding" were the most dominant terms in the categories of Biological Process and Molecular Function, respectively. This EST collection offers unprecedented opportunities for research on this unique nematode-bacterium symbiotic complex. The comparison of ESTs of H. bacteriophora TTO1 with those of AHPNs, FLNs, and PPNs resulted in the identification of 554 parasitic nematode-specific ESTs. These ESTs should be valuable for future research related to insect parasitism by these nematodes. We were able to identify a small number of ESTs involved in RNAi, among which is an EST encoding a Drosha homolog, suggesting structurally different RNAi pathway components from those in C. elegans. In addition, we have identified 157 microsatellite loci which may prove valuable once their polymorphisms are tested and validated. Overall, novel, parasitic nematode-specific, and C. elegans homologous genes have been identified in this EST study, greatly facilitating genome annotation, gene functional analysis, population genetic studies, and microarray development.
Methods
RNA isolation, cDNA library construction, and sequencing
Total RNA and poly(A) RNA were isolated from adult hermaphrodites of the isogenic line of Heterorhabditis bacteriophora TTO1-M31e strain propagated on a lawn of Photorhabdus luminescens bacterium. Poly(A) RNA was used for cDNA library construction with two different strategies. The first group of libraries were constructed using the CloneMiner™ cDNA Library Construction Kit (Invitrogen) following the manufacturer's instructions. Briefly, 2 μg single-stranded mRNA was converted into double stranded cDNA containing attB sequences on each end. Through site-specific recombination, attB-flanked cDNA was cloned into the attP-containing pDONR222 vector. The second group of libraries were constructed using SMART technology with modifications. Briefly, the double stranded cDNA was synthesized with SMART oligos from poly(A) RNA with SuperScript® III First-Strand Synthesis System (Invitrogen) and Advantage® High Fidelity 2 PCR kit (Clontech). The double-stranded cDNA was normalized with duplex-specific nuclease (Evrogen) and then was nebulized, end repaired with End Repair Kit (Lucigen), size separated, and ligated into pSMART hinc II Vector System (Lucigen). The cloning and sequencing of both pDONR222 and pSMART libraries were not directional, leading to the production of ESTs from both 5' and 3' ends. The sequences were generated by ABI 3730 machines from the cDNA libraries using and deposited in GenBank dbEST.
Contig assembly and analysis
EST sequences in FASTA format were downloaded from GenBank dbEST. The sequences were processed by removing vector sequences with Vector NTI Advance™ 10 program. The processed EST sequences were assembled into contigs (contiguous sequences) using the ContigExpress module embedded in Vector NTI Advance™ 10 (Invitrogen). These stringent parameters of assembly (overlap length cutoff of 40 and overlap percent identity cutoff of 95%) were used to assure proper assembly. The distinct EST sequences, including the contig consensus sequences and the singleton sequences, were searched against GenBank's nr database and Wormpep190 databases in a local Linux workstation using the BLASTx algorithm [36]. The E value cutoff of the BLASTx searches was 1e-5.
EST comparison
All nematode EST sequences were downloaded from GenBank dbEST to a local Linux workstation and formatted as a database for tBLASTx searches. Gene index (GI) numbers of all nematode EST sequences were extracted and grouped according to the categories of AHPNs, PPNs, and FLNs. The tBLASTx searches were performed in a local Linux workstation against the complete nematode EST database with the -l option enabled to restrict the database search to the list of GI's of the targeted group [7]. For example, when comparing H. bacteriophora ESTs to ESTs of FLNs, "-l fln.gi" was included in the command with fln.gi containing all GI numbers of EST entries from free-living nematodes. The BLAST outputs were parsed with in-house developed perl scripts to extract match information. ESTs with no significant matches to ESTs of FLNs were extracted and further searched against GenBank nr and Wormpep190 databases using BLASTx algorithm. EST entries with no significant matches to proteins of FLNs were designated parasitic nematode-specific ESTs, which were further characterized. The cutoff value of BLAST searches was 1e-5.
Gene Ontology annotation
For assignment of Gene Ontology terms, the distinct H. bacteriophora ESTs were searched using the BLASTx algorithm against the annotated sequences of FASTA format in the April 2008 release of GO database. The BLAST output was parsed and terms assigned with the assistance of in-house developed perl scripts accessing the MySQL database of "mygo" in a local Linux workstation. The distribution of GO terms in each of the main ontology categories, Biological Process, Cellular Component, and Molecular Function [37], was examined. The number of H. bacteriophora distinct ESTs assigned in a single GO category was considered as 100% [12,38].
Bioinformatics mining of microsatellite loci
The set of 10,886 H. bacteriophora distinct ESTs were searched for microsatellite loci using msatfinder v. 2.0.9 [39] in a local Linux workstation. The cut-off values of number of repeats were set to 6 for di-nucleotide loci and 5 for tri-, tetra-, penta-, and hexa-nucleotide loci. Primers were designed using Primer3 release 1.0 [40] in a local Linux workstation.
Authors' contributions
XB conducted vector sequence removal, assembly and analysis of EST sequences and prepared the manuscript. TAC provided the poly(A) RNA for cDNA library construction. RKW, SC, and JS led groups at the Genome Center at Washington University School of Medicine in St. Louis, MO that conducted cDNA library construction, sequencing, and data deposition. PSG, BJA, TAC, SC, RG, SAH, JS, and PWS initiated the study and obtained funding. PSG supervised the study and assisted in manuscript preparation. All authors read, edited, and approved the final manuscript.
Supplementary Material
ContigComponents. Components of H. bacteriophora assembled contigs. The table includes the components of H. bacteriophora assembled contigs, including contig names and their comma-delimited components in GenBank accession numbers.
defense_stress_response_genes. H. bacteriophora distinct ESTs similar to C. elegans genes involved in defense and stress responses. This table contains H. bacteriophora distinct ESTs matching to C. elegans genes that are involved in defense and stress responses. Information of each H. bacteriophora distinct EST includes matching C. elegans gene, its description, E value of the BLASTx search, alignment, and percentage similarity.
NematodeTaxa. Nematode species that ESTs used in the analysis came from. The table includes the nematode species in the categories of AHPNs, FLNs, and PPNs, the clade they belong to, and their corresponding number of ESTs in GenBank when the analysis was done.
panESTs. Summary of BLASTx identification of parasitic nematode-specific H. bacteriophora ESTs. The table describes the best BLASTx matches to GenBank's nr database of the 554 parasitic nematode-specific H. bacteriophora ESTs, including distinct EST ID, length, and annotation of the top BLASTx hit, including accession number, length, description, E value, bit score, frame, query_start, query_end, hit_start, hit_end, positives, and identities.
GOassignment. Summary of GO assignment of H. bacteriophora distinct ESTs. The table describes the GO assignment for each of 10,886 H. bacteriophora distinct ESTs, of which 4,201 have "no GO assignment". The GO terms separated by semicolons include accession numbers, term descriptions, and term types.
msat. Summary of microsatellite loci identified in H. bacteriophora distinct ESTs. The table describes the microsatellite loci identified in H. bacteriophora distinct ESTs, including the ID and length of H. bacteriophora distinct EST ID, the start and stop coordinates and motif units of the microsatellite loci, GC contents of the distinct ESTs and the flanking sequences, primer sequence, length, and Tm of the left and right primers, and the size of the amplification product from these primers. The asterisk (*) marking H. bacteriophora distinct EST IDs denotes that the primers designed for these loci amplify other adjacent loci too.
Acknowledgments
Acknowledgements
This project was supported by grants from the United States Department of Agriculture (USDA)/National Science Foundation (NSF) Microbial Genome Sequence Program and National Human Genome Research Institute (NHGRI) awarded to PSG, BJA, TAC, RG, and PWS. We thank teams of scientists led by Lucinda Fulton, Makedonka Mitreva, Kimberly Delehaunty, Michael Becker, and Brenda Theising at the Genome Center at Washington University School of Medicine in St. Louis, MO who produced and curated EST data.
Contributor Information
Xiaodong Bai, Email: bai.29@osu.edu.
Byron J Adams, Email: bjadams@byu.edu.
Todd A Ciche, Email: ciche@msu.edu.
Sandra Clifton, Email: sclifton@watson.wustl.edu.
Randy Gaugler, Email: gaugler@rci.rutgers.edu.
Saskia A Hogenhout, Email: Saskia.Hogenhout@bbsrc.ac.uk.
John Spieth, Email: jspieth@watson.wustl.edu.
Paul W Sternberg, Email: pws@caltech.edu.
Richard K Wilson, Email: rwilson@watson.wustl.edu.
Parwinder S Grewal, Email: grewal.4@osu.edu.
References
- Grewal PS, Ehlers RU, Shapiro-Ilan DI. Nematodes as Biocontrol Agents. Wallingford, UK: CABI Publishing; 2005. [Google Scholar]
- Ciche T. The biology and genome of Heterorhabditis bacteriophora (February 20, 2007) WormBook, ed. The C. elegans Research Community, WormBook; 2007. http://www.wormbook.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu SK, Jagdale GB, Hogenhout SA, Grewal PS. Comparative analysis of the expressed genome of the infective juvenile entomopathogenic nematode Heterorhabditis bacteriophora. Mol Biochem Parasitol. 2006;145:239–244. doi: 10.1016/j.molbiopara.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Grewal PS, Lewis EE, Gaugler R. Response of infective stage parasites (Nematoda: Steinernematidae) to volatile cues from infected hosts. J Chem Ecol. 1997;23:503–515. [Google Scholar]
- Ciche TA, Ensign JC. For the insect pathogen Photorhabdus luminescens, which end of a nematode is out? Appl Environ Microbiol. 2003;69:1890–1897. doi: 10.1128/AEM.69.4.1890-1897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Grewal PS, Hogenhout SA, Adams BJ, Ciche TA, Gaugler R, Sternberg PW. Expressed sequence tag analysis of gene representation in insect parasitic nematode Heterorhabditis bacteriophora. J Parasitol. 2007;93:1343–1349. doi: 10.1645/GE-1246.1. [DOI] [PubMed] [Google Scholar]
- Ciche TA, Sternberg PW. Postembryonic RNAi in Heterorhabditis bacteriophora: a nematode insect parasite and host for insect pathogenic symbionts. BMC Dev Biol. 2007;7:101. doi: 10.1186/1471-213X-7-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans sequencing consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003;1:E45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra D, et al. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317:1756–1760. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaud E, Rusniok C, Frangeul L, Buchrieser C, Givaudan A, Taourit S, Bocs S, Boursaux-Eude C, Chandler M, Charles JF, et al. The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat Biotechnol. 2003;21:1307–1313. doi: 10.1038/nbt886. [DOI] [PubMed] [Google Scholar]
- Quilang J, Wang S, Li P, Abernathy J, Peatman E, Wang Y, Wang L, Shi Y, Wallace R, Guo X, et al. Generation and analysis of ESTs from the eastern oyster, Crassostrea virginica Gmelin and identification of microsatellite and SNP markers. BMC Genomics. 2007;8:157. doi: 10.1186/1471-2164-8-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers A, Antoshechkin I, Bieri T, Blasiar D, Bastiani C, Canaran P, Chan J, Chen WJ, Davis P, Fernandes J, et al. WormBase 2007. Nucleic Acids Res. 2008:D612–617. doi: 10.1093/nar/gkm975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl EH, Thorne JL, McCarter JP, Bird DM. Horizontally transferred genes in plant-parasitic nematodes: a high-throughput genomic approach. Genome Biol. 2003;4:R39. doi: 10.1186/gb-2003-4-6-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Yan KS, Yan S, Farooq A, Han A, Zeng L, Zhou MM. Structure and conserved RNA binding of the PAZ domain. Nature. 2003;426:468–74. doi: 10.1038/nature02129. [DOI] [PubMed] [Google Scholar]
- Tasiemski A, Vandenbulcke F, Mitta G, Lemoine J, Lefebvre C, Sautiere PE, Salzet M. Molecular characterization of two novel antibacterial peptides inducible upon bacterial challenge in an annelid, the leech Theromyzon tessulatum. J Biol Chem. 2004;279:30973–30982. doi: 10.1074/jbc.M312156200. [DOI] [PubMed] [Google Scholar]
- Huckelhoven R. BAX Inhibitor-1, an ancient cell death suppressor in animals and plants with prokaryotic relatives. Apoptosis. 2004;9:299–307. doi: 10.1023/b:appt.0000025806.71000.1c. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Lam E. BAX inhibitor-1 modulates endoplasmic reticulum stress-mediated programmed cell death in Arabidopsis. J Biol Chem. 2008;283:3200–3210. doi: 10.1074/jbc.M706659200. [DOI] [PubMed] [Google Scholar]
- Lee GH, Kim HK, Chae SW, Kim DS, Ha KC, Cuddy M, Kress C, Reed JC, Kim HR, Chae HJ. Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression. J Biol Chem. 2007;282:21618–21628. doi: 10.1074/jbc.M700053200. [DOI] [PubMed] [Google Scholar]
- Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell. 2006;124:1039–1053. doi: 10.1016/j.cell.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Jones D, Dixon DK, Graham RW, Candido EP. Differential regulation of closely related members of the hsp16 gene family in Caenorhabditis elegans. DNA. 1989;8:481–490. doi: 10.1089/dna.1.1989.8.481. [DOI] [PubMed] [Google Scholar]
- Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan MW. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci USA. 2006;103:14086–14091. doi: 10.1073/pnas.0603424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togo SH, Maebuchi M, Yokota S, Bun-Ya M, Kawahara A, Kamiryo T. Immunological detection of alkaline-diaminobenzidine-negativeperoxisomes of the nematode Caenorhabditis elegans purification and unique pH optima of peroxisomal catalase. Eur J Biochem. 2000;267:1307–1312. doi: 10.1046/j.1432-1327.2000.01091.x. [DOI] [PubMed] [Google Scholar]
- Cho JH, Ko KM, Singaravelu G, Ahnn J. Caenorhabditis elegans PMR1, a P-type calcium ATPase, is important for calcium/manganese homeostasis and oxidative stress response. FEBS Lett. 2005;579:778–782. doi: 10.1016/j.febslet.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Hu PJ. Dauer (August 08, 2007) WormBook, ed. The C. elegans Research Community, WormBook; 2007. http://www.wormbook.org [Google Scholar]
- Caudy AA, Ketting RF, Hammond SM, Denli AM, Bathoorn AM, Tops BB, Silva JM, Myers MM, Hannon GJ, Plasterk RH. A micrococcal nuclease homologue in RNAi effector complexes. Nature. 2003;425:411–414. doi: 10.1038/nature01956. [DOI] [PubMed] [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 2002;16:2491–2496. doi: 10.1101/gad.1025202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Kim JK, Gabel HW, Kamath RS, Tewari M, Pasquinelli A, Rual JF, Kennedy S, Dybbs M, Bertin N, Kaplan JM, et al. Functional genomic analysis of RNA interference in C. elegans. Science. 2005;308:1164–1167. doi: 10.1126/science.1109267. [DOI] [PubMed] [Google Scholar]
- Anders KR, Grimson A, Anderson P. SMG-5, required for C. elegans nonsense-mediated mRNA decay, associates with SMG-2 and protein phosphatase 2A. EMBO J. 2003;22:641–650. doi: 10.1093/emboj/cdg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Dudley NR, Labbe JC, Goldstein B. Using RNA interference to identify genes required for RNA interference. Proc Natl Acad Sci USA. 2002;99:4191–4196. doi: 10.1073/pnas.062605199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002;295:2456–2459. doi: 10.1126/science.1068836. [DOI] [PubMed] [Google Scholar]
- Bai X, Saeb ATM, Michel A, Grewal PS. Isolation and characterization of microsatellite loci in the entomopathogenic nematode Heterorhabditis bacteriophora. Mol Ecol Resources. 2009;9:207–209. doi: 10.1111/j.1755-0998.2008.02369.x. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Gonzalez FJ, Suarez MB, Redondo J, Heinrich J, Delgado-Jarana J, Hermosa R, Gutierrez S, Monte E, Llobell A, et al. Generation, annotation and analysis of ESTs from Trichoderma harzianum CECT 2413. BMC Genomics. 2006;7:193. doi: 10.1186/1471-2164-7-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston MI, Field D. Msatfinder: detection and characterisation of microsatellites. Distributed by the authors at CEH Oxford, Mansfield Road, Oxford OX1 3SR; 2005. http://www.genomics.ceh.ac.uk/msatfinder/ [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editor. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Vol. 132. Totowa, NJ: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ContigComponents. Components of H. bacteriophora assembled contigs. The table includes the components of H. bacteriophora assembled contigs, including contig names and their comma-delimited components in GenBank accession numbers.
defense_stress_response_genes. H. bacteriophora distinct ESTs similar to C. elegans genes involved in defense and stress responses. This table contains H. bacteriophora distinct ESTs matching to C. elegans genes that are involved in defense and stress responses. Information of each H. bacteriophora distinct EST includes matching C. elegans gene, its description, E value of the BLASTx search, alignment, and percentage similarity.
NematodeTaxa. Nematode species that ESTs used in the analysis came from. The table includes the nematode species in the categories of AHPNs, FLNs, and PPNs, the clade they belong to, and their corresponding number of ESTs in GenBank when the analysis was done.
panESTs. Summary of BLASTx identification of parasitic nematode-specific H. bacteriophora ESTs. The table describes the best BLASTx matches to GenBank's nr database of the 554 parasitic nematode-specific H. bacteriophora ESTs, including distinct EST ID, length, and annotation of the top BLASTx hit, including accession number, length, description, E value, bit score, frame, query_start, query_end, hit_start, hit_end, positives, and identities.
GOassignment. Summary of GO assignment of H. bacteriophora distinct ESTs. The table describes the GO assignment for each of 10,886 H. bacteriophora distinct ESTs, of which 4,201 have "no GO assignment". The GO terms separated by semicolons include accession numbers, term descriptions, and term types.
msat. Summary of microsatellite loci identified in H. bacteriophora distinct ESTs. The table describes the microsatellite loci identified in H. bacteriophora distinct ESTs, including the ID and length of H. bacteriophora distinct EST ID, the start and stop coordinates and motif units of the microsatellite loci, GC contents of the distinct ESTs and the flanking sequences, primer sequence, length, and Tm of the left and right primers, and the size of the amplification product from these primers. The asterisk (*) marking H. bacteriophora distinct EST IDs denotes that the primers designed for these loci amplify other adjacent loci too.