

Abstract
The biological activity of connective tissue growth factor (CTGF, CCN2) is regulated at the level of intracellular signaling leading to gene expression, and by its extracellular interaction partners which determine the functional outcome of CCN2 action. In this overview, we summarize the data which provide evidence that one of the major signaling pathways, phosphatidylinositol-3 kinase (PI3K)–AKT signaling, shows a remarkable cell type-dependence in terms of regulation of CCN2 expression. In smooth muscle cells, fibroblasts, and epithelial cells, inhibition of this pathway either reduced CCN2 expression or was not involved in CCN2 gene expression depending on the stimulus used. In microvascular endothelial cells by contrast, activation of PI3K–AKT signaling was inversely related to CCN2 expression. Upregulation of CCN2 upon inhibition of PI3K–AKT was also observed in primary cultures of human endothelial cells (HUVEC) exposed to laminar flow in an in vitro flow-through system. In different types of endothelial cells, FoxO transcription factors, which are negatively regulated by AKT, were identified as potent activators of CCN2 gene expression. In HUVEC, we observed a correlation between enhanced nuclear localization of FoxO1 and increased synthesis of CCN2 protein in areas of non-uniform shear stress. These data indicate that FoxO proteins are key regulators of CCN2 gene expression which determine the effect of PI3K–AKT activation in terms of CCN2 regulation. Short summary Phosphatidylinositol-3 kinase (PI3K)–AKT signaling shows a remarkable cell type-dependence in terms of regulation of CCN2 expression. In endothelial cells activation of PI3K - AKT signaling was inversely related to CCN2 expression. FoxO transcription factors, which are negatively regulated by AKT, were identified as potent activators of CCN2 gene expression.
Electronic supplementary material
The online version of this article (doi:10.1007/s12079-009-0055-5) contains supplementary material, which is available to authorized users.
Keywords: AKT, Connective tissue growth factor, Phosphatidylinositol-3 kinase, FoxO, Endothelial cell
Introduction
Connective tissue growth factor (CTGF, CCN2) is a multifunctional protein, which is secreted and functions as a matricellular protein. Accordingly, the biological activity of CCN2 is regulated both at the level of intracellular signaling leading to gene expression, and by its extracellular interaction partners, which determine the functional outcome of CCN2 action. Multiple signaling pathways have been related to CCN2 gene expression, some of which, such as transforming growth factor (TGF)beta—Smad signaling, seem to work in different cell types, or are shared by multiple stimuli, such as activation of RhoA Rho-kinase signaling (Chaqour and Goppelt-Struebe 2006; Cicha & Goppelt-Struebe 2009). Other pathways leading to CCN2 induction seem to be rather specifically detected in a restricted number of cell types, for example, activation of Rac-1 has only been related to CCN2 expression in gingival fibroblasts and chondrocytes (Black and Trackman 2008; Woods et al. 2009). In this brief overview, we will concentrate on the impact of the activation of one of the major signaling pathways, which is operative in all cells, namely phosphatidylinositol-3 kinase (PI3K)–AKT signaling on CCN2 gene expression. We will summarize the published data and present evidence for cell type-specific regulation of CCN2 by PI3K–AKT signaling.
Activation of AKT may lead to the modulation of multiple pathways, giving rise to a highly complex signaling network (e.g. (Jiang and Liu 2008; Chaqour et al. 2006)). In smooth muscle cells either exposed to mechanical stretch (Chaqour et al. 2006) or activated by serum or sphingosine-1 phosphate (Chowdhury and Chaqour 2004), PI3K–AKT signaling contributed to CCN2 promoter activation. By contrast, inhibition of PI3K did not interfere with TGFbeta-induced CTGF induction in lung fibroblasts (Xie et al. 2005). In line with these data, we did not observe a role of PI3K–AKT signaling for CCN2 expression in tubular epithelial cells stimulated with hepatocyte growth factor, which is known to activate AKT (Kroening et al. 2009). In experiments with renal fibroblasts stimulated with the microtubule disrupting agent colchicine, we observed that the response of the cells depended on the cell culture conditions: When the cells were cultured on a rigid surface, upregulation of CCN2 gene expression was independent of PI3K–AKT signaling, whereas inhibitors of PI3K significantly reduced CTGF gene expression, when the cells were cultured in collagen gels (Graness et al. 2006). This differential reactivity may be related to the link between focal adhesion kinase and PI3K, which plays a role in the survival of fibroblasts in collagen gels (Xia et al. 2004). Analyzing VEGF-dependent induction of CCN2, Suzuma et al. observed inhibition of CCN2 gene expression upon interference with AKT signaling in bovine retinal endothelial cells (Suzuma et al. 2000). However, the downstream mediators of AKT signaling were not investigated in this or in any of the other studies. In recent studies with different microvascular endothelial cell lines we demonstrated that PI3K–AKT may also negatively regulate CCN2 expression as discussed in detail below (Samarin et al. 2009). To extend these findings, we investigated the regulation of CCN2 expression in primary cultures of human umbilical vein endothelial cells (HUVEC) cultured under flow conditions which more closely resemble the in vivo conditions compared to static culture conditions.
Methods
HUVEC were isolated from freshly delivered umbilical cords and grown on 0.1% gelatin-coated dishes as described (Muehlich et al. 2004). HUVEC were seeded in flow-through cell culture slides (Ibidi®, Munich, Germany) and exposed to steady laminar shear stress for 20 h at 10 dyne/cm2 (Cicha et al. 2008). Bifurcating slides were used to analyze cells exposed to non-uniform shear stress. Protein expression was detected by indirect immunocytochemistry with the following antibodies: polyclonal goat anti-CTGF (1:100; Santa Cruz), anti-goat IgG coupled to Alexa Fluor 488 (1:500; Molecular Probes), polyclonal rabbit anti-FoxO1 (1:100, Cell Signaling) and anti-rabbit IgG coupled to Alexa Fluor 555 (1:500, Molecular Probes). Images were obtained using an inverted fluorescence microscope combined with a digital camera and Meta-Morph software. For quantification, MetaVue software was used as described in detail in (Cicha et al. 2008).
To downregulate FoxO1 and FoxO3a expression, HUVEC were transfected with equal concentrations (50 nM each) of FoxO1 and FoxO3a siRNA or luciferase siRNA (Eurogentec, Seraing, Belgium) using Lipofectamine 2000 (Invitrogen). The following siRNAs were used: FoxO1 sense GCG-GGC-UGG-AAG-AAU-UCA-A; FoxO3a sense GCU-CUU-GGU-GGA-UCA-UCA-A. 24 h after transfection, the cells were exposed to flow.
Results
CCN2 expression in HUVEC cultured under static conditions is very high, in contrast to the low expression detectable in the endothelial lining of healthy vessels. Therefore, CCN2 expression was analyzed in cells pre-exposed to physiologic levels of laminar flow (10 dyne/cm2). Compared to cells cultured under static conditions, CCN2 is downregulated under these conditions (Fig. 1 and (Cicha et al. 2008)). Following 18 h pre-conditioning with laminar flow, the cells were treated with a specific inhibitor of PI3K, LY294002 (10 µM), for additional 2 h at flow. Inhibition of PI3K–AKT signaling led to a robust upregulation of CCN2 protein expression (Fig. 1). CCN2 expression was detectable in a perinuclear Golgi localization, as observed earlier (Samarin et al. 2009). These data thus showed that negative regulation of CCN2 by PI3K–AKT signaling was not restricted to microvascular endothelial cells but also observed in primary cultures of HUVEC studied under physiological flow conditions.
Fig. 1.

Inhibition of PI3K increases CCN2 expression in HUVEC exposed to laminar shear stress. a HUVEC were seeded in flow-through cell culture slides and exposed to steady laminar shear stress for 20 h at 10 dyne/cm2. For the last 2 h of flow, PI3K–AKT signaling was inhibited by treatment with LY294002 (10 µM). Protein expression was determined by immunofluorescence. Photos are representative of two independent experiments performed in duplicate. b Data quantification: six to seven images at objective magnification 20× were taken for every experiment, and analyzed using MetaVue software. The thresholded protein expression levels were expressed as arbitrary fluorescence units. ***P < 0.001; t-test vs. untreated controls, n = 2 with duplicate cell culture slides
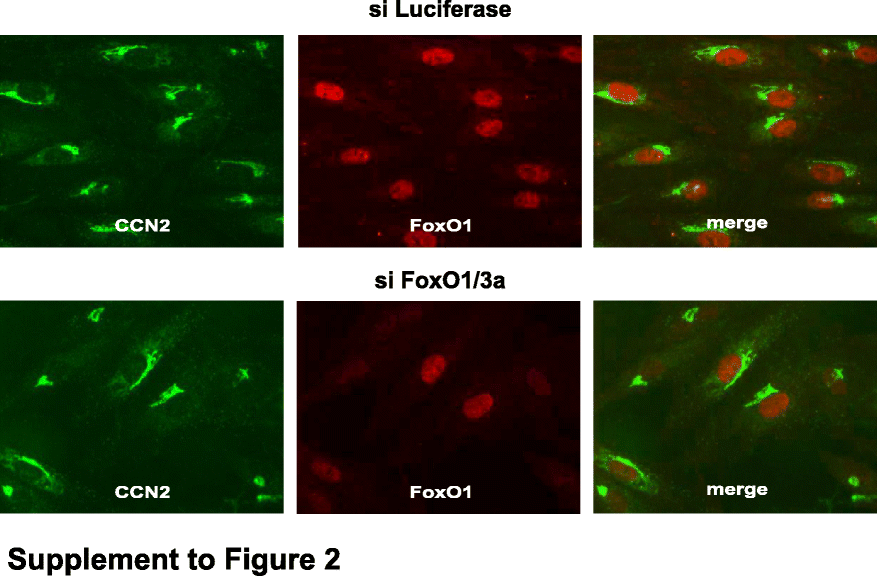
Transcription factors of the FoxO family are activated upon inhibition of PI3K–AKT signaling. Our previous data obtained in microvascular endothelial cells showed that these transcription factors are involved in the regulation of CCN2 expression (Samarin et al. 2009; Komorowsky et al. 2009). Therefore, we used an siRNA approach to analyze the role of FoxO transcription factors in HUVEC. FoxO1 was readily detectable in HUVEC by Western blot analysis and by immunocytochemistry (Fig. 2 and data not shown), whereas FoxO3a was barely detectable. However, it has been reported that these FoxO proteins can compensate one another (Potente et al. 2005). To avoid this potential compensating effect, we used a mixture of siRNAs directed against FoxO1 and FoxO3a.
Fig. 2.

FoxO1 is involved in CCN2 regulation in HUVEC. HUVEC were transfected with siRNA against luciferase or FoxO1 and FoxO3a 24 h after seeding. The next day the cells were exposed to laminar shear stress for 18 h and then treated with LY294002 (10 µM for 2 h). CCN2 and FoxO1 were detected by indirect immunofluorescence. Images showing nuclear FoxO1 expression were inverted. Arrows indicate cells with barely detectable FoxO1 expression and low levels of CCN2 expression. Color figures showing the merged images are provided as “Supplemental data”
Treatment of HUVEC exposed to laminar flow with the PI3K inhibitor LY294002 for 2 h led to nuclear localization of FoxO1 in all cells transfected with luciferase siRNA (Fig. 2, upper panel). In samples transfected with FoxO1/3a siRNA, nuclear localization of FoxO1 was only detectable in few cells (Fig. 2, lower panel). These cells expressed CCN2 at levels comparable to control HUVEC. By contrast, downregulation of FoxO1 by siRNA transfection was associated with a reduced expression of CCN2.
We have reported earlier that CCN2 is downregulated under laminar flow and upregulated when HUVEC are exposed to non-uniform shear stress (Cicha et al. 2008). Interestingly, FoxO1 was barely detectable in cells exposed to laminar shear stress, whereas it was localized to the nucleus in cells exposed to non-uniform shear stress (Fig. 3). Nuclear localization of FoxO1 correlated with an increased expression of CCN2 under in vivo-like hemodynamic conditions, suggesting a role of FoxO proteins in CCN2 regulation by non-uniform shear stress.
Fig. 3.

FoxO1 expression depends on flow conditions and is associated with CCN2 induction by non-uniform shear stress. HUVEC were seeded in bifurcating flow-through slides as described in detail in (Cicha et al. 2008). Cells were exposed to flow for 20 h. Photos were taken in areas of laminar shear stress (upper panel) or areas of non-uniform shear stress (lower panel). CCN2 and FoxO1 were detected by indirect immunofluorescence. Images showing nuclear FoxO1 expression were inverted. Color figures showing the merged images are provided as “Supplemental data”
Discussion
Regulation of CCN2 expression via PI3K–AKT signaling shows a remarkable cell type- and context-dependence. As outlined in the introduction, there is good evidence in the literature that activation of AKT is related to increased CCN2 expression. However, the molecular details of this activation remain to be defined. AKT activity is modulated by several binding partners which modulate signaling downstream of PI3K (Franke 2008). It is also unknown which of the multiple downstream mediators of AKT can actually lead to the increased CCN2 gene expression.
Our previously published data summarized in this report, provide evidence that PI3K–AKT negatively regulates CCN2 expression in endothelial cells. Upregulation rather than downregulation of CCN2 upon inhibition of PI3K–AKT signaling was observed in mouse microvascular endothelial cell lines obtained from heart or kidney (Samarin et al. 2009). This indicated that active AKT signaling prevented rather than simulated CCN2 gene expression in these cells. We now confirmed these observations in primary cultures of HUVEC exposed to laminar flow. Regarding the mechanisms of regulation, activated AKT can negatively modulate gene expression by recruiting FoxO transcription factors from the nucleus to the cytoplasm. FoxO transcription factors are important regulators of endothelial cell differentiation, angiogenesis, and maintenance of vascular homeostasis (Potente et al. 2005; Dejana et al. 2007). Interestingly, the function of FoxO proteins is highly context-specific and can differ even in endothelial cells obtained from different vascular beds (Paik et al. 2007), which may explain the apparently discrepant results obtained by Suzuma et al. (Suzuma et al. 2000). FoxO proteins localize to the cytosol upon phosphorylation, whereas dephosphorylated FoxO proteins localize to the nucleus and are active as transcription factors. Using an siRNA approach, we previously showed that these transcription factors play a central role in CCN2 induction in endothelial cells (Samarin et al. 2009; Komorowsky et al. 2009). Upregulation of CCN2 by inhibition of PI3K–AKT signaling was almost completely inhibited in the presence of siRNA against FoxO1 and FoxO3a in microvascular endothelial cells (Samarin et al. 2009) and, as shown in this study, in HUVEC. Furthermore, knock-down of FoxOs affected the induction of CCN2 by various stimuli, e.g. TGFbeta (Komorowsky et al. 2009) and hypoxia (unpublished observation).
FoxOs are not only phosphorylated by AKT but also by other kinases. AMP-activated protein kinase (AMPK) was shown to phosphorylate and thus inactivate FoxO1 in HUVEC exposed to laminar shear stress (Dixit et al. 2008). In line with these results FoxO1 was barely detectable in HUVEC exposed to laminar shear stress. However, nuclear localization was observed in HUVEC exposed to non-uniform shear stress. Whether this was related to an inhibition of AMPK remains to be investigated. Interestingly, there was a close correlation between nuclear FoxO1 localization and CCN2 expression, suggesting that the role of FoxOs in CCN2 regulation goes beyond PI3K–AKT signaling.
FoxO proteins as transcription factors have been shown to bind to coactivator and corepressor complexes. These interactions are modulated by the posttranscriptional modification state of FoxOs, which involves acetylation and ubiquitination besides phosphorylation (Vogt et al. 2005). There are several putative FoxO binding sites in the core promoter of CCN2 (Gomis et al. 2006), but the complexes formed with other transcripiton factors have not yet been analyzed in detail. The molecular details of FoxO-mediated upregulation of CCN2 thus remain to be investigated.
In accordance with our results, microarray data showed that upon overexpression of constitutively active AKT, CCN2 was downregulated, whereas active FoxO1 upregulated CCN2 in HUVEC (Daly et al. 2004). In human keratinocytes, CCN2 was identified as a member of a FoxO-Smad synergism group, i.e. proteins which were upregulated by TGFbeta in a Smad- and FoxO-dependent manner (Gomis et al. 2006). These data indicate that FoxO proteins may play an important role in the regulation of CCN2 in other cell types besides endothelial cells.
In summary, regulation of CCN2 expression via PI3K–AKT signaling varies depending on the cell type and the cell culture conditions. In endothelial cells, activation of PI3K–AKT suppresses CCN2 expression, whereas inhibition of AKT activates FoxO transcription factors, which serve as potent inducers of CCN2 gene expression. FoxO proteins are thus key regulators which determine the effects of the activation of PI3K–AKT signaling on CCN2 gene expression.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
{kind=link}
(GIF 142 kb)
{kind=link}
(GIF 174 kb)
Acknowledgement
This work was supported by the Interdisciplinary Center for Clinical Research (IZKF) at the University Hospital of the University of Erlangen-Nuremberg, project D5.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- CCN2
Connective tissue growth factor
- CTGF; HUVEC
Human umbilical vein endothelial cells
- PI3K
Phosphatidylinositol-3 kinase
- TGF
Transforming growth factor
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12079-009-0055-5) contains supplementary material, which is available to authorized users.
References
- Black SA Jr, Trackman PC (2008) Transforming growth factor-beta1 (TGFbeta1) stimulates connective tissue growth factor (CCN2/CTGF) expression in human gingival fibroblasts through a RhoA-independent, Rac1/Cdc42-dependent mechanism: statins with forskolin block TGFbeta1-induced CCN2/CTGF expression. J Biol Chem 283:10835–10847. doi:10.1074/jbc.M710363200 [DOI] [PMC free article] [PubMed]
- Chaqour B, Goppelt-Struebe M (2006) Mechanical regulation of the Cyr61/CCN1 and CTGF/CCN2 proteins. FEBS J 273:3639–3649. doi:10.1111/j.1742-4658.2006.05360.x [DOI] [PubMed]
- Chaqour B, Yang R, Sha Q (2006) Mechanical stretch modulates the promoter activity of the profibrotic factor CCN2 through increased actin polymerization and NF-kappa B activation. J Biol Chem 281:20608–20622. doi:10.1074/jbc.M600214200 [DOI] [PubMed]
- Chowdhury I, Chaqour B (2004) Regulation of connective tissue growth factor (CTGF/CCN2) gene transcription and mRNA stability in smooth muscle cells. Involvement of RhoA GTPase and p38 MAP kinase and sensitivity to actin dynamics. Eur J Biochem 271:4436–4450. doi:10.1111/j.1432-1033.2004.04382.x [DOI] [PubMed]
- Cicha I, Goppelt-Struebe M (2009) Connective tissue growth factor: Context-dependent functions and mechanisms of regulation. BioFactors 35(2):200–208 [DOI] [PubMed]
- Cicha I, Goppelt-Struebe M, Muehlich S, Yilmaz A, Raaz D, Daniel WG, Garlichs CD (2008) Pharmacological inhibition of RhoA signaling prevents connective tissue growth factor induction in endothelial cells exposed to non-uniform shear stress. Atherosclerosis 196:136–145. doi:10.1016/j.atherosclerosis.2007.03.016 [DOI] [PubMed]
- Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, Lai KM, Lin HC, Ioffe E, Yancopoulos GD, Rudge JS (2004) Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev 18:1060–1071. doi:10.1101/gad.1189704 [DOI] [PMC free article] [PubMed]
- Dejana E, Taddei A, Randi AM (2007) Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta 1775:298–312 [DOI] [PubMed]
- Dixit M, Bess E, Fisslthaler B, Hartel FV, Noll T, Busse R, Fleming I (2008) Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovasc Res 77:160–168. doi:10.1093/cvr/cvm017 [DOI] [PubMed]
- Franke TF (2008) Intracellular signaling by Akt: bound to be specific. Sci Signal 1:pe29. doi:10.1126/scisignal.124pe29 [DOI] [PubMed]
- Gomis RR, Alarcon C, He W, Wang Q, Seoane J, Lash A, Massague J (2006) A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci USA 103:12747–12752. doi:10.1073/pnas.0605333103 [DOI] [PMC free article] [PubMed]
- Graness A, Cicha I, Goppelt-Struebe M (2006) Contribution of Src-FAK signaling to the induction of connective tissue growth factor in renal fibroblasts. Kidney Int 68:1341–1349 [DOI] [PubMed]
- Jiang BH, Liu LZ (2008) PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim Biophys Acta 1784:150–158 [DOI] [PubMed]
- Komorowsky C, Ocker M, Goppelt-Struebe M (2009) Differential Regulation of Connective Tissue Growth Factor in Renal Cells by Histone Deacetylase Inhibitors. J Cell Mol Med. doi:10.1111/j.1582-4934.2008.00674.x [DOI] [PMC free article] [PubMed]
- Kroening S, Solomovitch S, Sachs M, Wullich B, Goppelt-Struebe M (2009) Regulation of connective tissue growth factor (CTGF) by hepatocyte growth factor in human tubular epithelial cells. Nephrol Dial Transplant 24:755–762. doi:10.1093/ndt/gfn530 [DOI] [PubMed]
- Muehlich S, Schneider N, Hinkmann F, Garlichs CD, Goppelt-Struebe M (2004) Induction of connective tissue growth factor (CTGF) in human endothelial cells by lysophosphatidic acid, sphingosine-1-phosphate, and platelets. Atherosclerosis 175:261–268. doi:10.1016/j.atherosclerosis.2004.04.011 [DOI] [PubMed]
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA (2007) FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128:309–323. doi:10.1016/j.cell.2006.12.029 [DOI] [PMC free article] [PubMed]
- Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S (2005) Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest 115:2382–2392. doi:10.1172/JCI23126 [DOI] [PMC free article] [PubMed]
- Samarin J, Rehm M, Krueger B, Waschke J, Goppelt-Struebe M (2009) Up-regulation of connective tissue growth factor in endothelial cells by the microtubule-destabilizing agent combretastatin A-4. Mol Cancer Res 7:180–188. doi:10.1158/1541-7786.MCR-08-0292 [DOI] [PubMed]
- Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP, King GL (2000) Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, flt1, and phosphatidylinositol 3-kinase- akt-dependent pathways in retinal vascular cells. J Biol Chem 275:40725–40731. doi:10.1074/jbc.M006509200 [DOI] [PubMed]
- Vogt PK, Jiang H, Aoki M (2005) Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 4:908–913 [DOI] [PubMed]
- Woods A, Pala D, Kennedy L, McLean S, Rockel JS, Wang G, Leask A, Beier F (2009) Rac1 signaling regulates CTGF/CCN2 gene expression via TGFbeta/Smad signaling in chondrocytes. Osteoarthritis Cartilage 17:406–413. doi:10.1016/j.joca.2008.07.002 [DOI] [PubMed]
- Xia H, Nho RS, Kahm J, Kleidon J, Henke CA (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem 279:33024–33034. doi:10.1074/jbc.M313265200 [DOI] [PubMed]
- Xie S, Sukkar MB, Issa R, Oltmanns U, Nicholson AG, Chung KF (2005) Regulation of TGF-beta 1-induced connective tissue growth factor expression in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 288:L68–L76. doi:10.1152/ajplung.00156.2004 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
(GIF 142 kb)
(GIF 174 kb)