

Abstract
Approximately one-third of human genetic diseases are caused by premature translation-termination codon (PTC)-generating mutations. These mutations in sodium channel and GABAA receptor genes have been associated with idiopathic generalized epilepsies, but the cellular consequences of the PTCs on the mutant channel subunit biogenesis and function are unknown. The PTCs could result in translation of a truncated subunit, or more likely, trigger mRNA degradation through nonsense-mediated mRNA decay (NMD), thus preventing or reducing production of mutant subunit at the transcriptional level. The GABAA receptor α1 subunit mutation, 975delC, S326fs328X, is an autosomal dominant mutation associated with childhood absence epilepsy that generates a PTC in exon 8 of the 9 exon GABRA1 gene that is 74 bp upstream of intron 8. Using an intron 8-inclusion minigene that supports NMD, we demonstrated that mutant mRNA was substantially reduced, but not absent. Loss of mutant transcripts was blocked by ribosome inhibition or by silencing the NMD-essential gene hUPF-1. In both neurons and non-neuronal cells, the PTC caused substantial loss of mutant α1(S326fs328X) subunit mRNA through NMD with a minor portion of the mRNA escaping NMD and producing a mutant protein. The translated mutant protein had reduced stability due to endoplasmic reticulum associated degradation (ERAD) and had enhanced association with molecular chaperones. This study suggests that loss of mRNA due to activation of NMD and activation of ERAD by the mutant protein may contribute to epileptogenesis. The molecular mechanisms outlined here delineate a model for the pathogenesis of many PTC-generating mutations.
Introduction
Several GABAAreceptor subunit missense and nonsense premature translation-termination codon (PTC)-generating mutations have been associated with idiopathic generalized epilepsies (IGEs) (Macdonald et al., 2006). Transcription of mutant genes produces mutant mRNA and translation of mutant mRNA produces mutant protein. Cellular mRNA surveillance mechanisms often degrade mutant PTC-containing mRNAs and endoplasmic reticulum (ER) protein quality control processes degrade misfolded or misrouted proteins. Trafficking deficient mutant subunits are often subject to ER retention and ER associated degradation (ERAD) after translation (Kang and Macdonald, 2004; Gallagher et al., 2005). The cellular fate of GABAA receptor subunits translated from PTC-containing mutant mRNAs is unknown.
PTC mutations are responsible for approximately one-third of inherited disorders including GABR subunit gene mutation associated epilepsies. In several of the genetic diseases caused by PTCs, mRNAs containing a PTC are often subject to degradation via a mechanism called nonsense mediated mRNA decay (NMD). NMD is a posttranscriptional, but translation-dependent, mRNA quality control mechanism that recognizes and selectively degrades mRNAs that contain a PTC that is 50–55 nt upstream from an exon–exon junction (Isken and Maquat, 2007) or mRNAs with aberrantly configured 3′untranslated regions (UTRs) (Amrani et al., 2004). NMD is translation-dependent since translating ribosomes recognize PTCs during the pioneering round of translation. NMD is also splicing-dependent and requires an exon junction complex (EJC) deposited during intron splicing at an exon–exon junction located downstream of the PTC. EJCs consist of mRNA decay factors including Upf-1 (rent1), which is an RNA helicase and essential factor to activate NMD. NMD rids cells of most transcripts containing PTCs and reduces intracellular levels of truncated and potentially deleterious proteins (Kuzmiak and Maquat, 2006). Most nonsense transcripts are reduced by cellular mRNA surveillance processes, including NMD, to ∼5–25% of wild-type levels (Kuzmiak and Maquat, 2006), with mutant mRNAs escaping NMD resulting in translation of truncated proteins that are often trafficking deficient, misfolded and misrouted and consequently subject to ERAD (Stephenson and Maquat, 1996). The mechanisms by which ERAD targets misfolded proteins includes the ubiquitin-proteasomal system (UPS) (Turnbull et al., 2007) and the autophagy/lysosome pathway (Cuervo, 2004). Inhibition of translation of mRNA by cycloheximide, the protein synthesis inhibitor, or silencing the key mRNA decay factor, Upf-1, reverse the mRNA loss produced by NMD.
The GABAA receptor α1 subunit mutation, 975delC, S326fs328X, is a rare autosomal dominant mutation associated with childhood absence epilepsy (CAE) (Maljevic et al., 2006). The deletion, 975delC, should cause a frameshift at residue S326 and create a PTC at residue L328, which is in the second to last exon (exon 8) of the 9 exon gene and 74 nt upstream of the last exon–exon junction. Based on either the “50 nucleotide boundary rule” (Maquat, 2005) or the “faux 3′-UTR model” (Amrani et al., 2004), the PTC likely would activate NMD, resulting in degradation of mutant transcripts (Shyu et al., 2008), but this has not been demonstrated. We used an intron-inclusion minigene approach to characterize the effect of the 975delC, S326fs328X mutation on α1 subunit mRNA and protein stability. This approach has been widely used to study aberrant splicing and NMD since it supports mRNA editing and splicing (Hefferon et al., 2002; Bühler et al., 2004; Busi and Cresteil, 2005). In addition to all 9 exons, the α1 minigene contained the entire intron 8, thus rendering mRNA produced from the minigene a candidate for NMD since splicing out intron 8 would create an exon–exon junction between exons 8 and 9. In contrast, mRNAs that contain PTCs derived from conventional intron-less cDNA constructs are not subject to NMD, since they do not contain an exon–exon junction. The α1 subunit mutation, 975delC, S326fs328X, is 74 nt upstream of the splicing-generated exon–exon junction in the mRNA, and thus, should elicit NMD. Here, we demonstrated that mutant α1 subunit mRNA was subject to substantial, but incomplete, NMD, and that the mutant subunit protein that was produced by the mRNA that escaped NMD was degraded by ERAD through the ubiquitin-proteasome system. The combination of mutant mRNA degradation by NMD and mutant protein degradation by ERAD may represent a molecular pathology model for many other PTC-generating mutations.
Materials and Methods
GABAA receptor subunit cDNA and minigene constructs.
The cDNAs encoding human, GABAA receptor β2, γ2S and wild-type and mutant α1 subunits were subcloned into the expression vector pcDNA3.1(+). For the remainder of the study, the mutant α1(975delC, S326fs328X) subunit will be referred to as the α1(S326fs328X) subunit. The FLAG (DYKDDDDK) epitope was inserted between amino acids 4 and 5 in the N terminus of the mature peptide to create the α1FLAG and α1(S326fs328X)FLAG subunit cDNAs. The wild-type α1 subunit minigene was generated by including the entire intron 8 of the GABAA receptor α1 subunit (nm_000806), which was extracted from human genomic DNA. The α1(S326fs328X), α1(A322D) and α1(Y368X) subunit mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing.
Rat neuronal culture.
Rat cortical neurons were dissected from the brains of embryonic 17–18-d-old Sprague Dawley rat embryos. Dissociation of cells and culturing and transfection procedures were performed as described previously with minor modifications (Kang et al., 2006).
Transfection, siRNA treatment.
Cortical neurons were transfected by Fugene or nucleofection (Amaxa) with program 0–03 following the manufacturer's protocols. HeLa cells were transfected with Fugene (Roche Molecular Systems), and HEK 293T cells were transfected with the calcium phosphate precipitation (Calcium) method unless otherwise specified. Cells were cotransfected with 1–2 μg of each subunit plasmid for each 60 mm2 dish and 0.33 μg for each 35 mm2 dish. To silence the hUPF-1 gene, a factor essential for NMD, cells were transfected with siRNA targeting nucleotides of hUPF-1 (5′-AAGATGCAGTTCCG-CTCCATTTT-3′) (Ambion) with Oligofectamine (Invitrogen) as described before (Mendell et al., 2002). The same cells were retransfected with the wild-type and mutant α1 minigene constructs or in addition to β2 and γ2S subunits 48 h later and harvested 2 additional days later for RT-PCR and Western blot.
Biotinylation and Western blot analysis.
The procedures of cell surface receptor biotinylation and Western blot were as described previously (Kang et al., 2004). After SDS-PAGE, the membranes were incubated with specific primary antibody overnight at 4°C with gentle rotation. The monoclonal anti-human α1 antibody (BD24) was purchased from Millipore Bioscience Research Reagents. The polyclonal anti-human Upf-1 (hUpf-1) antibody was from Abgent primary antibody company. Polyclonal rabbit anti-Bip/GRP78 and monoclonal anti-Bip(GRP79) were purchased from Abcam and BD Transduction Laboratories, respectively. After washing, the membranes were incubated with horseradish peroxidase conjugated secondary antibody [goat anti-mouse IgG or goat anti-rabbit IgG (1:7500; Jackson ImmunoResearch Laboratories)]. The antibody-reactive bands were revealed by chemiluminescence. The Western blots were quantified with ChemiImager AlphaEaseFC.
Confocal microscopy.
Rat cortical neurons were plated on poly-d-lysine-coated, glass-bottom imaging dishes at the density of 1–2 × 105 cells. Rat cortical neurons were transfected with wild-type α1 minigene and β2 and γ2S subunits at a ratio of 1:1:1 (wt); mutant α1 minigene (S326fs328X) and β2 and γ2S subunits and mutant α1(Y368X) mini gene and β2 and γ2S subunits for 6 d. The neurons were fixed, permeabilized and stained with anti monoclonal human α1 subunit antibody conjugated with fluorophore Alexa-647. The neurons were then visualized under confocal microscopy as described previously (Kang et al., 2004).
Electrophysiology.
HEK 293T cells were cotransfected with 2 μg of each subunit plasmid and 1 μg of the pHook-1 cDNA (Invitrogen) using a modified calcium phosphate precipitation method and selected 24 h after transfection by magnetic hapten coated beads (Greenfield et al., 1997). Recordings were obtained from the cells 2 d later using methods described previously (Kang and Macdonald, 2004).
RNA extraction, PCR, quantitative real-time PCR for mRNA determination.
Total RNAs were extracted by using Versagene RNA Cell kit (Gentra Systems) or by RNAeasy Micro kit (Qiagen) following the manufacturer's protocol. The transcribed cDNA products were probed with either human GABAA receptor α1 subunit probes or 18 s with 1:10 dilution. Quantitative real-time PCR experiments were performed with these specific primers and Taqman probes (sequence available upon request) in a 5 μl final volume and normalized to endogenous 18 s rDNA (acetyl-H3 CHIP). Relative RNA abundance was quantified by evaluating threshold cycle (Ct) values, according to the “relative standard curve” method (ABI Prism 7700 Manual).
35S radiolabeling metabolic pulse-chase assays.
The protocol was modified from our previously published protocol (Gallagher et al., 2007). Briefly, 48 h after transfection, the cells were replenished with the starving medium which lacked methionine and cysteine (Invitrogen), and incubated at 37°C for 30 min. The starving medium was then replaced by 1.5 ml 35S radionuclide methionine [100–250 μCi/ml (1 Ci = 37GBq); PerkinElmer] labeling medium for 20 min at 37°C. The labeling medium was then changed to the chase medium for a series of different time points. The FLAG-tagged α1 subunits were then immunoprecipitated from radio-labeled lysates with an anti-FLAG M2-agarose affinity gel by rotating at 4°C overnight. The immunoprecipitated products were then eluted from the beads with FLAG peptide (Sigma-Aldrich). The immunopurified subunits were then analyzed by 12.5% SDS-PAGE and exposed on a digital PhosphorImager (GE Healthcare).
Data analysis.
Macroscopic currents were low-pass filtered at 2 kHz, digitized at 10 kHz, and analyzed using pClamp9 software suite (Axon Instruments). Except for the pulse-chase assays, proteins were quantified by ChemiImager AlphaEaseFC software, and data were normalized to either wild-type subunits or loading controls. Data from pulse-chase experiments were quantified using Quantity One software (Bio-Rad). Numerical data were expressed as mean ± SEM. When wild-type data were arbitrarily taken as 1, column statistics were used. Statistical significance, using Student's unpaired t test (GraphPad Prism), was taken as p < 0.05.
Results
The cellular fate of mutant α1(S326fs328X) subunits was studied using a minigene strategy
The minigene strategy has been used to study transcript alteration and NMD in a number of diseases, including cystic fibrosis, since it contains introns that support mRNA editing and splicing. We thus used this approach to study the intracellular processing of human wild-type and mutant α1 subunits. The α1 subunit mutation, 975delC, S326fs328X, is located in the middle of TM3, and the PTC produced by the deletion would result in a loss of the C-terminal 141 aa, thus producing a truncated α1(S326fs328X) subunit (Fig. 1A). The human α1 subunit gene, GABRA1, has 9 exons and 8 introns. Minigenes were constructed by including the entire GABRA1 intron 8 (1242 bp) between exon 8 and 9 of the wild-type and mutant α1 subunit cDNA constructs (Fig. 1Ba,b). The stop codon for the wild-type α1 subunit minigene is located at the end of exon 9 (Fig. 1Ba), and the PTC for the mutant α1 subunit minigene α1(S326fs328X) subunit minigene is located in exon 8, which is 128 bp downstream from intron 7 and 74 bp upstream from intron 8 (Fig. 1Bb). We also generated an α1(A322D) subunit minigene as a control (Fig. 1A,Bc). The α1 subunit mutation, A322D, associated with juvenile myoclonic epilepsy, is a missense mutation four residues upstream of the 975delC, S326fs328X frameshift mutation and should not elicit NMD. In addition, we generated an artificial α1 subunit nonsense mutation, Y368X, in the middle of the last exon (exon 9) as an NMD-insensitive control (Fig. 1Bd).
Figure 1.
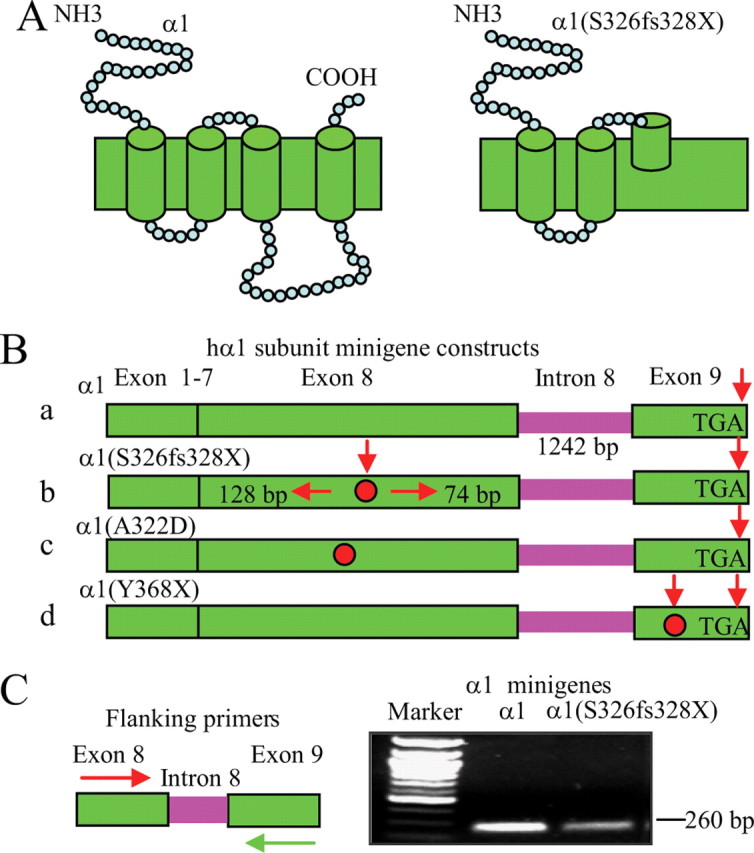
A minigene strategy was used to study the α1 subunit mutation, 975delC, S326fs328X. A, Schematic topologies of GABAA receptor wild-type (left) and mutant α1(S326fs328X) subunit (right). B, The α1 subunit gene, GABRA1, has 9 exons and 8 introns. Wild-type and mutant α1 subunit minigenes were constructed by including intron 8 (1242 bp). The stop codons of the wild-type minigene (α1) (Ba) and α1(A322D) (red dot) (Bc) are located at the end of exon 9, and the nonsense stop codon caused by the α1 subunit mutation, 975delC, S326fs328X, is located at the middle of exon 8 (Bb). An NMD-incompetent control minigene had a nonsense stop codon inserted in the last (ninth) exon of the α1 subunit gene α1(Y368X) (Bd). C, With primers in exon 8 and 9 that flank intron 8, both wild-type and mutant α1 subunit minigenes displayed a band at 260 bp, suggesting correct splicing in the reverse transcribed cDNA products from RNAs of α1 and α1(S326fs328X) subunit minigenes.
In HEK 293T cells, wild-type and mutant α1(S326fs328X) subunit minigenes were cotransfected with β2 and γ2 subunit cDNAs. In this and all other experiments, the Calcium transfection technique was used unless otherwise noted. Total RNAs were extracted 48 h after transfection, and equal amounts of total RNA were transcribed to cDNA. With flanking primers in exon 8 and 9, equal amounts of cDNAs from wild-type and mutant α1 subunit minigenes were used for a template in the PCR amplification, and both minigenes displayed a band at 260 bp, suggesting correct splicing in the reverse transcribed cDNA products from α1 and α1(S326fs328X) subunit minigene mRNAs (Fig. 1C). The mutant α1 subunit minigene band integrated density values (IDVs), however, were only ∼20% of the wild-type α1 subunit minigene band IDVs, suggesting reduction of mutant transcript levels.
Expression of the NMD competent α1(S326fs328X) subunit minigenes in heterologous cells and rat cortical neurons produced reduced mutant protein
It is unknown how robust the endogenous NMD machinery is in different cell types, and if NMD in a given cell type can degrade mRNAs equally when transfection methods with different efficiencies are used. We thus first used the less efficient Calcium transfection technique and then the more efficient Fugene transfection technique (see Materials and Methods). With Calcium transfection, we cotransfected HEK 293T cells with β2 and γ2S subunit and wild-type and/or mutant α1 subunit cDNAs (Fig. 2A,B, left) or minigenes (Fig. 2A,B, middle) at a cDNA ratio of 1:1:1 for wild-type α1 subunits and mutant α1(S326fs328X) subunits and at a cDNA ratio of 1:1:0.5:0.5 for mixed α1/α1(S326fs328X) subunits. Wild-type α1 subunits should migrate at ∼50 KDa, and mutant α1(S326fs328X) subunits should migrate at ∼37 KDa (Fig. 2A, left, green arrows). Our data are consistent with the original report that mutant α1(S326fs328X) subunit protein was reduced in both mixed and mutant conditions, suggesting that the mutant protein was degraded (Fig. 2B, left) (0.328 ± 0.063 mutant vs 0.765 ± 0.082 wt; p = 0.002). By flow cytometry, the total amount of mutant protein was <3% of wild-type protein (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Interestingly, the ratio of wild-type to mutant protein with mixed expression was larger than the ratio of wild-type to mutant protein with wt and mut expression, suggesting that the presence of the wild-type α1 subunit or a failure to compete with the wild-type subunit during assembly somehow facilitated degradation of the mutant protein.
Figure 2.
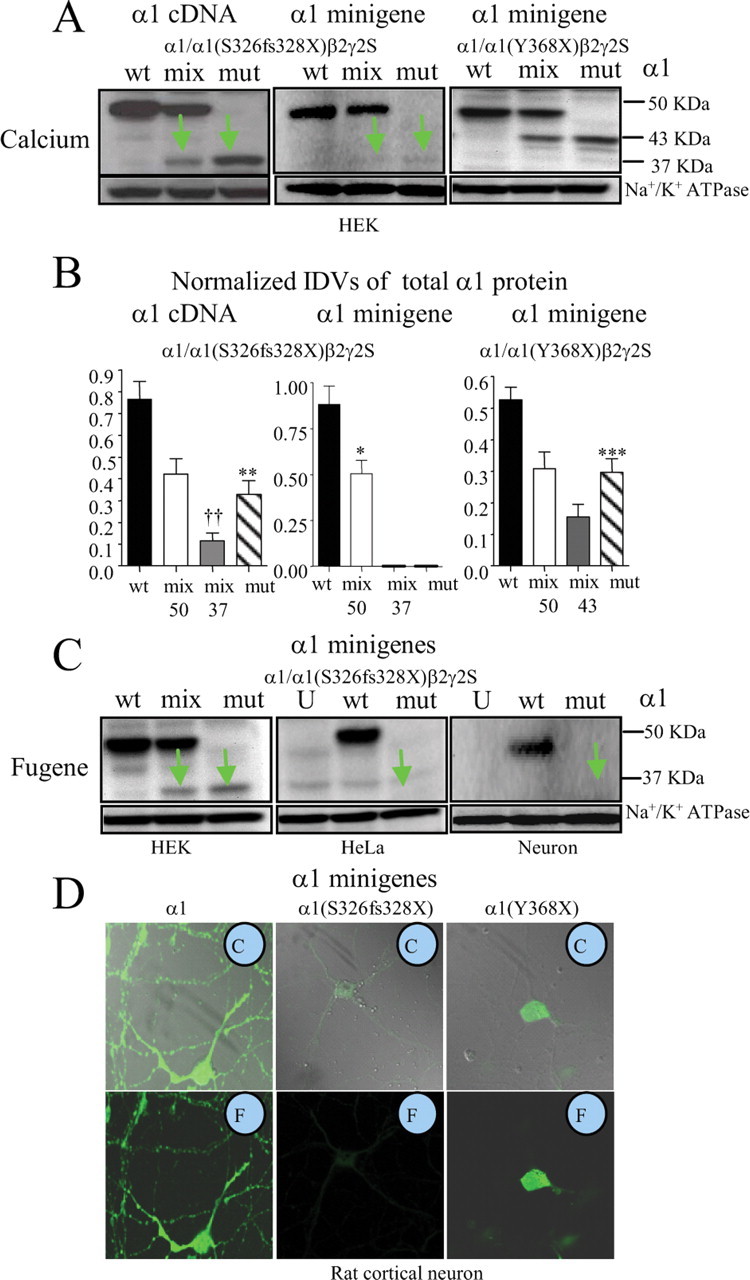
Expression of the NMD competent α1(S326fs328X) subunit minigene produced minimal mutant protein. A, Total lysates from HEK 293T cells cotransfected with β2 and γ2S subunit cDNAs and α1 subunit cDNAs (left) or minigenes (middle and right) with wild-type (wt) subunits (ratio of 1:1:1), mixed (mix) α1/α1(S326fs328X) or α1/α1(Y368X) subunits (0.5:0.5:1:1) and mutant (mut) α1(S326fs328X) or α1/α1(Y368X) subunits (1:1:1) with the Calcium phosphate precipitation method (Calcium). The total lysates were analyzed by immunoblot. Na+/K+ ATPase was used for a loading control. Wild-type α1 subunits migrated at ∼50 KD, α1(S326fs328X) subunits migrated at 37 KD, and α1(Y368X) subunits migrated at 43 KD. B, The relative amount of total wt, mix and mut α1 subunit protein from A was plotted. Mix 50 stands for the wild-type subunit level detected at 50 KDa in the mixed transfection condition, mix 37 or 43 stands for the mutant truncated subunits detected at 37 or 43 KDa in the mixed condition (**p < 0.01, ***p < 0.001 vs wild-type, ††p < 0.01 vs mix 50). C, HEK 293T cells, HeLa cells and rat cortical neurons were cotransfected with β2 and γ2S subunit cDNAs and wild-type α1, mixed α1/α1(S326fs328X) or mutant α1(S326fs328X) subunit minigenes (1:1:1) with Fugene transfection. In A and C, the green arrows indicate the mutant subunit. In C, U stands for untransfected. wt, mix, and mut are as described in A. D, Rat cortical neurons were transfected with β2 and γ2S subunit cDNAs and the wild-type α1 subunit minigene (left), the mutant α1 subunit minigene (S326fs328X) (middle) or the mutant α1(Y368X) subunit minigene (right) at a cDNA ratio of 1:1:1. After 6 d in culture, the neurons were permeabilized and stained with anti-monoclonal human α1 subunit antibody conjugated with fluorophore Alexa-647. In the insets, C refers to combined transmitted and fluorescent images, and F refers to fluorescent images. In C and D, neurons were transfected by nucleofection.
Second, HEK 293T cells were cotransfected with β2 and γ2S subunit cDNAs and wild-type and mutant α1 subunit minigenes (wt, cDNA ratio of 1:1:1), mixed α1/α1(S326fs328X) subunit minigenes (mix, cDNA ratio of 1;1:0.5:0.5) or mutant α1(S326fs328X) subunit minigene (cDNA ratio of 1:1:1) (Fig. 2A,B, middle). The wild-type α1 subunit minigene displayed a robust 50 KDa band in both wild-type and mixed conditions. With mixed or mutant expression of the minigene constructs, however, mutant protein (37 KDa) was substantially reduced (Fig. 2A, middle, green arrows) (Fig. 2B, middle), suggesting an additional mechanism for loss of mutant protein.
Finally, HEK 293T cells were cotransfected with β2 and γ2S subunit cDNAs and the wild-type α1 subunit minigene (wt, cDNA ratio of 1:1:1), mixed α1/α1(Y368X) subunit minigenes (mix, cDNA ratio of 1:1:0.5:0.5) or mutant α1(Y368X) subunit minigene (cDNA ratio of 1:1:1) minigenes (Fig. 2A,B, right). Expression of the NMD-insensitive α1(Y368X) subunit minigene in both mixed and mutant conditions produced truncated proteins but at reduced levels that migrated at the predicted molecular mass of 43 KDa (Fig. 2A,B, right).
We next extended our study to include other cell types including HeLa cells and rat cortical neurons. Because these cells are less efficiently transfected, we used the more efficient Fugene transfection technique, and included Fugene transfection of HEK 293T cells for comparison. In HEK 293T cells with Fugene transfection of wild-type and/or mutant α1 subunit minigenes, mutant α1(S326fs328X) subunit protein was present, although substantially reduced compared with wild-type subunit protein in both mixed and mutant conditions (Fig. 2C, left), despite being almost absent with Calcium transfection (Fig. 2A, middle). However, the mutant protein was not detectable with Fugene transfection in either HeLa cells (Fig. 2C, middle) or rat cortical neurons (Fig. 2C, right), despite robust expression of wild-type α1 subunit protein.
Consistent with the biochemical data, similar data were obtained with immunocytochemistry and confocal microscopy for total human α1 subunit protein expression (Fig. 2D). In rat cortical neurons expressing wild-type α1β2γ2S subunits, 6 d after transfection of β2 and γ2S subunit cDNAs and wild-type α1 subunit minigenes (Fig. 2D, left), there was robust signal in both the cell bodies and dendrites, but in neurons expressing α1(S326fs328X)β2γ2S subunits, there was only a minimal fluorescent signal in the soma (Fig. 2D, middle). In contrast, a significant amount of fluorescence was detected in the cell somata of neurons expressing α1(Y368X)β2γ2S subunits (Fig. 2D, right). The levels of mutant relative to wild-type subunit protein seemed to vary somewhat with different transfection reagents, different cell types and the presence or absence of the β2 and γ2 subunits (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). It is likely that the more efficient transfection technique (Fugene) and use of efficiently transfected cell types (HEK 293T cells) were able to partially overwhelm the NMD machinery and ER quality control mechanisms due to high minigene copy number. Nonetheless, reduction of mutant protein following minigene expression occurred with all transfection techniques and in all cell types, suggesting common and consistent pathways for loss of mutant protein, likely NMD and ERAD.
Peak current amplitudes and surface α1 subunit levels were reduced with mixed expression of α1/α1(S326fs328X)β2γ2S subunits
We recorded currents evoked by 6 (Fig. 3A,B) or 28 (Fig. 3C,D) sec applications of 1 mm GABA from HEK 293T cells cotransfected with β2 and γ2S subunit cDNAs and the wild-type α1 subunit minigene (1:1:1 cDNA ratio; wt, black), half the amount of wild-type α1 subunit minigene as a haploinsufficiency control (1:1:0.5 cDNA ratio; hc, silver), mixed α1/α1(S326fs328X) subunit minigenes (1:1:0.5:0.5 cDNA ratio; mix, green) or mutant α1(S326fs328X) subunit minigene (1:1:1 cDNA ratio; mut, gray). Peak haploinsufficiency control and mixed receptor currents were smaller than wild-type currents, and there was no significant difference between them (p = 0.65) (Fig. 3A,B). No currents were recorded from cells following mutant subunit expression. Compared with wild-type type peak currents (2490 ± 189.3 pA), peak amplitudes of both haploinsufficiency control (1461 ± 294.7 pA; p = 0.01) and mixed receptor currents (1299 ± 158.2 pA; p = 0.0004) were significantly reduced (Fig. 3A,B) without change in macroscopic kinetic properties. When normalized to the peak wild-type receptor current, there was no difference in the steady state/peak current ratio between wild-type and haploinsufficiency control or wild-type and the receptor from mixed expression of wild-type and mutant receptors (Fig. 3C,D), suggesting that the functional receptors on the cell surface were likely wild-type receptors.
Figure 3.
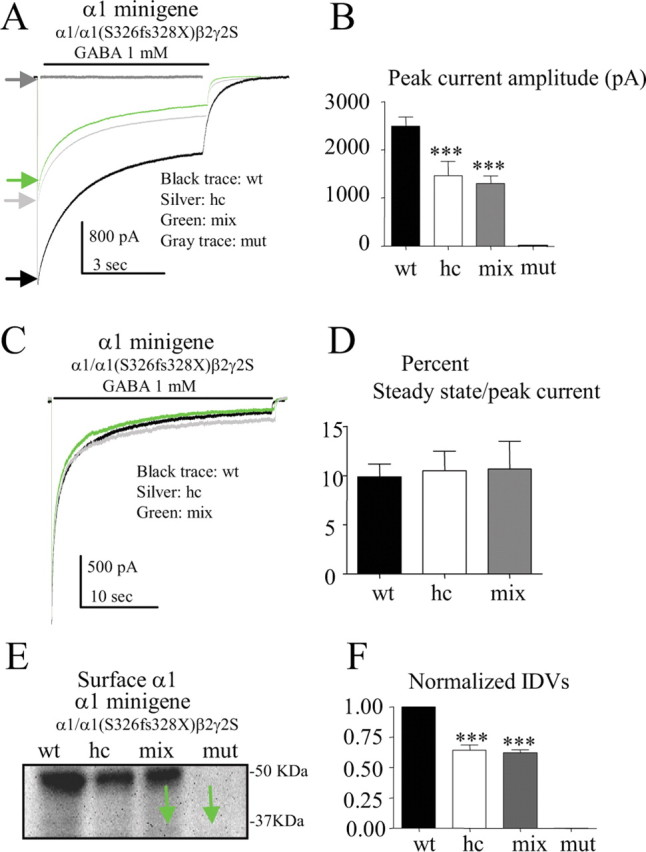
Mixed α1/α1(S326fs328X)β2γ2S receptors had reduced peak current amplitudes and reduced α1 subunit surface expression. A–D, Human GABAA receptor currents were obtained from HEK 293T cells cotransfected with β2 and γ2S subunit cDNAs and wild-type α1 subunit minigenes for wild-type (wt α1:β2:γ2S 1:1:1 cDNA ratio, black) or for haploinsufficiency control (0.5:1:1, hc, silver) or with mixed α1/α1(S326fs328X) minigenes (0.5:0.5:1:1, mix, green) and mutant (mut) α1(S326fs328X) minigene (1:1:1, mut, gray) with 1 mm GABA applied for 6 s (A) or 28 s (C). In A, arrows indicate the peak of each trace. B, The mean peak amplitude of each group was plotted (n = 7 for wt, n = 8 for hc, n = 7 for mix). C, The peak haploinsufficiency control and mixed receptor currents were normalized to the wild-type receptor current. D, The ratios of steady-state current/peak amplitude of both haploinsufficiency control and mixed receptor currents were similar to wild-type current (n = 7 for each group). In A and C, lifted whole cells were voltage-clamped at −50 mV. E, Equal amounts of membrane-bound protein were biotinylated and immunoblotted with anti-α1 antibody. The green arrows indicate the mutant protein. F, The relative amounts of surface wild-type, haploinsufficiency control, mixed and mutant α1 subunit protein were plotted (n = 6). In B and F, ***p < 0.001 versus wild-type.
Results from cell surface receptor biotinylation and immunoblotting were consistent with the electrophysiological results. HEK 293T cells were cotransfected with β2 and γ2S subunit cDNAs and the wild-type α1 subunit minigene (1:1:1 cDNA ratio; wt), half the amount of wild-type α1 subunit minigene as a haploinsufficiency control (1:1:0.5 cDNA ratio; hc), mixed α1/α1(S326fs328X) subunit minigenes (1:1:0.5:0.5 cDNA ratio; mix) or mutant α1(S326fs328X) subunit minigene (1:1:1 cDNA ratio; mut) (Fig. 3E). Compared with transfection of wild-type subunits, transfection of haploinsufficiency control or mixed subunits resulted in reduced wild-type α1 subunit surface levels. There was no mutant α1(S326fs328X) subunit on the surface with either mixed or mutant expression. When normalized to wild-type α1 subunit surface levels, α1 subunit levels were reduced with expression of either haploinsufficiency control (0.64 ± 0.04, p < 0.0001) or mixed (0.62 ± 0.03, p < 0.0001) subunits, but there was no significant difference in α1 subunit surface levels between the haploinsufficiency control and mixed subunits (p = 0.67) (Fig. 3F). A similar pattern of surface α1 subunit protein expression was obtained by a more quantitative flow cytometry (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material).
Mutant α1(S326fs328X) subunits were not significantly “rescued” by proteasome inhibition, and mutant protein expressed was subject to ERAD
The mutant α1(S326fs328X) subunit that was detected was likely retained in the ER and degraded. To confirm this, following coexpression of β2 and γ2S subunits with either wild-type α1 subunit minigenes or mutant α1(S326fs328X) subunit cDNAs, total lysates from HEK 293T cells were either undigested (U) or digested with endoglycosidase H (Endo-H) (Fig. 4A,H) or peptide-N-glycosidase F (PNGase-F) (Fig. 4A,F). Since only small amounts of protein were produced with the mutant minigene expression, the mutant cDNA construct was used for this experiment. Endo-H removes N-linked high-mannose oligosaccharides attached in the ER, but not complex oligosaccharides attached in the trans-Golgi region. In contrast, PNGase-F removes N-linked oligosaccharides attached in both ER and trans-Golgi region. Therefore, protein retained inside the ER should have similar molecular mass with either Endo-H or PNGase-F digestion. Wild-type α1 subunits migrated at a higher molecular mass after Endo-H digestion and migrated at a lower molecular mass after PNGase-F digestion as we described previously (Gallagher et al., 2005). The mutant protein, however, migrated at a similar molecular mass after either Endo-H or PNGase-F digestion. The different shifts in molecular mass of wild-type and mutant α1 subunits after Endo-H or PNGase-F digestion suggested that the majority of the wild-type α1 subunits were trafficked beyond the ER, and that the mutant subunits were retained in the ER.
Figure 4.

The truncated mutant α1(S326fs328X) subunit protein was subject to ERAD. A, Total lysates of HEK 293T cells coexpressing β2 and γ2S subunit cDNAs and wild-type α1 subunit minigenes or mutant α1(S326fs328X) subunit cDNAs were undigested (U) or digested with Endo-H (H) or PNGase-F (F). The relatively high level of nonspecific signal with mutant expression of the mutant subunit was due to overexposing the gel to detect the very low level of mutant subunit protein that was present when the mutant subunit minigene was used. B, Total lysates of HEK 293T cells expressing β2 and γ2S subunit cDNAs and wild-type α1 subunit minigene (wt) or half amount of wild-type α1 subunit minigene (haploinsufficiency control; hc), mixed α1/α1(S326fs328X) subunit minigenes (mix) and mutant α1(S326fs328X) subunit minigene (mut) with or without lactacystin (Lac,10 μm) treatment for 6 h were analyzed by immunoblot. The arrows indicate the mutant protein. Calcium stands for Calcium phosphate precipitation transfection while Fugene stands for Fugene transfection. C, The total α1 subunit protein in each condition before lactacystin was quantified and normalized to the total α1 subunit expression of wild-type receptors (n = 8, ***p < 0.001 vs wild-type, Calcium; n = 4, *p < 0.05, ***p < 0.001 vs wild-type, Fugene). D, The histogram displays the ratio of increased α1 subunit protein from Fugene-transfected HEK 293T cells with lactacystin (10 μm) treatment over α1 subunit protein without treatment (**p < 0.01 vs wild-type, n = 4). E, HEK 293T or HeLa cells cotransfected with β2 and γ2S subunit cDNAs and α1 subunit cDNAs were treated with or without lactacystin, and the cell lysates were analyzed by immunoblot. F, The graph was plotted with the ratios of the mean IDVs ± SEM obtained with lactacystin treatment divided by results obtained without treatment (n = 7; *p < 0.05, ***p < 0.001 vs wild-type).
Loss of mutant protein could have been due to rapid degradation after protein synthesis, NMD of the PTC-containing transcripts before protein synthesis or both. If the mutant protein was mainly subject to ERAD after synthesis, proteasomal inhibition should “rescue” the mutant protein. Therefore, we applied lactacystin (Lac, 10 μm) and MG-132 (3 μm) (data not shown), two structurally different proteasome inhibitors, for 6 h to HEK 293T cells cotransfected with β2 and γ2S subunit cDNAs and wild-type, haploinsufficiency control, mixed or mutant α1/α1(S326fs328X) subunit minigenes (Fig. 4B). With either Lac or MG-132 (data not shown) treatment, the total mutant α1(S326fs328X) subunit proteins from both less efficient Calcium or more efficient Fugene transfections were increased, but still minimal, compared with the wild-type subunit (Fig. 4B). Relative to the wild-type α1 subunits, the magnitude of the total protein reduction in the haploinsufficiency control, mixed or mutant condition was greater with Calcium transfection (Fig. 4B, top) than with Fugene transfection (Fig. 4B, bottom) but was significantly reduced with both transfection techniques (Fig. 4C). The mutant α1(S326fs328X) subunit protein, but not wild-type, haploinsufficiency control or mixed α1 subunit proteins, were increased by lactacystin treatment when normalized to the subunits with lactacystin treatment, suggesting enhanced degradation of the mutant subunit (Fig. 4D). There was no difference between the ratio of the total protein of the mixed and wild-type subunit proteins before or after lactacystin, probably because the mutant protein was minimal compared with the wild-type protein in the mixed condition (Fig. 4B, Fugene; D), thus obscuring any increase in degradation of the mutant subunit. The NMD-insensitive control α1(Y368X) subunit proteins were all significantly increased by both proteasomal inhibitors (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The data suggest that the loss of the mutant α1(S326fs328X) subunit was mainly due to NMD while the loss of α1(Y368X) subunit was mainly due to ERAD. The mutant α1(S326fs328X) subunit, however, was partially rescued by blocking protein degradation, thus suggesting that any mutant subunit produced would be subject to degradation by ERAD.
We then used intronless cDNA constructs, which would not be subject to NMD, and Fugene transfection to produce more mutant protein (Fig. 4E). Mutant α1(S326fs328X) subunit levels in both HEK 293T and HeLa cells were reduced compared with wild-type α1 subunit levels but were increased by proteasomal inhibition to a greater extent than wild-type α1 subunits (1.24 ± 0.12 vs 1.99 ± 0.11, p = 0.0007 for HEK 293T cells; 1.66 ± 0.20 vs 2.36 ± 0.23%, p = 0.046 for HeLa cells) (Fig. 4F). The increased production of mutant α1(S326fs328X) subunit protein with transfection of intronless cDNA constructs and with Fugene transfection of minigene constructs (Fig. 4B,D) also suggested that any mutant protein produced would be subject to rapid degradation by ERAD through the ubiquitin-proteasome system.
Mutant α1(S326fs328X) subunits were degraded faster than the wild-type α1 subunits
To further investigate the cellular fate of the mutant protein, we performed 35S methionine radio-labeling pulse-chase experiment to determine if the mutant protein had an altered rate of degradation. We used HEK 293T cells since they had highly efficient protein expression, which produced higher levels of mutant subunit. The cells were transfected with either wild-type α1 or mutant α1(S326fs328X) subunits alone with Fugene and then used for study after 48 h. After radionuclide labeling for 20 min, the cells were lysed immediately and analyzed as time 0 or as “chased” for 30, 60, 90, 120, 180 and 240 min (Fig. 5A). Compared with the amount of protein at time 0, the remaining amounts of wild-type and mutant subunit protein were unchanged at 30 min, but the mutant subunit degraded faster than wild-type subunit from 60 min to 240 min. From 60 to 240 min of chase, the mutant subunit was significantly reduced compared with wild-type α1 subunit. The half-life of the mutant subunit was 64 min, and the half-life of wild-type α1 subunits was 194 min, which was >3-fold higher than that of the mutant subunit. To exclude the possibility that the increased mutant protein after proteasomal inhibition was due to increased synthesis, we applied cycloheximide (CHX) (100 μg/ml) to block protein synthesis (data not shown) and found that the mutant protein was degraded more rapidly either with or without CHX treatment.
Figure 5.

Mutant α1(S326fs328X) subunit protein had a reduced half-life. A, HEK 293T cells containing pulse-chase 35S methionine radio-labeled wild-type (W), and mutant α1 (S326fs328X) (M) subunits were lysed, immunopurified and analyzed by 12.5% SDS-PAGE. After 20 min labeling, the cells were chased for the indicated times (n = 5). The same amount of total protein (800 μg) from each sample was used for immunopurification. The percentage of radioactivity remaining is plotted versus the amount of radioactivity measured at time 0 for either the wild-type or mutant subunits (*p < 0.05, **p < 0.01, ***p < 0.001 vs wild-type). B, HEK 293T cells expressing wild-type (W) and mutant α1 (S326fs328X) (M) were pulse-labeled for 0, 5, 10, 15, and 20 min and lysed for immunopurification and SDS-PAGE (n = 4). The percentage of radioactivity incorporated during subunit synthesis was plotted by normalizing to the value obtained at 20 min for either the wild-type or mutant subunits.
The reduced mutant protein could also be due to the reduced synthesis. To determine the rate of synthesis of wild-type and mutant subunits, we pulse-labeled cells transfected with α1FLAG or α1(S326fs328X)FLAG subunits with 35S methionine for 0, 5, 10, 15, and 20 min. We used intronless α1 subunit cDNA constructs since they were used for degradation pulse chase experiments above. Normalized to the total wild-type or mutant protein labeled for 20 min, there were no significant differences in the amount of wild-type or mutant α1 subunits, demonstrating no difference in the rate of synthesis of these subunits.
Mutant α1(S326fs328X) subunits had an increased association with the chaperone Bip
Molecular chaperones are a group of proteins that mediate protein folding, assembly, trafficking and degradation (Hartl, 1996). Our data above suggested that the mutant protein was subject to ERAD, which is known to be mediated by chaperones. We thus tested the association of the mutant protein with Bip, which is an ER luminal chaperone that binds misfolded or unfolded, unassembled secretory proteins and ensures proper maturation and movement of proteins from the ER to the Golgi apparatus (Connolly et al., 1996). The cells were transfected with either wild-type α1FLAG or mutant α1(S326fs328X)FLAG subunits alone with Fugene and studied after 48 h. The total lysates were purified with FLAG antibody conjugated beads and probed with either anti-FLAG antibody for α1FLAG subunits or anti-Bip antibody (Fig. 6). The ratio of Bip to α1FLAG subunit IDV was then determined. Consistent with the accelerated degradation of the mutant subunit, there was less mutant than wild-type subunit protein. Also, more Bip protein was pulled down with the mutant subunit compared with the wild-type subunit. Thus the ratio of Bip to mutant subunit was enhanced substantially (sevenfold) compared with the wild-type subunit (wt 0.12 ± 0.011 vs mut 0.8430 ± 0.053) (n = 4, p < 0.001), consistent with an increased association of Bip with the mutant subunit.
Figure 6.

Mutant α1(S326fs328X) subunit protein had increased association with the ER resident chaperone Bip. HEK 293T cells expressing wild-type and mutant FLAG-tagged α1 cDNA subunits were lysed and immunopurified with FLAG antibody conjugated beads. The immunoblots show the expression level of the Flag tagged α1 subunit and its association with the chaperone Bip.
Mutant α1(S326fs328X) subunit expression was increased by ribosomal or hUPF-1 inhibition, suggesting that loss of mutant protein was due also to NMD
Was loss of mutant α1(S326fs328X) subunit due also to loss of mRNA triggered by NMD? To determine this, we first quantified wild-type and mutant α1 subunit mRNAs in HeLa and HEK 293T cells and rat cortical neurons transfected with wild-type α1 and mutant α1(S326fs328X) subunit minigenes. Total RNAs were extracted 2–4 d after transfection, and mRNAs were analyzed by real-time PCR. In HEK 293T and HeLa cells and in neurons, mutant α1(S326fs328X) transcripts were substantially reduced (Fig. 7A). When normalized to wild-type α1 subunit mRNA levels, mutant α1(S326fs328X) subunit mRNA levels were reduced to 39.1 ± 7.3% (p = 0.0001) in HEK 293T cells, to 16.6 ± 4% (p = 0.006) in rat cortical neurons and to 23.8 ± 3.6% (p < 0.0001) in HeLa cells.
Figure 7.
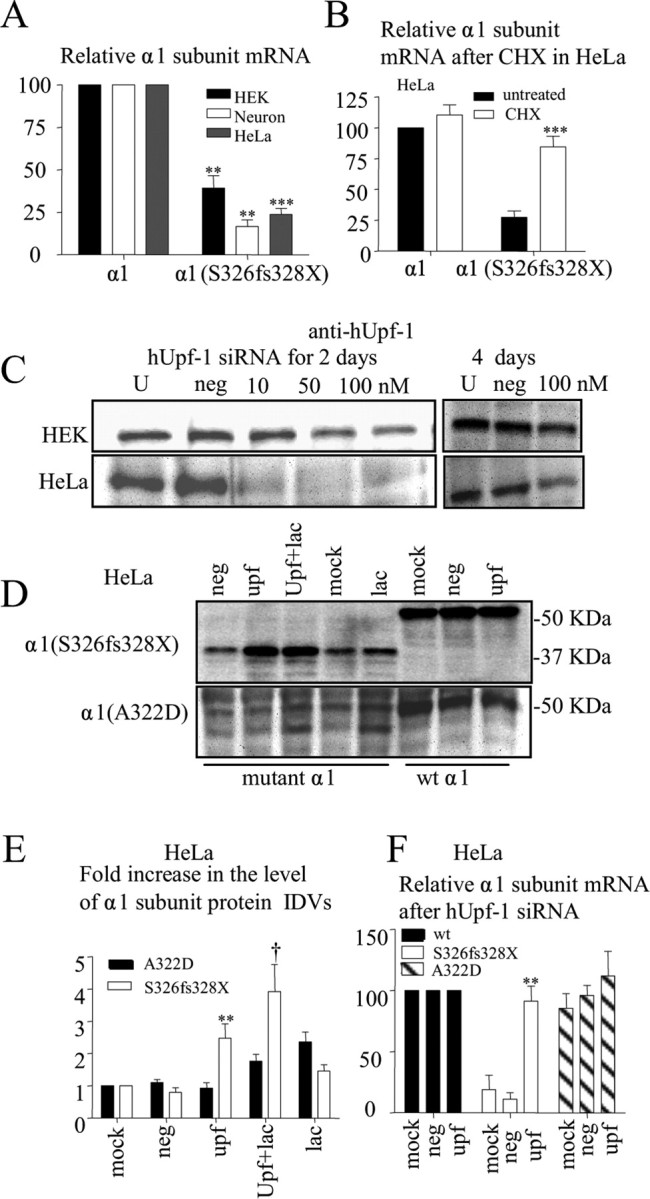
Mutant α1(S326fs328X) subunit mRNA was reduced and reversed by ribosome inhibition or by silencing the NMD essential factor hUpf-1. A, The relative amount of wild-type α1 and α1(S326fs328X) subunit mRNAs were measured by quantitative RT-PCR in different cells expressing α1 and α1(S326fs328X) minigenes (**p < 0.01, ***p < 0.001 vs wt; n = 15 for HEK, n = 7 for neuron, n = 11 for HeLa). B, HeLa cells expressing α1 and α1(S326fs328X) subunit minigenes were either treated or untreated with cycloheximide (CHX) (100 μg/ml) for 6 h before the total RNA extraction. The mRNAs were measured by RT-PCR and were normalized to wild-type mRNAs without CHX treatment (***p < 0.001 vs mutant α1(S326fs328X) subunit without CHX; n = 7). C, Total cell lysates of HEK 293T or HeLa cells untreated (U) or treated with negative control siRNA (100 nm, neg) or transfected with hUpf-1 siRNA (10 nm, 50 and 100 nm) for 2–4 d were immunoblotted with hUpf-1 antibody. D, Wild-type α1 or mutant α1(A322D) minigenes with β2 and γ2S subunits or wild-type and α1(S326fs328X) minigenes alone were transfected into sister cultures of the HeLa cells from the experiment in C 2 d later after siRNA treatment. The total proteins were harvested 48–56 h later with or without 6 h of lactacystin (10 μm) (Lac) treatment and then analyzed. Equal amounts of protein (90 μg) were loaded, and α1 subunits (3 μg) were expressed to better visualize the α1(S326fs328X) protein. E, The total mutant α1 subunit protein IDVs with different treatments were normalized to their internal control and then normalized to the mock treated group (**p < 0.01 vs neg, †p < 0.05 vs lac; n = 4 for A322D, n = 8–11 for S326fs328X). F, Sister cultures from the experiment in E were harvested and analyzed for RT-PCR. The mRNA levels from mock, negative control or Upf-1 were normalized to the results from wild-type cells treated with the same conditions (**p < 0.01 vs neg; n = 6). In D–F, mock stands for siRNA mock transfection, neg for negative control siRNA transfection, Upf for human Upf-1 specific siRNA, Upf+lac for both human Upf-1 specific siRNA and Lactacystin treated and lac for only Lactacystin treated.
Treatment with the protein synthesis inhibitor cycloheximide (CHX) or silencing of the core component of the NMD machinery, Upf-1 (also known as Rent1), are widely used to determine if transcripts are subject to NMD (Montfort et al., 2006). CHX is an indirect NMD inhibitor since NMD activation is posttranscriptional, but translation-dependent, and ribosomal association is required during the pioneer round of translation. Upf-1 silencing is believed to produce direct suppression of NMD. The RNA helicase, Upf-1, is a central effector of NMD that links the translation-termination event to the assembly of a surveillance complex. Upf2 and Upf3 are believed to recruit and activate Upf1 on NMD substrates (Lykke-Andersen et al., 2000). Although the discrimination of PTCs from normal termination codons and the molecular links that trigger NMD are still unclear, it is well established that the core components of the NMD machinery are the conserved proteins, Upf-1, Upf-2 and Upf-3. PTC-containing mRNAs marked by the Upf complex are finally degraded by decay factors (Conti and Izaurralde, 2005). A recent study reported that Upf-1 phosphorylation triggers translation repression, which is a key step preceding mRNA decay (Isken et al., 2008). With CHX (100 μg/ml) treatment for 6 h, mutant α1(S326fs328X) subunit mRNA transcripts were increased in HeLa cells from 27.4 ± 5.2% to 84.5 ± 8.5% of wild-type α1 subunit mRNA after CHX treatment (Fig. 7B). Addition of CHX reversed the downregulation of the mutant transcripts, having no discernible effect on wild-type mRNA transcripts (Fig. 7B).
We then investigated the role of Upf-1 in reducing α1(S326fs328X) subunit levels. We transfected cells with synthetic short-interfering RNA (siRNA) duplexes targeting the human Upf-1 (hUpf-1) gene (Mendell et al., 2002; Chan et al., 2007) and harvested the cells 48 h after transfection. Western blotting of whole-cell lysates demonstrated significant loss of hUpf-1 protein in cells treated with hUpf-1 siRNA in HEK 293T (from 50 to 100 nm) and HeLa cells (from 10 to 100 nm) (Fig. 7C). Treatment with 100 nm siRNA for either 2 or 4 d significantly reduced hUpf-1 protein levels in both HEK 293T and HeLa cells, but the siRNA appeared to be more effective in HeLa cells at both time points (Fig. 7C). We thus used HeLa cells in these experiments. A similar effect, but to a lesser magnitude, was observed with HEK 293T cells (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Wild-type or mutant α1 subunit minigene constructs were transfected into HeLa cells treated 48 h earlier with siRNA. The cells were harvested after an additional 48 h for protein (Fig. 7D,E) and transcript (Fig. 7F) assays. All results were normalized to the cells treated only with siRNA delivery reagents (mock). Cells treated with siRNA (100 nm) against hUpf-1 (Upf) had increased expression of the mutant α1(S326fs328X) subunit compared with cells treated with the negative control (neg) siRNA (Fig. 7D,E) in both HEK 293T cells (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) and HeLa cells (0.80 ± 0.35 vs 2.48 ± 0.45; p = 0.006). Cells treated with lactacystin, in addition to the hUpf-1 specific siRNA, had increased expression of the mutant protein compared with cells treated with lactacystin alone in both cell lines [3.93 ± 0.83 (Upf + lac) vs 1.46 ± 0.20 (lac) (p = 0.013)]. There was no significant change in expression of wild-type α1 subunit proteins (Fig. 7D) with hUpf-1 specific siRNAs treatment (100 nm). In contrast, the mutant α1(A322D) subunit protein was similarly increased when treated with lactacystin in addition to the hUpf-1 specific siRNA or with lactacystin alone (Fig. 7D,E). The data suggest that the α1(S326fs328X) subunit protein was reduced mainly due to mRNA degradation, and that the α1(A322D) subunit protein was reduced mainly due to protein degradation by ERAD (Gallagher et al., 2007).
Similarly, normalized to mRNA levels in cells expressing wild-type α1 subunits with mock treatment, expression of mutant α1(S326fs328X) subunit transcripts was increased in cells treated with siRNA against hUpf-1 (Fig. 7F) compared with expression of mutant α1(S326fs328X) subunit transcripts in cells treated with negative control siRNA (11.3 ± 5 vs 91.4 ± 12; p = 0.002) in HeLa cells. There was no significant change in the wild-type (data not shown) or α1(A322D) (p = 0.28) subunit transcripts after hUpf-1 siRNA treatment (Fig. 7F). Together, our data suggest that elimination of the mutant α1(S326fs328X) subunit was due primarily to loss of transcripts due to mRNA surveillance (NMD), but that ERAD also played a role in reducing mutant protein levels.
Differential molecular mechanisms underlying two GABRA1 epilepsy mutations, 975delC, S326fs328X) and A322D
Although both GABRA1 epilepsy mutations, A322D and 975delC, S326fs328X, are located in the same domain of the α1 subunit (TM3) and both mutations produced haploinsufficiency, the molecular bases for the functional consequences are different. The α1(A322D) subunit mutation is a missense mutation, and the mutant mRNAs are translated and produce an unstable mutant protein, which is subject to rapid ERAD. The α1 subunit mutation, 975delC, S326fs328X, is a PTC-generating mutation and activated NMD. However, NMD is not complete; it degrades the majority of the mutant mRNA, but a small portion of mutant mRNA escapes NMD. The surviving mutant mRNA would result in translation of a mutant, truncated protein, which is not stable and would be subject to ERAD like the other mutant α1(A322D) subunit. The loss of mutant message and protein from both molecular pathways would directly contribute to the loss of the functional α1 subunit-containing GABAA receptors, and thus, the loss of inhibition that would result in epilepsy (Fig. 8).
Figure 8.

Different molecular pathways contribute to epileptogenesis of a missense and a frameshift PTC-generating epilepsy GABRA1 gene mutation. Although GABAA receptor α1 subunit mutations, 975delC, S326fs328X and A322D, are located in the same transmembrane domain (TM3), the two mutations caused haploinsufficiency through different molecular pathways. The α1 subunit mutation, A322D, caused mutant protein degradation through proteasome-ubiquitin pathway (ERAD). The α1 subunit mutation, 975delC, S326fs328X, activated NMD, resulting in loss of majority mutant mRNAs and the portion of mutant mRNA that escaped NMD translated mutant truncated protein that was subject to rapid degradation through the ubiquitin-proteasome pathway.
Discussion
Decreased expression of mutant α1(S326fs328X) subunits was due primarily to NMD of the mutant mRNA but also due to degradation of mutant protein by ERAD
Several GABAA receptor subunit gene mutations that should generate PTCs have been associated with IGEs and include three nonsense γ2 subunit mutations, Q1X, Q351X and W390X, and a γ2 subunit intron splice donor site mutation, IVS 6 + 2T->G, in addition to the present α1 subunit mutation, 975delC, S326fs328X (Harkin et al., 2002; Kananura et al., 2002; Hirose, 2006; Sun et al., 2008). The epilepsy phenotypic spectrum produced by these mutations is wide and includes febrile seizures, GEFS+, Dravet syndrome and CAE. For unknown reasons there are also substantial intrafamilial variations in seizure phenotype for individual mutations such as the γ2 subunit mutations, Q1X and Q351X (Macdonald et al., 2006). Studies of PTC-generating mutations in sodium channels indicated that mutation mosaicism was associated with the intrafamilial phenotypic variations (Marini et al., 2006; Morimoto et al., 2006). However, the bases for phenotypic variations in patients harboring GABAA receptor PTC-generating mutations have never been explored.
NMD is a conserved mRNA surveillance pathway in all eukaryotes, and our study indicated that PTC-generating mutations in a CNS ion channel gene would be subject to the same cellular fate as seen with other genes and in other cell types. The role of NMD has been confirmed in a number of human diseases such as Marfan's syndrome and cystic fibrosis. NMD is rarely complete, however, with ∼5–25% of the mutant mRNA escaping NMD (Kuzmiak and Maquat, 2006a). Similarly, our data demonstrated that the α1 subunit mutation, 975delC, S326fs328X, activated NMD, resulting a substantial loss of the mutant mRNA (∼80% in HeLa cells and rat cortical neurons), but NMD was not complete, and small amounts of mutant protein were produced. The mutant protein was not stable and was likely subject to rapid proteasomal degradation, resulting in a reduced half-life (64 min) compared with that of wild-type α1 subunits (194 min).
The intracellular processing of two GABRA1 mutations, 975delC, S326fs328X and A322D, is different
Our data suggested that the functional defect caused by the α1 subunit mutation, 975delC, S326fs328X, was due to degradation of the mutant mRNA and protein. The mRNA reduction was reversed by treatment with either the indirect NMD inhibitor CHX or by suppression of the NMD machinery core factor, Upf-1. After suppression of Upf-1, mutant α1(S326fs328X) subunit protein was increased to over twofold of the mock treated cells and increased nearly fourfold in cells with Upf-1 suppression in addition to proteasomal inhibition. In contrast, proteasomal inhibition alone only increased mutant protein to 145% of the mock treated cells. This suggested that a substantial portion of mutant protein reduction was due to loss of mutant mRNA and that mutant protein produced was subject to rapid proteasomal degradation. In contrast, the mutant α1(A322D) subunit was degraded through the ubiquitin-proteasomal system (Gallagher et al., 2007). However, it is unclear why the α1 subunit mutation, A322D, was associated with juvenile myoclonic epilepsy while the α1 subunit mutation, S326fs328X, was associated with CAE, although both mutations produced functional haploinsufficiency in vitro. The differential intracellular processing of these two different mutations may at least partially contribute to their phenotypic variations. For example, α1(S326fs328X) subunits were lost mainly through NMD at the transcriptional level while α1(A322D) subunits were lost through ERAD after translation. It is unclear if there are any negative effects imposed by activation of the quality control mechanisms, NMD or ERAD, themselves. In addition, our data suggested that both NMD and ERAD efficiency varies among cell types and with different mutant α1 subunit copy numbers. Future studies focusing on the efficiency of NMD and ERAD in different neuronal cell types and during different developmental time windows may further clarify this discrepancy.
Residual mutant α1(S326fs328X) subunit protein activated ERAD and increased Bip association
The functional consequence of the α1 subunit mutation, del975C, S326fs328X, is likely to be due, at least in part, to haploinsufficiency of the GABRA1 gene. Based on our data in HEK 293T cells, there was ∼59% of total wild-type α1 subunit produced in the haploinsufficiency control condition and 56% of total wild-type α1 protein in the mixed α1/α1(S326fs328X) subunit condition. There was approximately half of the total wild-type current amplitude produced in the mixed condition, which was similar to the current produced in the haploinsufficiency control condition. There were no current kinetic differences among these three groups, suggesting that the functional receptors on the cell surface were recorded mainly from wild-type α1β2γ2S receptors.
We cannot exclude, however, a small dominant negative effect of truncated α1(S326fs328X) subunits that escape NMD and have a dominant negative effect on GABAA receptor assembly. We demonstrated that the mutant protein was degraded rapidly, likely through the ubiquitin-proteasomal pathway. The stress of removal of mutant misfolded protein would cause an imbalance in the load of unfolded proteins that enter the ER and the folding capacity of the ER environment (Hartl, 1996). Given the fact that heterozygous α1(+/−) subunit knock-out mice do not develop epilepsy, there may be a subtle difference in functional and cellular consequences between α1(+/−) knock-outs and mixed α1(S326fs328X) subunit mutants. For example, the enhanced chaperone activity of Bip and an increased association with mutant subunits in affected patients would not occur in α1(+/−) knock-out mice. This subtle difference in vivo may be enough to cause a clinical phenotype during a restricted developmental period.
NMD attenuates disease phenotype but the presence of small amounts of mutant protein produces dominant negative effects in other human diseases
Activation of NMD can rid cells of most transcripts containing PTCs and reduce synthesis of the truncated proteins that have potentially deleterious effects inside cells. This may reduce the manifestation of some genetic diseases if the single wild-type allele is sufficient for physiological function or may only produce a mild phenotype compared with some C-terminal truncation mutations that have dominant-negative effects on wild-type proteins. Triggering NMD and escaping NMD may cause distinct disease phenotypes (Inoue et al., 2004). In osteogenesis imperfecta, Stickler syndrome and Marfan's syndrome, it has been postulated that NMD moderates the phenotype compared with that produced by missense mutations. Marfan's syndrome is an autosomal dominant systemic disorder of connective tissue caused by mutations in the extracellular matrix protein fibrillin 1 (Dietz et al., 1993). A genotype-phenotype difference has been noticed in Marfan's syndrome; patients with low levels of mutant FBN1 due to NMD often exhibit milder phenotypes that fall outside the clinical criteria required for a Marfan's syndrome diagnosis. It was also suggested that the minimal mutant transcript levels of ∼7–10% of wild-type levels may produce sufficient amounts of truncated protein to produce dominant-negative interference with the wild-type allele leading to severe Marfan's syndrome (Montgomery et al., 1998).
A molecular mechanism similar to that in Marfan's syndrome may help explain why mutations in the same gene cause different clinical phenotypes and with different severities in GABAA receptor gene mutations associated with IGEs. Namely, how complete the NMD is in the individual may be associated with the intrafamilial phenotypic variation among family members carrying the same mutation. A similar discrepancy in genotype-phenotype correlation has been reported for the GABAA receptor γ2(+/−) knock-out mouse that displayed only a hyperanxiety phenotype, and in humans an early exon PTC-causing splice-donor site mutation produced CAE and febrile seizures (Kananura et al., 2002) and a C-terminal γ2 subunit truncation, Q351X, caused the more severe GEFS+ epilepsy syndrome (Harkin et al., 2002). The pathogenesis of the GABAA receptor α1 subunit mutation, 975delC, S326fs328X, is due mainly to NMD with a small portion of mutant transcripts escaping NMD and generating a truncated mutant protein that is degraded by ERAD. However, based on our observation in vitro and on previous reports, although the mRNA surveillance is conserved, the efficiency of NMD for a certain mutant transcript is cell-specific (Linde et al., 2007). The efficiency is likely dependent on the capacity of the NMD machinery, such as expression of the NMD core components such as UPF factors and the relative abundance of the mutant transcripts in a given cell. Thus, there may be a different balance of NMD reduction of the mutant mRNA or ERAD reduction of the mutant protein in neurons in different regions of the brain, during different developmental stages or among different individuals.
Remaining challenges and potential therapeutic strategies
Our present work suggests that ion channel PTC mutations can cause NMD, and that neurons are likely to share common mRNA surveillance mechanisms present in other cell types, thus extending the molecular underpinning of and therapeutic strategy to develop treatment for ion channel diseases. For example, promotion of read-through with aminoglycoside treatment has been used in patients with cystic fibrosis (Linde et al., 2007). The same strategy may be useful for some patients with epilepsy who harbor PTC-generating mutations if the mutant protein is functional. From a therapeutic point of view, since the α1 subunit, 975delC, S326fs328X, mutation causes a frameshift, attempted promotion of read-through with aminoglycoside treatment to restore α1 subunit expression (Howard et al., 2004) would not be feasible. However, our data suggest that the pathology of this mutation is likely to be a combination of reduction of channel function and disturbance of cellular homeostasis due to the presence of small amounts of mutant protein. Thus, a therapeutic strategy to eliminate production of mutant protein using siRNA targeting of the mutant transcripts might be a useful approach (Rodriguez-Lebron and Paulson, 2006).
Footnotes
This work was supported by National Institutes of Health Grant R01 NS51590 to R.L.M. and a CURE (Citizens United for Research in Epilepsy) grant to J.-Q.K. We thank Drs. Miles F. Wilkinson and Martin Gallagher for their constructive advice, Hannah Yan for technical assistance, and Mengnan Tian for helpful discussions.
References
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–118. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- Bühler M, Paillusson A, Mühlemann O. Efficient downregulation of immunoglobulin mu mRNA with premature translation-termination codons requires the 5′-half of the VDJ exon. Nucleic Acids Res. 2004;32:3304–3315. doi: 10.1093/nar/gkh651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busi F, Cresteil T. CYP3A5 mRNA degradation by nonsense-mediated mRNA decay. Mol Pharmacol. 2005;68:808–815. doi: 10.1124/mol.105.014225. [DOI] [PubMed] [Google Scholar]
- Chan WK, Huang L, Gudikote JP, Chang YF, Imam JS, MacLean JA, 2nd, Wilkinson MF. An alternative branch of the nonsense-mediated decay pathway. EMBO J. 2007;26:1820–1830. doi: 10.1038/sj.emboj.7601628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ. Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J Biol Chem. 1996;271:89–96. doi: 10.1074/jbc.271.1.89. [DOI] [PubMed] [Google Scholar]
- Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Dietz HC, McIntosh I, Sakai LY, Corson GM, Chalberg SC, Pyeritz RE, Francomano CA. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics. 1993;17:468–475. doi: 10.1006/geno.1993.1349. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Shen W, Song L, Macdonald RL. Endoplasmic reticulum retention and associated degradation of a GABAA receptor epilepsy mutation that inserts an aspartate in the M3 transmembrane segment of the alpha 1 subunit. J Biol Chem. 2005;280:37995–38004. doi: 10.1074/jbc.M508305200. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Ding L, Maheshwari A, Macdonald RL. The GABAA receptor alpha1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc Natl Acad Sci U S A. 2007;104:12999–13004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield LJ, Jr, Sun F, Neelands TR, Burgard EC, Donnelly JL, Macdonald RL. Expression of functional GABAA receptors in transfected L929 cells isolated by immunomagnetic bead separation. Neuropharmacology. 1997;36:63–73. doi: 10.1016/s0028-3908(96)00150-5. [DOI] [PubMed] [Google Scholar]
- Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F, Wallace RH, Richards MC, Williams DA, Mulley JC, Berkovic SF, Scheffer IE, Petrou S. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–536. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hefferon TW, Broackes-Carter FC, Harris A, Cutting GR. Atypical 5′ splice sites cause CFTR exon 9 to be vulnerable to skipping. Am J Hum Genet. 2002;71:294–303. doi: 10.1086/341664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 2006;70(Suppl 1):S206–S217. doi: 10.1016/j.eplepsyres.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, Mancias P, Butler IJ, Wilkinson MF, Wegner M, Lupski JR. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133:314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, Rebstock J, Heils A, Steinlein OK. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–1141. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Macdonald RL. The GABAA receptor γ2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of α1β2γ2S receptors in the endoplasmic reticulum. J Neurosci. 2004;24:8672–8677. doi: 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci. 2006;26:2590–2597. doi: 10.1523/JNEUROSCI.4243-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Linde L, Boelz S, Neu-Yilik G, Kulozik AE, Kerem B. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur J Hum Genet. 2007;15:1156–1162. doi: 10.1038/sj.ejhg.5201889. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen J, Shu MD, Steitz JA. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell. 2000;103:1121–1131. doi: 10.1016/s0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ, Feng HJ. GABA(A) receptor mutations associated with generalized epilepsies. Adv Pharmacol. 2006;54:147–169. doi: 10.1016/s1054-3589(06)54007-4. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, Bufler J, Lerche H, Heils A. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay in mammals. J Cell Sci. 2005;118:1773–1776. doi: 10.1242/jcs.01701. [DOI] [PubMed] [Google Scholar]
- Marini C, Mei D, Helen CJ, Guerrini R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia. 2006;47:1737–1740. doi: 10.1111/j.1528-1167.2006.00675.x. [DOI] [PubMed] [Google Scholar]
- Mendell JT, ap Rhys CM, Dietz HC. Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science. 2002;298:419–422. doi: 10.1126/science.1074428. [DOI] [PubMed] [Google Scholar]
- Montfort M, Chabás A, Vilageliu L, Grinberg D. Analysis of nonsense-mediated mRNA decay in mutant alleles identified in Spanish Gaucher disease patients. Blood Cells Mol Dis. 2006;36:46–52. doi: 10.1016/j.bcmd.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Montgomery RA, Geraghty MT, Bull E, Gelb BD, Johnson M, McIntosh I, Francomano CA, Dietz HC. Multiple molecular mechanisms underlying subdiagnostic variants of Marfan syndrome. Am J Hum Genet. 1998;63:1703–1711. doi: 10.1086/302144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto M, Mazaki E, Nishimura A, Chiyonobu T, Sawai Y, Murakami A, Nakamura K, Inoue I, Ogiwara I, Sugimoto T, Yamakawa K. SCN1A mutation mosaicism in a family with severe myoclonic epilepsy in infancy. Epilepsia. 2006;47:1732–1736. doi: 10.1111/j.1528-1167.2006.00645.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Paulson HL. Allele-specific RNA interference for neurological disease. Gene Ther. 2006;13:576–581. doi: 10.1038/sj.gt.3302702. [DOI] [PubMed] [Google Scholar]
- Shyu AB, Wilkinson MF, van Hoof A. Messenger RNA regulation: to translate or to degrade. EMBO J. 2008;27:471–481. doi: 10.1038/sj.emboj.7601977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson LS, Maquat LE. Cytoplasmic mRNA for human triosephosphate isomerase is immune to nonsense-mediated decay despite forming polysomes. Biochimie. 1996;78:1043–1047. doi: 10.1016/s0300-9084(97)86728-4. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhang Y, Liang J, Liu X, Ma X, Wu H, Xu K, Qin J, Qi Y, Wu X. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J Hum Genet. 2008;53:769–774. doi: 10.1007/s10038-008-0306-y. [DOI] [PubMed] [Google Scholar]
- Turnbull EL, Rosser MF, Cyr DM. The role of the UPS in cystic fibrosis. BMC Biochem. 2007;8(Suppl 1):S11. doi: 10.1186/1471-2091-8-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]