

Abstract
Prevotella ruminicola 23 is an obligate anaerobic bacterium in the phylum Bacteroidetes that contributes to hemicellulose utilization within the bovine rumen. To gain insight into the cellular machinery that this organism elaborates to degrade the hemicellulosic polymer xylan, we identified and cloned a gene predicted to encode a bifunctional xylanase-ferulic acid esterase (xyn10D-fae1A) and expressed the recombinant protein in Escherichia coli. Biochemical analysis of purified Xyn10D-Fae1A revealed that this protein possesses both endo-β-1,4-xylanase and ferulic acid esterase activities. A putative glycoside hydrolase (GH) family 3 β-d-glucosidase gene, with a novel PA14-like insertion sequence, was identified two genes downstream of xyn10D-fae1A. Biochemical analyses of the purified recombinant protein revealed that the putative β-d-glucosidase has activity for pNP-β-d-xylopyranoside, pNP-α-l-arabinofuranoside, and xylo-oligosaccharides; thus, the gene was designated xyl3A. When incubated in combination with Xyn10D-Fae1A, Xyl3A improved the release of xylose monomers from a hemicellulosic xylan substrate, suggesting that these two enzymes function synergistically to depolymerize xylan. Directed mutagenesis studies of Xyn10D-Fae1A mapped the catalytic sites for the two enzymatic functionalities to distinct regions within the polypeptide sequence. When a mutation was introduced into the putative catalytic site for the xylanase domain (E280S), the ferulic acid esterase activity increased threefold, which suggests that the two catalytic domains for Xyn10D-Fae1A are functionally coupled. Directed mutagenesis of conserved residues for Xyl3A resulted in attenuation of activity, which supports the assignment of Xyl3A as a GH family 3 β-d-xylosidase.
β-1,4-linked xylopyranose is the principal component of plant cell wall hemicellulose, which represents the second largest reservoir of fixed carbon in the biosphere (11, 30, 34, 38, 42, 72). The catabolic breakdown of hemicellulose thus represents a critical step in the recycling of carbon in nature and has been targeted as a subject of intense research with respect to renewable energy resources. Two enzymes of principal importance for recycling hemicellulosic material are endo-1,4-β-xylanases (EC 3.2.1.8), which cleave the xylan backbone, and β-d-xylosidases (EC 3.2.1.37), which cleave xylose monomers from the nonreducing end of xylo-oligosaccharides (17).
Prevotella ruminicola 23 is an important member of the anaerobic rumen microbiota (60) that contributes to the utilization of noncellulosic polysaccharides, such as starch and xylan (15, 25, 35, 55). Despite its documented importance in rumen physiology, relatively little is known about the cellular machinery that P. ruminicola 23 employs to harvest energy from hemicellulosic substrates. Several studies have explored the carbohydrate active enzymes from the related taxon Prevotella bryantii B14 (previously classified as P. ruminicola B14) (25, 27, 28, 50, 52, 66, 73); however, less is known about the xylanolytic system that P. ruminicola 23 elaborates. These two species share 88.9% nucleotide identity in their 16S rRNA genes (1,473 nucleotides aligned); however, there is evidence that these two species exhibit differences in their xylanolytic systems (2). Two xylanases from P. ruminicola 23 have been characterized: a 66-kDa xylanase with significant sequence identity to glycoside hydrolase (GH) family 5 endoglucanases (69, 70) and an 80-kDa xylanase related to GH family 10 endoxylanases which possesses a 260-amino-acid insertion within the putative catalytic domain (25). Recently, the draft genome sequence for P. ruminicola 23 has been obtained (http://www.tigr.org/tdb/rumenomics/), and a large number of GHs have been identified. Consistent with the predicted role of P. ruminicola 23 in hemicellulose degradation, many of these genes appear to encode enzymes important for hemicellulose depolymerization and utilization.
In this study, we report the biochemical properties for two GHs with unique domain architectures from P. ruminicola 23. These two enzymes function synergistically to release monomeric sugars from a complex hemicellulosic substrate, xylan. This study, therefore, provides important insight into the molecular strategies for xylan depolymerization by P. ruminicola 23.
MATERIALS AND METHODS
Materials.
P. ruminicola 23 (ATCC 19189) was obtained from our culture collection preserved in liquid nitrogen vapor and housed at the Department of Animal Sciences, University of Illinois at Urbana-Champaign. Escherichia coli JM109 and BL21-CodonPlus (DE3) RIL competent cells were acquired from Stratagene (La Jolla, CA). The pGEM-T TA-cloning vector was purchased from Promega (Madison, WI). The pET-28a expression vector was obtained from Novagen (San Diego, CA). NdeI and XhoI restriction endonucleases were purchased from New England Biolabs (Ipswich, MA). ExTaq high-fidelity DNA polymerase was obtained from TaKaRa Bio (Madison, WI). The UltraClean DNA isolation kit was obtained from Mo Bio Laboratories, Inc. (Carlsbad, CA). Xylo-oligosaccharides (X2 to X6) were obtained from Megazyme (Bray, Ireland). Methyl ferulate was obtained from Apin Chemicals Ltd. (Abingdon, United Kingdom). Gel filtration standards were obtained from Bio-Rad (Hercules, CA). All other reagents were of the highest possible purity and were purchased from Sigma-Aldrich (St. Louis, MO).
Sequencing, cloning, expression, and purification of xyn10D-fae1A and xyl3A.
The genome of P. ruminicola 23 was sequenced by the North American Consortium for Fibrolytic Rumen Bacteria in collaboration with The Institute for Genomic Research. Autoannotation of the P. ruminicola genome identified ORF02827 (xyn10D-fae1A) as a gene encoding a bifunctional xylanase-ferulic acid esterase and ORF02829 (xyl3A) as a gene encoding a putative β-glucosidase.
P. ruminicola 23 was maintained and grown in a standard anaerobically prepared medium as described previously (32). DNA was isolated from mid-log-phase cultures and was purified with an UltraClean DNA isolation kit. The DNA sequences corresponding to the entire ORFs of xyn10D-fae1A and xyl3A were amplified from P. ruminicola 23 genomic DNA by PCR using the primers ORF02827-F (5′-CATATGAAGAAACTATTAGTAGCGTTATCG-3′) and ORF02827-R (5′-CTCGAGTTACTTAAAGAGACTCTGAGCCATCTTTTC-3′) for xyn10D-fae1A and ORF02829-F (5′-CATATGAAATATCAACTATTCTTATCATTGGC-3′) and ORF02829-R (5′-CTCGAGCTATAAAGTGATGGTCAGTTTTTTCAGATC-3′) for xyl3A. The primer sets were engineered to incorporate 5′ NdeI and 3′ XhoI restriction sites for subsequent directional cloning. The resulting amplicons were then cloned into the pGEM-T vector via TA cloning and subcloned in frame with the hexahistidine-encoding sequence of a modified pET-28a expression vector by replacing the NdeI-XhoI polylinker. Thus, recombinant expression of the gene products results in N-terminal polyhistidine fusion proteins. The pET-28a vector was modified to replace the gene for kanamycin resistance with that for ampicillin resistance (9). The integrity of the cloned xyn10D-fae1A and xyl3A genes was confirmed by DNA sequencing (W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign).
The resulting plasmid constructs, pET28-xyn10D-fae1A and pET28-xyl3A, were introduced into E. coli BL-21 CodonPlus (DE3) RIL competent cells by heat shock and grown overnight in lysogeny broth (4, 5) (LB) (10 ml) supplemented with ampicillin (100 μg/ml) and chloramphenicol (50 μg/ml) at 37°C and with aeration. After 12 h, the starter cultures were diluted into fresh LB (1 liter) supplemented with ampicillin and chloramphenicol and cultured at 37°C with aeration until the absorbance at 600 nm reached 0.3. The culture was then incubated at 16°C with aeration, and protein expression was induced by the addition of isopropyl β-d-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM. After 16 h, the cells were harvested by centrifugation (4,000 × g, 15 min, 4°C), resuspended in 35 ml lysis buffer (50 mM sodium phosphate, 300 mM NaCl [pH 7.0]), and ruptured by two passages through a French pressure cell (American Instrument Company Inc., Silver Spring, MD). The cell lysate was then clarified by centrifugation at 30,000 × g for 30 min at 4°C and purified utilizing Talon polyhistidine tag purification resin from Clontech (Mountain View, CA) as described by the manufacturer. Aliquots of eluted fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using the method described by Laemmli (43), and protein bands were visualized by staining with Coomassie brilliant blue G-250. Elution fractions were pooled and dialyzed against storage buffer (50 mM Tris-HCl, 200 mM NaCl, 1 mM dithiothreitol [pH 7.0]), and assays were performed immediately.
The protein concentrations were determined by a kit from Bio-Rad (Hercules, CA) based on the Bradford method (7) with bovine serum albumin as the standard.
Size exclusion chromatography.
Size exclusion chromatography was carried out with an AKTA Purifier 900 fast protein liquid chromatography system equipped with a Superdex 200 10/300 GL size exclusion column and a UV detector, all obtained from GE Healthcare (Piscataway, NJ). Xyn10D-Fae1A (1 mg/ml; 100 μl), Xyl3A (1 mg/ml; 100 μl), or a gel filtration standard mixture was injected onto the column, which had been pre-equilibrated with a Tris buffer (50 mM Tris·HCl, 150 mM NaCl; pH 8.0) liquid phase at a flow rate of 0.5 ml/min. Standard curves were generated by plotting the logarithm of the molecular weights of the gel filtration standards versus retention times. Experimental retention times for three independent experiments were used to calculate the apparent molecular weights of Xyl3A and Xyn10D-Fae1A from the standard curve.
Evaluation of xylanase and esterase activities on agar plates.
Enzyme-catalyzed hydrolysis of plant cell wall polysaccharides was evaluated by spotting purified Xyn10D-Fae1A (5 μg) onto agar plates (0.8%) containing oat spelt xylan (OSX) (0.1%), carboxymethyl cellulose (0.1%), or locust bean gum (0.1%). The plates were then incubated at 37°C for 16 h and were stained by incubation with Congo red (0.1%) for 5 min followed by destaining with NaCl (1 M) (63). Esterase activity was assessed by adapting the method described by Lanz and Williams to an agar plate (45). Briefly, purified Xyn10D-Fae1A (14 μg, 28 μg, or 56 μg) was spotted onto an agar plate (0.8%) containing 1-napthyl butyrate (5 mM) and Tween 80 (2%). The plate was then incubated at 37°C for 16 h, stained with fast garnet GBC sulfate (5 mg/ml in 10% SDS) for 10 min, and then washed with Tween 80 (2%).
Hydrolysis of xylo-oligosaccharides and OSX.
The capacity of P. ruminicola Xyn10D-Fae1A and Xyl3A to hydrolyze xylo-oligosaccharides was assessed by resolving and detecting the hydrolysis products utilizing thin-layer chromatography (TLC). Initial biochemical studies had revealed that the optimum pHs for activity for Xyn10D-Fae1A and Xyl3D were 5.0 and 6.0, respectively (data not shown). Thus, for measuring the hydrolysis of xylo-oligosaccharides, reaction mixtures (10 μl) were prepared in phosphate buffer (50 mM sodium phosphate, 100 mM NaCl; pH 6.0 [Xyn10D-Fae1A] or pH 5.0 [Xyl3A]) with X2 to X6 (100 to 150 μg), and reactions were initiated by the addition of Xyn10D-Fae1A (10 μM, final concentration) or Xyl3A (12 μM, final concentration). Following incubation at 37°C for 7.5 h, 2 volumes of ethanol was added, and the fluid was evaporated using a SC110A Speed Vac concentrator (Savant, Ramsey, MN). The dry matter was then dissolved in 2.5 μl of H2O and spotted onto a DC-Plastikfolien silica gel 60 F254 TLC plate from Merck (Whitehouse Station, NJ) to resolve the products (40).
To identify the products of hydrolysis of OSX following incubation with Xyn10D-Fae1A or Xyl3A, TLC was utilized. Reaction mixtures (250 μl) were prepared in phosphate buffer (50 mM sodium phosphate, 100 mM NaCl; pH 6.0 [Xyn10D-Fae1A], pH 5.0 [Xyl3A], or pH 5.5 [Xyn10D-Fae1A plus Xyl3A]) with OSX (1%), and reactions were initiated by the addition of Xyn10D-Fae1A (0.50 μM), Xyl3A (0.50 μM), or both enzymes (0.50 μM each). Following incubation at 37°C for 14 h, the reaction mixtures were centrifuged, and then 2.5 μl or 5.0 μl was spotted onto TLC plates.
Monomeric xylose (X1) and xylo-oligosaccharides (X2 to X6) (25 μg each) were used as standards, and the TLC plates were developed in an acetone-ethyl acetate-acetic acid (2:1:1, vol/vol/vol) mobile phase. The products were then visualized by spraying the plates with a 1:1 (vol/vol) mixture of methanolic orcinol (0.2% wt/vol) and sulfuric acid (20% vol/vol), and then the plates were heated at 100°C for 5 min (33, 53).
To quantify the amount of reducing equivalents that were released following incubation of Xyn10D-Fae1A, Xyl3A, or both with OSX, a para-hydroxybenzoic acid hydrazide (PAHBAH) assay was performed as described previously by Lever (47).
Hydrolysis of pNP-linked sugars.
Enzyme-catalyzed hydrolysis of para-nitrophenyl (pNP)-linked monosaccharide substrates was assayed using a thermostated Cary 300 UV-visible-spectrum spectrophotometer from Varian Inc. (Palo Alto, CA). pNP substrates (10 mM) in acetate buffer (115 μl; 50 mM sodium acetate, 100 mM NaCl; pH 5.0) were incubated at 37°C in the presence or absence of Xyl3A (0.74 μM) for 10 min, and the level of pNP release was determined by continuously monitoring the absorbance at 400 nm. The extinction coefficient for pNPl at pH 5.0 and a wavelength of 400 nm was measured as 319 M−1 cm−1.
Analysis of methyl ferulate hydrolysis.
To evaluate the capacity of Xyn10D-Fae1A and the Xyn10D-Fae1A site-directed mutants (see below) to hydrolyze the ester linkage of methyl ferulate, we utilized an HPLC-based detection system. Reaction mixtures (2 ml) were prepared with methyl ferulate (5 mM) in phosphate buffer (50 mM sodium phosphate, 100 mM NaCl; pH 6.0), and reactions were initiated by the addition of wild-type or mutant Xyn10D-Fae1A (1.2 μM). Following incubation at 37°C for 30 min, an equal volume of ethyl acetate was added to the mixture, vortexed, and then centrifuged at 6,000 × g for 10 min. The ethyl acetate layer was then transferred to a new tube and evaporated under N2 gas, and the extracted ferulic acid was dissolved in 1 ml of 50% methanol. The concentration of ferulic acid was then determined by HPLC as described by Wang et al. (67).
Site-directed mutagenesis.
Mutagenesis was performed using a QuikChange Multi site-directed mutagenesis kit from Stratagene (La Jolla, CA). First, mutagenic primers were engineered with the desired mutation in the center of the primer and ∼15 bases of correct sequence on either side (Table 1). Reaction mixtures were prepared as described in the QuikChange protocol with pGEMT-xyn10D-fae1A and pGEMT-xyl3A as the DNA templates for generation of the Xyn10D-Fae1A and Xyl3A mutants, respectively. After cycling of the reaction mixture 18 times in a PCR thermal cycler, the mixture was digested with DpnI, and the resulting DNA was transformed into chemically competent E. coli JM109 cells by heat shock. The mutant genes were then subcloned into the pET-28a vector and sequenced to ensure that the appropriate mutations were introduced, while the rest of the gene sequences remained unchanged. Expression and purification of the mutant recombinant proteins were performed as described above for wild-type Xyn10D-Fae1A and Xyl3A.
TABLE 1.
Primer sequences used for mutagenesis
| Protein | Desired mutationa | Primer nameb | Primer sequence (5′→3′)c |
|---|---|---|---|
| Xyn10D-Fae1A | Glu280Ser | xyn10D-fae1A-E280S | 5′-ACTATCCACATTAC ATCGCTGGATCTGCGTACC-3′ |
| Xyn10D-Fae1A | Ser629Ala | xyn10D-fae1A-S629A | 5′-CGTGCGATGGCAGGTCTGGCGATGGGTGGTATGGAAACCAAG-3′ |
| Xyl3A | Arg149Leu | xyl3A-R149L | 5′-GACCCTCGTTGGGGACTTGGACAGGAAACCTAT-3′ |
| Xyl3A | Lys188Leu | xyl3A-K188L | 5′-GCTCTGGGCCTGTGCCCTACACTATGCCGTGCACA-3′ |
| Xyl3A | Tyr237Phe | xyl3A-Y237F | 5′-GAGGTGATGTGTGCCTTTCAGCGTCTGGACGAT-3′ |
| Xyl3A | Asp269Ala | xyl3A-D269A | 5′-TACCTCGTTGTGAGCGCATGTGGTGCTGTTAGC-3′ |
| Xyl3A | Asp269Asn | xyl3A-D269N | 5′-TACCTCGTTGTGAGCAACTGTGGTGCTGTTAGC-3′ |
| Xyl3A | Glu594Gln | xyl3A-E594Q | 5′-AATATCGATTATCAACAGACCATCGCCCAGCTG-3′ |
| Xyl3A | Glu616Ala | xyl3A-E616A | 5′-GCTCCCAGTCTGGCGGGCGAGGAGATG-3′ |
Residues were selected for conservative mutagenesis due to their conservation as predicted by alignment of the primary amino acid sequences for family 3 GHs with biochemically defined catalytic activities.
Primers were designed in accordance with the QuikChange Multi protocol by Stratagene (La Jolla, CA) and were synthesized by Integrated DNA Technologies (Coralville, IA).
Underlining indicates engineered codon mutations.
Steady-state kinetic measurements for wild-type and mutant Xyl3A.
Kinetic studies of the wild type and mutant Xyl3A proteins were performed using a Cary 300 UV-visible-spectrum spectrophotometer equipped with a circulating water bath from Varian Inc. (Palo Alto, CA). pNP β-d-xylopyranoside (pNPX) (0 to 10 mM) was incubated in acetate buffer (50 mM sodium acetate, 100 mM NaCl; pH 5.0) at 37°C in a 1-cm-path-length quartz cuvette, and reactions were initiated by the addition of Xyl3A (0.74 μM). Hydrolysis of pNPX was continuously monitored by recording the UV signal at 400 nm. Initial rate data were then plotted against the substrate concentration, and kinetic values were estimated by applying a nonlinear curve fit using GraphPad Prism v5.01 from GraphPad Software (San Diego, CA). The extinction coefficient for para-nitrophenol at pH 5.0 and a wavelength of 400 nm was measured as 319 M−1 cm−1.
For mutants with very low activities, kcat(apparent) was determined at 10 mM pNPX and an enzyme concentration of 7.4 μM. Each kcat determination was performed in triplicate, and the rates of spontaneous pNPX hydrolysis (absence of enzyme) were subtracted from the rates of enzyme-catalyzed reactions.
Nucleotide sequence accession numbers.
The gene sequences for xyn10D-fae1A and xyl3A have been deposited in the NCBI GenBank database with accession numbers FJ713437.1 and FJ713438.1, respectively.
RESULTS
Identification, cloning, and expression of P. ruminicola 23 Xyn10D-Fae1A.
To identify potential xylanolytic gene clusters in P. ruminicola 23, the genome sequence of the bacterium was scanned for putative GH family 10 or 11 endo-β-1,4-xylanases, which generate xylo-oligosaccharides from a xylan source. This scan revealed the presence of three putative GH 10 endoxylanases; however, no GH 11 endoxylanases were identified. One of these putative endoxylanase genes (ORF02827) was predicted to encode a bifunctional polypeptide, composed of a GH 10 domain in the N-terminal half and a carbohydrate esterase (CE) family 1 ferulic acid esterase domain in the C-terminal half (10). Furthermore, ORF02827 was identified near a gene (ORF02829) encoding a putative GH family 3 β-glucosidase (Fig. 1). Since a family 10 xylanase, XynC, has already been identified in this bacterium, the gene encoding the putative bifunctional protein was designated xyn10D-fae1A. The gene was amplified by PCR and cloned into an expression vector for recombinant protein expression in E. coli. Xyn10D-Fae1A was expressed as an N-terminal hexahistidine fusion protein to facilitate purification by metal affinity chromatography. The predicted molecular mass for the hexahistidine fusion protein, His-Xyn10D-Fae1A, is 84 kDa, which is in agreement with the size of the purified protein estimated by comparison with molecular weight markers through SDS-PAGE analysis (Fig. 2A). To evaluate the quaternary structure of Xyn10D-Fae1A, the apparent molecular mass of purified Xyn10D-Fae1A was determined by size exclusion chromatography. The molecular mass of Xyn10D-Fae1A was calculated from the retention times of the peak absorbance versus calibration standards of known molecular masss. The apparent molecular mass of Xyn10D-Fae1A was calculated to be 124 ± 1.7 kDa, and since the molecular mass of a monomer of His-Xyn10D-Fae1A is ∼84 kDa, this result suggested a protein existing as either a monomer or homodimer in solution (Fig. 2B).
FIG. 1.

Identification of a putative xylanase-esterase gene (xyn10D-fae1A) from the genome of P. ruminicola 23. The genome of P. ruminicola 23 was sequenced by the North American Consortium for Fibrolytic Rumen Bacteria in collaboration with The Institute for Genomic Research. ORF02827 was annotated as a putative bifunctional xylanase-esterase. Downstream of the xylanase-esterase are genes predicted to encode a hypothetical protein, a β-d-glucosidase, an ABC transporter, and a hybrid two-component system regulator.
FIG. 2.
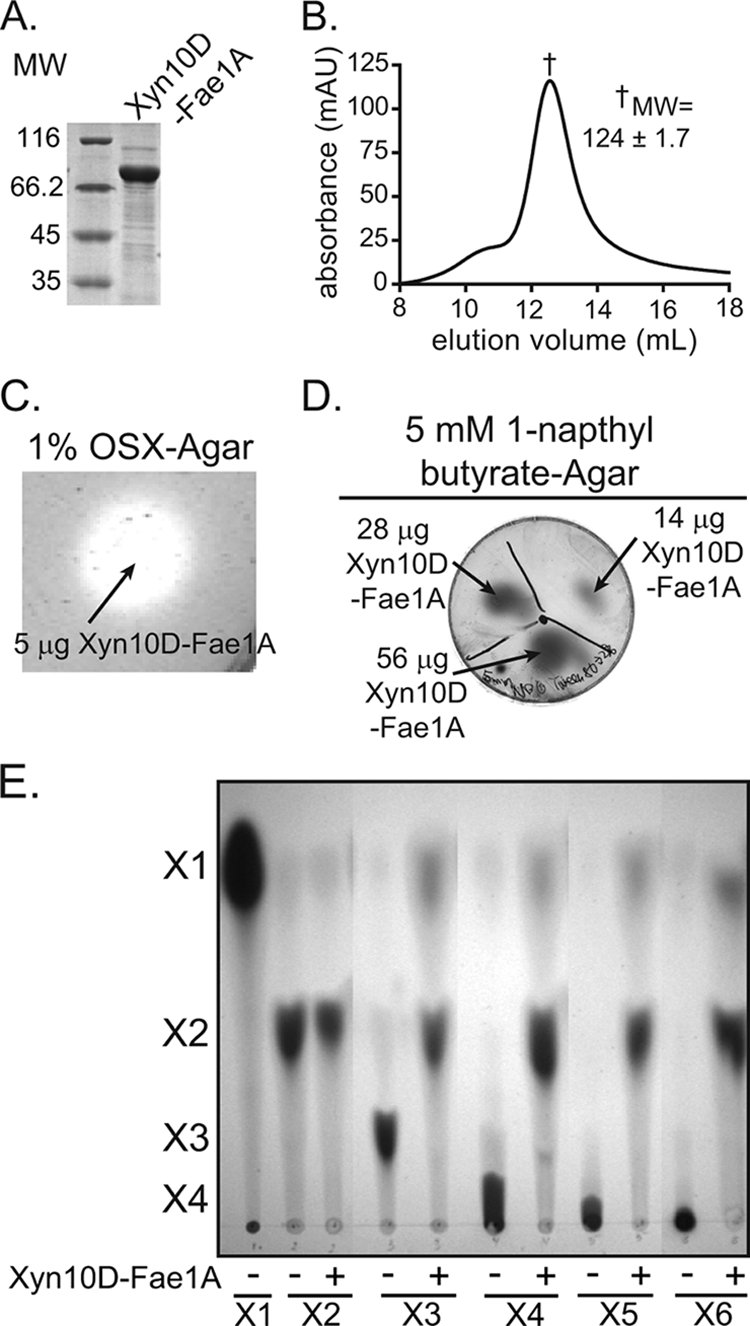
xyn10D-fae1A encodes a bifunctional xylanase-esterase. (A) Purification of recombinant Xyn10D-Fae1A. The eluate from cobalt chelate chromatography was analyzed by 12% SDS-PAGE, followed by Coomassie brilliant blue G-250 staining. MW, molecular weight (in thousands). (B) Gel filtration chromatography. The size of purified Xyn10D-Fae1A was estimated by size exclusion chromatography. The molecular weight of Xyn10D-Fae1A was calculated from the retention time of the peak absorbance by comparison with calibration standards having known molecular weights. Molecular weight is reported as the mean ± standard deviation from three independent experiments. mAU, milli-absorbance units. (C) Depolymerization of OSX. Xyn10D-Fae1A was assessed for its capacity to depolymerize OSX by incubating the protein on an agar plate infused with OSX followed by staining with Congo red. (D) Hydrolysis of 1-napthyl butyrate. Xyn10D-Fae1A was assessed for its capacity to hydrolyze 1-naphtyl butyrate by incubating the protein on an agar plate infused with 1-NB followed by development with fast garnet GBC sulfate. (E) Hydrolysis of xylo-oligosaccharides. Xyn10D-Fae1A-catalyzed hydrolysis of xylo-oligosaccharides (X2 to X6) was assessed by incubating the enzyme with each substrate and then resolving the products by TLC followed by staining with methanolic orcinol.
Xyn10D-Fae1A possesses both xylanase and esterase activities.
The gene sequence for xyn10D-fae1A is predicted to encode a polypeptide with two distinct catalytic properties (xylanase and esterase activities). To evaluate whether Xyn10D-Fae1A possesses endoxylanase activity, agar plates containing 0.1% either OSX, carboxymethyl cellulose, or locust bean gum were prepared. Purified Xyn10D-Fae1A (5 μg) was then spotted onto each plate, and plates were incubated overnight at 37°C. Upon staining of agar plates containing OSX with Congo red followed by destaining with 1 M NaCl, a clearing zone, representing hydrolysis of the polysaccharide substrate, was observed (Fig. 2C). In contrast, clearing zones were not seen in the case of the plates containing carboxymethyl cellulose and locust bean gum. These results suggested that Xyn10D-Fae1A possesses xylanase activity but not endoglucanase or mannanase activities. To evaluate whether Xyn10D-Fae1A possesses esterase activity, Xyn10D-Fae1A in increasing amounts (14, 28, or 56 μg) was spotted onto agar plates infused with 1-naphthyl butyrate, and plates were incubated overnight at 37°C and stained with fast garnet GBC sulfate (45). These experiments revealed that incubation with increasing amounts of Xyn10D-Fae1A led to a concomitant increase in 1-naphthol production, signified by the intensity and size of the spots developed from staining with GBC sulfate (Fig. 2D). The results, therefore, suggested that in addition to xylanase activity, Xyn10D-Fae1A possesses esterase activity. To determine the relevance of the Xyn10D-Fae1A to plant cell wall hydrolysis in the rumen, however, the specificity of the esterase activity needed determination.
Endo-β-1,4-xylanases hydrolyze the β-1,4 linkage between xylose units within the backbone of xylan polymers. To evaluate the size of xylose products that Xyn10D-Fae1A releases from xylo-oligosaccharides of different degrees of polymerization, we incubated Xyn10D-Fae1A with xylo-oligosaccharides (X2 to X6) and resolved the products by TLC. In the presence of Xyn10D-Fae1A, xylobiose hydrolysis to xylose was not detected (Fig. 2E); however, all xylo-oligosaccharides with higher degrees of polymerization (X3 to X6) were hydrolyzed mostly to xylobiose, with some xylose also being released (Fig. 2E). These results suggest that Xyn10D-Fae1A is a functional endo-β-1,4-xylanase. Furthermore, these data indicate that the smallest xylo-oligosaccharide that Xyn10D-Fae1A can effectively hydrolyze is xylotriose.
Identification, cloning, and expression of P. ruminicola 23 ORF02829.
Xyn10D-Fae1A encodes a functional xylanase-esterase (Fig. 2C and 2D); however, the major product released from xylo-oligosaccharides appears to be xylobiose (Fig. 2E). In order for P. ruminicola 23 to utilize xylose from longer-chain xylo-oligosaccharides or xylan, the bacterium requires an enzyme that releases xylose or monomeric sugars from the products of Xyn10D-Fae1A. We therefore cloned and expressed the two genes downstream of the Xyn10D-Fae1A-encoding gene. While no catalytic activity has been detected yet for ORF02828 (hypothetical protein), the product of ORF02829, expressed as an N-terminal hexahistidine fusion protein, exhibited robust catalytic activity as described below. The gene product was purified by metal affinity chromatography, and the predicted molecular mass of the hexahistidine fusion protein (98 kDa) was in agreement with the size of the purified protein estimated by comparison with molecular weight markers resolved by SDS-PAGE (Fig. 3A). To evaluate the quaternary structure for this fusion protein, the apparent molecular mass was determined by size exclusion chromatography. As shown in Fig. 3B, the elution volume of the putative β-glucosidase was 8.22 ml, which was just after the exclusion volume of the column (∼7.8 ml), indicating that this protein exists as a large polymeric species in solution. This peak had a shorter retention time than that for the highest-molecular-weight standard tested (Fig. 3B, inset). Therefore, the apparent molecular mass for the protein was estimated by extrapolation of the standard curve. Based on our calculation, the apparent molecular mass for this protein is 868 ± 2.2 kDa, which is close to the value expected for a nonamer (886 kDa).
FIG. 3.

xyl3A encodes a functional β-xylosidase. (A) Purification of recombinant Xyl3A. The eluate from cobalt chelate chromatography was analyzed by 12% SDS-PAGE, followed by Coomassie brilliant blue G-250 staining. MW, molecular weight (in thousands). (B) Gel filtration chromatography. The size of purified Xyl3A was estimated by size exclusion chromatography. The molecular weight (MW, in thousands; reported as the mean ± standard deviation from three independent experiments) of Xyl3A was calculated from the retention time of the peak absorbance by comparison with calibration standards having known molecular weights. mAU, milli-absorbance units. (C) Hydrolysis of pNP-linked sugars. Xyl3A was assessed for its capacity to hydrolyze several pNP-linked sugars by UV spectroscopy. Values are means ± standard deviations from three independent experiments. (D) Hydrolysis of xylo-oligosaccharides. Xyl3A-catalyzed hydrolysis of xylo-oligosaccharides (X2 to X6) was assessed by incubating the enzyme with each substrate and then resolving the products by TLC followed by staining with methanolic orcinol.
ORF02829 encodes a bifunctional β-xylosidase/α-l-arabinofuranosidase.
The gene sequence for ORF02829 is predicted to encode a family 3 GH and was annotated as a β-d-glucosidase. To determine the substrate specificity for the gene product of ORF02829, the recombinant protein was screened for activity with a library of α- and β-para-nitrophenol-linked monosaccharides. The putative β-d-glucosidase was incubated in the presence of the derivatized sugars, and the reactions were monitored spectrophotometrically. These experiments revealed that upon incubation with pNP-β-d-glucopyranoside, release of pNP was not detectable (Fig. 3C). In contrast, hydrolysis of pNPX was readily observed in the presence of the protein (Fig. 3C). In addition, low activity was observed with pNP-α-l-arabinofuranoside; however, this activity represented a fraction of the pNPX activity (Fig. 3C). These results indicated that ORF02829 encodes a functional GH that exhibits primarily β-xylosidase activity, although it also possesses the capacity to hydrolyze pNP-α-l-arabinofuranoside with lower activity. Based on these results, which indicated that ORF02829 encodes the first GH family 3 xylosidase to be characterized from P. ruminicola 23, the gene was designated xyl3A.
To evaluate whether Xyl3A possesses the capacity to hydrolyze β-1,4-linked xylo-oligosaccharides of various lengths, Xyl3A (0.42 μM) was incubated with the substrates (X2 to X6), and the products were resolved by TLC followed by staining with methanolic orcinol. Upon incubation of the enzyme with each of the β-1,4-linked xylo-oligosaccharides, the sole products released were xylose monomers (Fig. 3D). These experiments revealed that Xyl3A is capable of hydrolyzing the glycosidic linkages of β-1,4-xylo-oligosaccharides with various lengths.
Taken together, these results suggest that xyl3A does not encode an enzyme with β-d-glucosidase activity as previously annotated; rather, this gene encodes a bifunctional β-d-xylosidase/α-l-arabinofuranosidase that can hydrolyze the glycosidic linkage of β-1,4-xylo-oligosaccharides.
Xyn10D-Fae1A and Xyl3A function synergistically to release xylose from xylan.
To evaluate whether the enzymatic properties of Xyn10D-Fae1A and Xyl3A may function coordinately in the depolymerization of xylan, the two enzymes were incubated independently or in combination with OSX and the products of hydrolysis were resolved by TLC. In the absence of either enzyme, no depolymerization of the xylan substrate was detected (Fig. 4A). When OSX was incubated with Xyn10D-Fae1A, depolymerization of the xylan substrate was detected (Fig. 4A). By comparison with xylo-oligosaccharide standards, the major hydrolysis products are predicted to be xylobiose and several other undefined products. When OSX was incubated with Xyl3A, a relatively small amount of depolymerization was observed (Fig. 4B), and the only detectable product was predicted to be xylose (Fig. 4A). Incubation of OSX with both Xyn10D-Fae1A and Xyl3A resulted in a xylan depolymerization pattern similar to that observed for Xyn10D-Fae1A alone (Fig. 4A); however, conversion of the hydrolytic products to the monosaccharide, xylose, was increased (Fig. 4B).
FIG. 4.
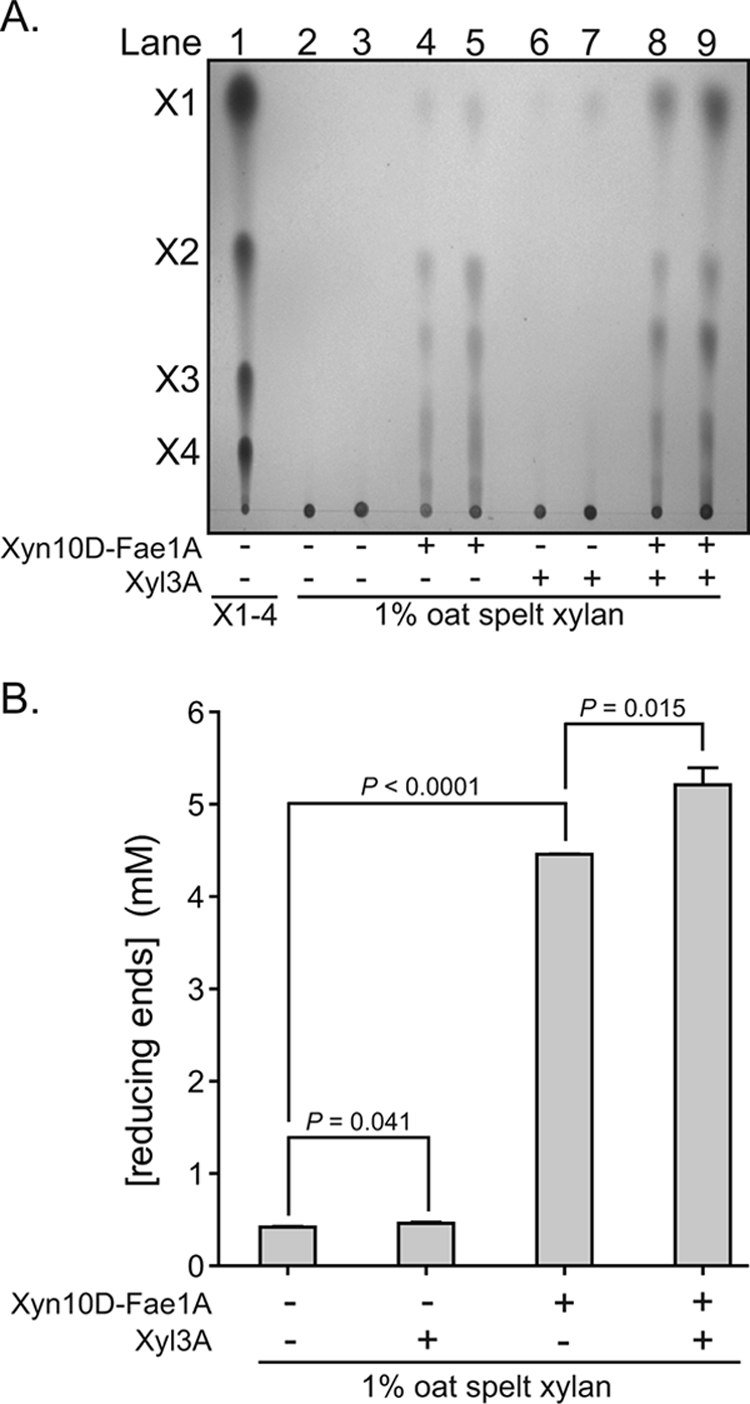
Xyn10D-Fae1A and Xyl3A function synergistically to release xylose from xylan. (A) TLC separation of products released from OSX by Xyn10D-Fae1A. Xyn10D-Fae1A (0.50 μM) or Xyl3A (0.50 μM) was incubated with OSX (1%), and the products were resolved by TLC followed by staining with methanolic orcinol. Xylo-oligosaccharide standards X1 to X4 were spotted onto the plate to serve as markers (lane 1) for the identification of hydrolysis products. The indicated enzymes were incubated with OSX at 37°C for 14 h, and 2.5 μl (lanes 2, 4, 6, and 8) or 5.0 μl (lanes 3, 5, 7, and 9) of the reaction mixtures was spotted onto the TLC plate. (B) Reducing sugars released from OSX by Xyn10D-Fae1A and Xyl3A. Wild-type or mutant (E280S or S629A) Xyn10D-Fae1A was incubated with OSX (1%), and the reducing sugars were detected by using the PAHBAH assay. The reducing sugar concentrations were calculated from the absorbance at 410 nm with comparison to a standard curve generated with known concentrations of glucose. Two-tailed P values were determined by performing an unpaired Student's t test.
These experiments revealed that when incubated together, Xyn10D-Fae1A and Xyl3A release more xylose from OSX than each individual enzyme. This result indicates that the catalytic properties of Xyn10D-Fae1A and Xyl3A are synergistic and that the two enzymes function to release monosaccharides from OSX, a complex substrate.
Mutational analyses of Xyn10D-Fae1A maps the two catalytic domains to discrete regions of the polypeptide sequence.
To test our prediction that Xyn10D-Fae1A possesses two distinct catalytic modules, amino acid substitutions were introduced in the putative catalytic residues for these two domains. Based upon alignment with the catalytic nucleophile (Glu298) identified for the extracellular xylanase from Bacillus stearothermophilus (29, 64), we predicted that Glu280 may be the catalytic nucleophile for the xylanase domain of Xyn10D-Fae1A (Fig. 5A). Amino acid alignments also revealed that a catalytic triad (Ser-His-Glu) that is crucial for catalysis in ferulic acid esterase enzymes (6, 58) is present and that S629 may be the catalytic nucleophile for the esterase domain of Xyn10D-Fae1A (Fig. 5A).
FIG. 5.
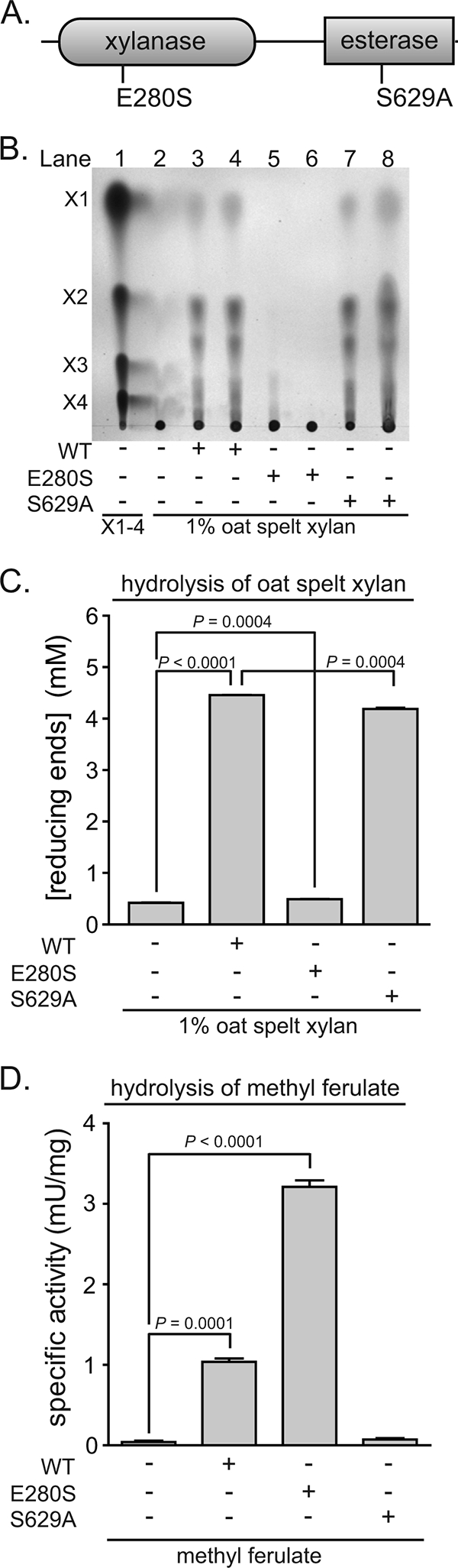
Mutational analyses of Xyn10D-Fae1A map the two catalytic domains to discrete regions of the polypeptide sequence. (A) Domain architecture for Xyn10D-Fae1A. Functional domains were assigned utilizing the Pfam online server (24). Amino acid substitutions were independently introduced into the putative catalytic residue for the xylanase domain (Glu280) and the ferulic acid esterase domain (Ser629) by site-directed mutagenesis. (B) Depolymerization of OSX by wild-type and mutant (E280S or S629A) Xyn10D-Fae1A. Wild-type or mutant (E280S or S629A) Xyn10D-Fae1A was incubated with OSX (1%), and the products were resolved by TLC followed by staining with methanolic orcinol. Xylo-oligosaccharide standards X1 to X4 were spotted on the plate in lane 1 to serve as markers for the identification of hydrolysis products. The wild-type enzyme, E280S mutant, or S629A mutant (0.93 μM in lanes 3, 5, and 7, respectively, and 1.9 μM in lanes 4, 6, and 8, respectively) was incubated with OSX at 37°C for 21 h, and 2.5 μl of the reaction mixtures was spotted onto the TLC plate. (C) Reducing sugars released from OSX by wild-type and mutant (E280S or S629A) Xyn10D-Fae1A. Wild-type or mutant (E280S or S629A) Xyn10D-Fae1A was incubated with OSX (1%), and the reducing sugars were detected by using the PAHBAH assay. The reducing sugar concentrations were calculated from the absorbance at 410 nm with comparison to a standard curve generated with known concentrations of glucose. (D) Hydrolysis of methyl ferulate by wild-type and mutant (E280S or S629A) Xyn10D-Fae1A. Methyl ferulate (5 mM) was incubated with wild-type, E280S, or S629A Xyn10D-Fae1A (1.2 μM, final concentration). Following incubation at 37°C for 30 min, ferulic acid was determined by HPLC as described by Wang et al. (67). For panels C and D, values are means plus standard deviations from three independent experiments. Two-tailed P values were determined by performing an unpaired Student's t test.
When the putative nucleophile for the xylanase domain was mutated (Glu280Ser), the capacity for Xyn10D-Fae1A to depolymerize OSX was attenuated (Fig. 5B and C). Similarly, when the putative nucleophile for the esterase domain was mutated (Ser629Ala), the ferulic acid esterase activity for Xyn10D-Fae1A was attenuated (Fig. 5D). These results suggest that for Xyn10D-Fae1A, Glu280 is an important residue for xylanase activity and Ser629 is an important residue for ferulic acid esterase activity. Furthermore, these results indicate that these distinct catalytic activities (xylanase and esterase) map to distinct regions on the polypeptide.
To evaluate whether these independent mutations influenced the catalytic activity of the adjacent domains, the Glu280Ser mutant was monitored for ferulic acid esterase activity and the Ser629Ala mutant was characterized for xylanase activity. When incubated with OSX, the Ser629Ala mutant depolymerized xylan to an extent similar to the that of wild-type Xyn10D-Fae1A, although a statistically significant decrease was detected for the mutant protein (Fig. 5B and C). However, when the Glu280Ser mutant was evaluated for ferulic acid esterase activity, the specific activity was increased threefold over that of wild-type Xyn10D-Fae1A (Fig. 5D).
Xyl3A is a GH family 3 β-xylosidase with a unique domain architecture.
Comparison of the domain architectures of Xyl3A and other biochemically characterized members of the GH family 3 utilizing Pfam (http://www.sanger.ac.uk/Software/Pfam/) (24) suggested that Xyl3A possesses two domains, as with most members of this family. However, the analysis also predicted that Xyl3A, which belongs to cluster B, has a PA14 domain insertion in the C-terminal GH family 3 domain (Table 2). It is predicted that the PA14-like insertion sequence may represent a novel carbohydrate binding module (57).
TABLE 2.
Activities and domain architecture for experimentally characterized family 3 GHs
 |
aGH family 3 clusters are assigned based on phylogenetic relationships inferred by Cornoyer and Faure (12).
bFunctional domains were assigned utilizing the Pfam online server (24). Domain hits were included if the expect value (E value) was smaller than 1×10−8.
cEnzymatic activities were assigned based on results from kinetic assays with purified enzymes as reported in the corresponding cited manuscripts.
In order to identify conserved amino acids that may play important roles in catalysis among members of the GH family 3, amino acid sequences for biochemically characterized GH family 3 β-glycosidases were aligned (data not shown). Several conserved motifs were identified, and conserved residues shown to reside within the active site of ExoI, a family 3 GH from Hordeum vulgare (barley) (65), were chosen for site-directed mutagenesis (Table 2).
Steady-state kinetic analysis of wild-type and mutant Xyl3A.
To compare the catalytic properties for wild-type Xyl3A and six mutant forms (R149L, Y237F, D269A, D269N, E594Q, and E616A), we analyzed the steady-state kinetic values of these enzymes in the presence of pNPX. Wild-type or mutant Xyl3A was incubated in a sodium acetate buffer in the presence of various concentrations of pNPX, and the change in magnitude of the A400 signal was monitored by UV spectroscopy. Initial rate data for Xyl3A and the mutants were obtained for each of the substrate concentrations, and plots of the initial velocity of pNPX hydrolysis (nmol/s) versus substrate concentration (μM) were generated (data not shown). For mutants that exhibited very low rates, the kcat(apparent) was determined at 10 mM substrate. Steady-state kinetic values for wild-type and mutant Xyl3A were obtained by applying a nonlinear curve fit to the data (Table 3). These results suggested that the E594Q mutation has no significant effect on catalysis for Xyl3A. Conversely, the Y237F and E616A mutations have moderate effects on catalysis for Xyl3A, and the R149L, D269A, and D269N mutations have severe effects on catalysis for Xyl3A. The circular dichroism spectra for the wild type and Xyl3A mutants were collected and analyzed as described by Dodd et al. (18). Comparison of the circular dichroism spectra for the wild type and Xyl3A mutants revealed that they possess similar overall secondary structures (data not shown).
TABLE 3.
Steady-state kinetic measurements for wild-type and mutant Xyl3A with pNPX
| Protein | kcat (s−1) | Km (mM) | kcat/Km (mM−1s−1) | rkcat (%)c |
|---|---|---|---|---|
| Wild typea | 9.7 ± 0.3 | 1.8 ± 0.2 | 5.4 ± 0.6 | 100 |
| R149Lb | (2.5 ± 0.2) × 10−2 | ND | ND | 0.26 |
| Y237Fa | 1.6 ± 0.08 | 3.9 ± 0.5 | 0.41 ± 0.06 | 16 |
| D269Ab | (1.2 ± 0.5) × 10−2 | ND | ND | 0.12 |
| D269Nb | (2.2 ± 1.0) × 10−1 | ND | ND | 2.3 |
| E594Qa | 10 ± 0.2 | 2.0 ± 0.1 | 5.0 ± 0.3 | 103 |
| E616Aa | 3.3 ± 0.1 | 18 ± 1 | 0.2 ± 0.01 | 34 |
Error estimates are from propagating errors from nonlinear regression analysis utilizing GraphPad Prism v5.01.
The reported kcat value is kcat(apparent), as Km values were not determined; rates for these two mutants were determined at 10 mM pNPX concentration and thus represent a lower boundary on kcat. Each kcat determination was performed in triplicate, and data are means ± standard deviations from the mean.
rkcat, residual kcat.
DISCUSSION
The rumen harbors a complex community of microbes which catalyzes the depolymerization of cellulose and hemicellulose to soluble sugars which are then fermented to short-chain fatty acids. These end products of fermentation are utilized by the mammalian host as the major source of energy (3, 41). Among the bacterial genera in the rumen, Prevotella has been reported to be one of the most numerous (8, 60). P. ruminicola 23 is one of the most commonly isolated species (8) from rumen contents and possesses the capacity to degrade plant cell wall polysaccharides, proteins, and peptides (61). Therefore, this species is thought to play an important role in the utilization of xylans and other hemicelluloses within the rumen.
Two xylan-hydrolyzing enzymes have been previously characterized from P. ruminicola: a bifunctional GH family 5 xylanase/endoglucanase cloned from the type strain, P. ruminicola 23T (70), and a GH family 10 xylanase with an interrupted catalytic domain cloned from the related strain P. ruminicola D31d (71). The recently sequenced genome for P. ruminicola 23 harbors genes corresponding to these two previously characterized xylanases (data not shown) and also harbors two additional putative xylanase genes that were not previously characterized. One of these genes was annotated as a bifunctional xylanase-esterase (designated xyn10D-fae1A). To provide further insight into the molecular basis for xylan depolymerization by P. ruminicola 23, we cloned this gene and characterized the recombinant protein. Our results show that xyn10D-fae1A encodes a bifunctional GH family 10 xylanase-ferulic acid esterase. Furthermore, directed mutagenesis studies mapped the two catalytic functionalities to distinct regions on the polypeptide. A unique result that was obtained during the course of these experiments was that mutation of the putative nucleophile for the xylanase domain (E280S) led to an approximately threefold increase in ferulic acid esterase activity. While a clear explanation for these data will require further biophysical experiments, these results indicate that the xylanase and esterase domains may be functionally coupled.
The recently sequenced genomes for Bacteroides intestinalis DSM 17393 (GenBank accession no. EDV07678.1) and Bacteroides eggerthii DSM 20697 (GenBank accession no. ABVO01000038.1, nucleotides 298769 to 296631) harbor the only putative proteins in the GenBank database with significant sequence identity (64% for both) across the entire protein sequence for Xyn10D-Fae1A. Two genes from Clostridium thermocellum (xynY and xynZ) encode proteins with both GH family 10 xylanase (26, 31) and ferulic acid esterase (6) activities; however, in addition, the clostridial proteins have carbohydrate binding modules and dockerin domains. Despite the presence of these accessory domains in C. thermocellum XynY and XynZ, the natural occurrence of the GH 10 xylanase and CE 1 ferulic acid esterase domains within a single polypeptide sequence from different organisms suggests that this linkage may impart a selective advantage for xylan degradation over the two enzymatic functionalities in isolation. Another rumen bacterium, Ruminococcus flavefaciens 17, harbors a gene predicted to encode a protein with GH 11 xylanase and CE 1 esterase domains (1); however, the biochemical properties of this protein have not been characterized. Thus, it is not clear whether the CE 1 domain possesses ferulic acid esterase activity, as is the case for Xyn10D-Fae1A.
A number of different exoglycosidase functions have been identified for GH family 3 enzymes, including β-d-glucosidase, β-d-xylosidase, α-l-arabinofuranosidase, and N-acetyl-β-d-glucosaminidase (21). When we cloned the putative β-d-glucosidase gene (ORF02829) and characterized the recombinant protein, we found that it releases pNP from pNPX and pNP-α-l-arabinofuranoside but not pNP-β-d-glucopyranoside. The hydrolysis of both pNPX and pNP-α-l-arabinofuranoside (Xyl/Ara) has been reported for some members of GH family 3 (20, 46, 51, 74). With one exception (68), all of the characterized family 3 β-d-xylosidases possess substrate specificity for Xyl/Ara and do not possess β-d-glucosidase activity. The crystal structure of a family 3 β-d-glucosidase from barley (H. vulgare) revealed that Asp120 forms a hydrogen bond to the 6′OH group of the glucose substrate bound at the −1 subsite (36). Those authors found that Asp120 was highly conserved among biochemically defined family 3 β-d-glucosidases and also observed that the Xyl/Ara-active enzymes did not possess this conserved aspartate residue (36). The pentose sugars (β-d-xylopyranose and α-l-arabinofuranose) do not possess the additional CH2OH substituent from C-5 that is present in glucose; thus, it is possible that the larger glutamate residue could function to discriminate between hexoses and pentoses on the basis of steric interactions. It is more difficult to explain why the structurally distinct six-membered ring of xylopyranose and the five-membered ring of arabinofuranose are permitted into the active site (36, 44). To identify a primary amino acid sequence motif that might distinguish the Xyl/Ara-active enzymes from the glucose-active enzymes, we constructed amino acid sequence alignments for biochemically defined members of these two distinct groups. These alignments revealed a conserved motif (WWSEAL) for the Xyl/Ara enzymes that was not found in the β-d-glucosidase enzymes (data not shown). This motif may be useful for distinguishing between GH family 3 β-d-glucosidases and Xyl/Ara-active enzymes based solely on primary sequence analysis.
Xyl3A possesses a PA14 domain that forms an insertion within the C-terminal GH family 3 domain (Table 2). The precise function for the PA14 domain remains poorly defined; however, its presence in bacterial, archaeal, and eukaryotic proteins, including glycosidases, glycosyltransferases, proteases, amidases, adhesins, and bacterial toxins (57), and its predominantly β-sheet structure (56) suggest a carbohydrate-binding function. Alignment of the amino acid sequences for Xyl3A with other GH family 3 β-d-xylosidases/α-l-arabinofuranosidases suggests that the PA14 domain may be inserted within the C-terminal domain (data not shown). The predicted proximity of this region to the active site supports the possibility that the PA14 domain may aid in binding to oligosaccharide substrates. Thus, insertion of the PA14 domain in this region could influence the substrate specificity for the enzyme. A recent report identified a loop within the PA14 domain of two fungal adhesins from Candida glabrata that is responsible for determining the binding specificity (14, 75) to eukaryotic membrane-anchored glycoproteins. The amino acid sequence for the PA14 domain of Xyl3A aligns with the ligand specificity determinants for the fungal cell surface adhesins Epa6 and Epa7 (data not shown), which supports the possibility that the PA14 domain could facilitate anchoring of Xyl3A to sugars. Identification of the true functional role for the PA14 domain within Xyl3A will require further studies.
There are a number of residues that have been shown to make hydrogen bond contacts to hydroxyl groups of the glycosyl substrate within the active site for the β-d-glucan glucohydrolase (ExoI) from H. vulgare (65), and directed-mutagenesis studies in the β-glucosidase from Flavobacterium meningosepticum have confirmed the importance of these residues for catalysis (49). We identified analogous residues in Xyl3A by amino acid sequence alignments, and based on these alignments, we constructed site-directed mutants that are predicted to have important roles in catalysis (residues indicated in Table 2). The significant attenuation in activity for the mutants tested in this study (Table 3) provides support for the classification of Xyl3A as a family 3 GH.
In the gene cluster identified in this study, there is a protein just upstream of xyl3A that was annotated as a hypothetical protein. Based on the short intergenic space (31 nucleotides) between this gene and xyl3A, it is possible that a single polycistronic mRNA may encode these two gene products. Comparison of the hypothetical protein with other proteins in the GenBank database revealed that this putative protein shares significant sequence identity (30%) with an α-1,2-fucosidase from Bifidobacterium bifidum (39, 54). It is unclear whether this protein may function synergistically with Xyn10D-Fae1A and Xyl3A to hydrolyze xylan; however, studies of the substrate specificity of this protein are under way in our laboratory.
Acknowledgments
This work was conducted under Sponsored Research Agreement no. 2005-2619-00 with Archer Daniels Midland Company, Decatur, IL. Partial support was also provided by the USDA Cooperative State Research, Education and Extension Service, Hatch project ILLU 538-364 (to I.K.O.C.) and the USDA National Research Initiative (proposal no. AG 2008-35206-18784; R.I.M., I.K.O.C., and M.A.S.).
Footnotes
Published ahead of print on 20 March 2009.
REFERENCES
- 1.Aurilia, V., J. C. Martin, S. I. McCrae, K. P. Scott, M. T. Rincon, and H. J. Flint. 2000. Three multidomain esterases from the cellulolytic rumen anaerobe Ruminococcus flavefaciens 17 that carry divergent dockerin sequences. Microbiology 1461391-1397. [DOI] [PubMed] [Google Scholar]
- 2.Avgustin, G., H. J. Flint, and T. R. Whitehead. 1992. Distribution of xylanase genes and enzymes among strains of Prevotella (Bacteroides) ruminicola from the rumen. FEMS Microbiol. Lett. 78137-143. [DOI] [PubMed] [Google Scholar]
- 3.Bergman, E. N. 1990. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 70567-590. [DOI] [PubMed] [Google Scholar]
- 4.Bertani, G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186595-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertani, G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blum, D. L., I. A. Kataeva, X. L. Li, and L. G. Ljungdahl. 2000. Feruloyl esterase activity of the Clostridium thermocellum cellulosome can be attributed to previously unknown domains of XynY and XynZ. J. Bacteriol. 1821346-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 8.Bryant, M. P., N. Small, C. Bouma, and H. Chu. 1958. Bacteroides ruminicola n. sp. and Succinimonas amylolytica; the new genus and species; species of succinic acid-producing anaerobic bacteria of the bovine rumen. J. Bacteriol. 7615-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cann, I. K., S. Ishino, M. Yuasa, H. Daiyasu, H. Toh, and Y. Ishino. 2001. Biochemical analysis of replication factor C from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 1832614-2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantarel, B. L., P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard, and B. Henrissat. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37D233-D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coughlan, M. P. 1985. Cellulose hydrolysis: the potential, the problems and relevant research at Galway. Biochem. Soc. Trans. 13405-406. [DOI] [PubMed] [Google Scholar]
- 12.Cournoyer, B., and D. Faure. 2003. Radiation and functional specialization of the family-3 glycoside hydrolases. J. Mol. Microbiol. Biotechnol. 5190-198. [DOI] [PubMed] [Google Scholar]
- 13.Dan, S., I. Marton, M. Dekel, B. A. Bravdo, S. He, S. G. Withers, and O. Shoseyov. 2000. Cloning, expression, characterization, and nucleophile identification of family 3, Aspergillus niger beta-glucosidase. J. Biol. Chem. 2754973-4980. [DOI] [PubMed] [Google Scholar]
- 14.de Groot, P. W., and F. M. Klis. 2008. The conserved PA14 domain of cell wall-associated fungal adhesins governs their glycan binding specificity. Mol. Microbiol. 68535-537. [DOI] [PubMed] [Google Scholar]
- 15.Dehority, B. A. 1969. Pectin-fermenting bacteria isolated from the bovine rumen. J. Bacteriol. 99189-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deshpande, V., K. E. Eriksson, and B. Pettersson. 1978. Production, purification and partial characterization of 1,4-beta-glucosidase enzymes from Sporotrichum pulverulentum. Eur. J. Biochem. 90191-198. [DOI] [PubMed] [Google Scholar]
- 17.Dodd, D., and I. K. O. Cann. 2009. Enzymatic deconstruction of xylan for biofuel production. Global Change Biol. Bioenergy 12-17. [DOI] [PMC free article] [PubMed]
- 18.Dodd, D., J. G. Reese, C. R. Louer, J. D. Ballard, M. A. Spies, and S. R. Blanke. 2007. Functional comparison of the two Bacillus anthracis glutamate racemases. J. Bacteriol. 1895265-5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eneyskaya, E. V., H. Brumer III, L. V. Backinowsky, D. R. Ivanen, A. A. Kulminskaya, K. A. Shabalin, and K. N. Neustroev. 2003. Enzymatic synthesis of beta-xylanase substrates: transglycosylation reactions of the beta-xylosidase from Aspergillus sp. Carbohydr. Res. 338313-325. [DOI] [PubMed] [Google Scholar]
- 20.Eneyskaya, E. V., D. R. Ivanen, K. S. Bobrov, L. S. Isaeva-Ivanova, K. A. Shabalin, A. N. Savel'ev, A. M. Golubev, and A. A. Kulminskaya. 2007. Biochemical and kinetic analysis of the GH3 family beta-xylosidase from Aspergillus awamori X-100. Arch. Biochem. Biophys. 457225-234. [DOI] [PubMed] [Google Scholar]
- 21.Faure, D. 2002. The family-3 glycoside hydrolases: from housekeeping functions to host-microbe interactions. Appl. Environ. Microbiol. 681485-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faure, D., J. Desair, V. Keijers, M. A. Bekri, P. Proost, B. Henrissat, and J. Vanderleyden. 1999. Growth of Azospirillum irakense KBC1 on the aryl beta-glucoside salicin requires either salA or salB. J. Bacteriol. 1813003-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faure, D., B. Henrissat, D. Ptacek, M. A. Bekri, and J. Vanderleyden. 2001. The celA gene, encoding a glycosyl hydrolase family 3 beta-glucosidase in Azospirillum irakense, is required for optimal growth on cellobiosides. Appl. Environ. Microbiol. 672380-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finn, R. D., J. Mistry, B. Schuster-Bockler, S. Griffiths-Jones, V. Hollich, T. Lassmann, S. Moxon, M. Marshall, A. Khanna, R. Durbin, S. R. Eddy, E. L. Sonnhammer, and A. Bateman. 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34D247-D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flint, H. J., T. R. Whitehead, J. C. Martin, and A. Gasparic. 1997. Interrupted catalytic domain structures in xylanases from two distantly related strains of Prevotella ruminicola. Biochim. Biophys. Acta 1337161-165. [DOI] [PubMed] [Google Scholar]
- 26.Fontes, C. M., G. P. Hazlewood, E. Morag, J. Hall, B. H. Hirst, and H. J. Gilbert. 1995. Evidence for a general role for non-catalytic thermostabilizing domains in xylanases from thermophilic bacteria. Biochem. J. 307151-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gasparic, A., R. Marinsek-Logar, J. Martin, R. J. Wallace, F. V. Nekrep, and H. J. Flint. 1995. Isolation of genes encoding beta-d-xylanase, beta-d-xylosidase and alpha-l-arabinofuranosidase activities from the rumen bacterium Prevotella ruminicola B1(4). FEMS Microbiol. Lett. 125135-141. [DOI] [PubMed] [Google Scholar]
- 28.Gasparic, A., J. Martin, A. S. Daniel, and H. J. Flint. 1995. A xylan hydrolase gene cluster in Prevotella ruminicola B(1)4: sequence relationships, synergistic interactions, and oxygen sensitivity of a novel enzyme with exoxylanase and beta-(1,4)-xylosidase activities. Appl. Environ. Microbiol. 612958-2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gat, O., A. Lapidot, I. Alchanati, C. Regueros, and Y. Shoham. 1994. Cloning and DNA sequence of the gene coding for Bacillus stearothermophilus T-6 xylanase. Appl. Environ. Microbiol. 601889-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert, H. J., and G. P. Hazlewood. 1993. Bacterial cellulases and xylanases. J. Gen. Microbiol. 139187-194. [Google Scholar]
- 31.Grepinet, O., M. C. Chebrou, and P. Beguin. 1988. Purification of Clostridium thermocellum xylanase Z expressed in Escherichia coli and identification of the corresponding product in the culture medium of C. thermocellum. J. Bacteriol. 1704576-4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griswold, K. E., and R. I. Mackie. 1997. Degradation of protein and utilization of the hydrolytic products by a predominant ruminal bacterium, Prevotella ruminicola B1(4). J. Dairy Sci. 80167-175. [DOI] [PubMed] [Google Scholar]
- 33.Han, S. O., H. Yukawa, M. Inui, and R. H. Doi. 2004. Isolation and expression of the xynB gene and its product, XynB, a consistent component of the Clostridium cellulovorans cellulosome. J. Bacteriol. 1868347-8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henrissat, B. 1992. Analysis of hemicellulases sequences. Relationships to other glycanases, p. 97-110. In J. Visser, G. Beldman, M. A. Kusters-van Someren, and A. G. J. Voragen (ed.), Xylans and xylanases, vol. 7. Elsevier, Amsterdam, The Netherlands. [Google Scholar]
- 35.Hespell, R. B., D. E. Akin, and B. A. Dehority. 1997. Bacteria, fungi, and protozoa of the rumen., p. 59-141. In R. I. Mackie, B. A. White, and R. E. Isaacson (ed.), Gastrointestinal microbiology. Chapman and Hall, New York, NY.
- 36.Hrmova, M., R. De Gori, B. J. Smith, J. K. Fairweather, H. Driguez, J. N. Varghese, and G. B. Fincher. 2002. Structural basis for broad substrate specificity in higher plant beta-d-glucan glucohydrolases. Plant Cell 141033-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hrmova, M., A. J. Harvey, J. Wang, N. J. Shirley, G. P. Jones, B. A. Stone, P. B. Hoj, and G. B. Fincher. 1996. Barley beta-d-glucan exohydrolases with beta-d-glucosidase activity. Purification, characterization, and determination of primary structure from a cDNA clone. J. Biol. Chem. 2715277-5286. [DOI] [PubMed] [Google Scholar]
- 38.Jeffries, T. W. 1990. Biodegradation of lignin-carbohydrate complexes. Biodegradation 1163-176. [Google Scholar]
- 39.Katayama, T., A. Sakuma, T. Kimura, Y. Makimura, J. Hiratake, K. Sakata, T. Yamanoi, H. Kumagai, and K. Yamamoto. 2004. Molecular cloning and characterization of Bifidobacterium bifidum 1,2-α-l-fucosidase (AfcA), a novel inverting glycosidase (glycoside hydrolase family 95). J. Bacteriol. 1864885-4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiyohara, M., Y. Hama, K. Yamaguchi, and M. Ito. 2006. Structure of beta-1,3-xylooligosaccharides generated from Caulerpa racemosa var. laete-virens beta-1,3-xylan by the action of beta-1,3-xylanase. J. Biochem. (Tokyo) 140369-373. [DOI] [PubMed] [Google Scholar]
- 41.Krause, D. O., S. E. Denman, R. I. Mackie, M. Morrison, A. L. Rae, G. T. Attwood, and C. S. McSweeney. 2003. Opportunities to improve fiber degradation in the rumen: microbiology, ecology, and genomics. FEMS Microbiol. Rev. 27663-693. [DOI] [PubMed] [Google Scholar]
- 42.Kuhad, R. C. 1993. Lignocellulose biotechnology: current and future prospects. Crit. Rev. Biotechnol. 13151-172. [Google Scholar]
- 43.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 44.Langston, J., N. Sheehy, and F. Xu. 2006. Substrate specificity of Aspergillus oryzae family 3 beta-glucosidase. Biochim. Biophys. Acta 1764972-978. [DOI] [PubMed] [Google Scholar]
- 45.Lanz, W. W., and P. P. Williams. 1973. Characterization of esterases produced by a ruminal bacterium identified as Butyrivibrio fibrisolvens. J. Bacteriol. 1131170-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee, R. C., M. Hrmova, R. A. Burton, J. Lahnstein, and G. B. Fincher. 2003. Bifunctional family 3 glycoside hydrolases from barley with alpha-l-arabinofuranosidase and beta-d-xylosidase activity. Characterization, primary structures, and COOH-terminal processing. J. Biol. Chem. 2785377-5387. [DOI] [PubMed] [Google Scholar]
- 47.Lever, M. 1972. A new reaction for colorimetric determination of carbohydrates. Anal. Biochem. 47273-279. [DOI] [PubMed] [Google Scholar]
- 48.Li, Y. K., J. Chir, and F. Y. Chen. 2001. Catalytic mechanism of a family 3 beta-glucosidase and mutagenesis study on residue Asp-247. Biochem. J. 355835-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, Y. K., J. Chir, S. Tanaka, and F. Y. Chen. 2002. Identification of the general acid/base catalyst of a family 3 beta-glucosidase from Flavobacterium meningosepticum. Biochemistry 412751-2759. [DOI] [PubMed] [Google Scholar]
- 50.Matsushita, O., J. B. Russell, and D. B. Wilson. 1991. A Bacteroides ruminicola 1,4-β-d-endoglucanase is encoded in two reading frames. J. Bacteriol. 1736919-6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minic, Z., C. Rihouey, C. T. Do, P. Lerouge, and L. Jouanin. 2004. Purification and characterization of enzymes exhibiting beta-d-xylosidase activities in stem tissues of Arabidopsis. Plant Physiol. 135867-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyazaki, K., H. Miyamoto, D. K. Mercer, T. Hirase, J. C. Martin, Y. Kojima, and H. J. Flint. 2003. Involvement of the multidomain regulatory protein XynR in positive control of xylanase gene expression in the ruminal anaerobe Prevotella bryantii B(1)4. J. Bacteriol. 1852219-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morag, E., E. A. Bayer, and R. Lamed. 1990. Relationship of cellulosomal and noncellulosomal xylanases of Clostridium thermocellum to cellulose-degrading enzymes. J. Bacteriol. 1726098-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagae, M., A. Tsuchiya, T. Katayama, K. Yamamoto, S. Wakatsuki, and R. Kato. 2007. Structural basis of the catalytic reaction mechanism of novel 1,2-alpha-l-fucosidase from Bifidobacterium bifidum. J. Biol. Chem. 28218497-18509. [DOI] [PubMed] [Google Scholar]
- 55.Osborne, J. M., and B. A. Dehority. 1989. Synergism in degradation and utilization of intact forage cellulose, hemicellulose, and pectin by three pure cultures of ruminal bacteria. Appl. Environ. Microbiol. 552247-2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petosa, C., R. J. Collier, K. R. Klimpel, S. H. Leppla, and R. C. Liddington. 1997. Crystal structure of the anthrax toxin protective antigen. Nature 385833-838. [DOI] [PubMed] [Google Scholar]
- 57.Rigden, D. J., L. V. Mello, and M. Y. Galperin. 2004. The PA14 domain, a conserved all-beta domain in bacterial toxins, enzymes, adhesins and signaling molecules. Trends Biochem. Sci. 29335-339. [DOI] [PubMed] [Google Scholar]
- 58.Schubot, F. D., I. A. Kataeva, D. L. Blum, A. K. Shah, L. G. Ljungdahl, J. P. Rose, and B. C. Wang. 2001. Structural basis for the substrate specificity of the feruloyl esterase domain of the cellulosomal xylanase Z from Clostridium thermocellum. Biochemistry 4012524-12532. [DOI] [PubMed] [Google Scholar]
- 59.Reference deleted.
- 60.Stevenson, D. M., and P. J. Weimer. 2007. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 75165-174. [DOI] [PubMed] [Google Scholar]
- 61.Stewart, C. S., H. J. Flint, and M. P. Bryant. 1997. The rumen bacteria, p. 10-72. In P. N. Hobson and C. S. Stewart (ed.), The rumen microbial ecosystem, 2nd ed. Elsevier, New York, NY.
- 62.Sumida, T., N. Sueyoshi, and M. Ito. 2002. Molecular cloning and characterization of a novel glucocerebrosidase of Paenibacillus sp. TS12. J. Biochem. 132237-243. [DOI] [PubMed] [Google Scholar]
- 63.Teather, R. M., and P. J. Wood. 1982. Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl. Environ. Microbiol. 43777-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teplitsky, A., A. Mechaly, V. Stojanoff, G. Sainz, G. Golan, H. Feinberg, R. Gilboa, V. Reiland, G. Zolotnitsky, D. Shallom, A. Thompson, Y. Shoham, and G. Shoham. 2004. Structure determination of the extracellular xylanase from Geobacillus stearothermophilus by selenomethionyl MAD phasing. Acta Crystallogr. D 60836-848. [DOI] [PubMed] [Google Scholar]
- 65.Varghese, J. N., M. Hrmova, and G. B. Fincher. 1999. Three-dimensional structure of a barley beta-d-glucan exohydrolase, a family 3 glycosyl hydrolase. Structure 7179-190. [DOI] [PubMed] [Google Scholar]
- 66.Vercoe, P. E., and K. Gregg. 1992. DNA sequence and transcription of an endoglucanase gene from Prevotella (Bacteroides) ruminicola AR20. Mol. Gen. Genet. 233284-292. [DOI] [PubMed] [Google Scholar]
- 67.Wang, X., X. Geng, Y. Egashira, and H. Sanada. 2004. Purification and characterization of a feruloyl esterase from the intestinal bacterium Lactobacillus acidophilus. Appl. Environ. Microbiol. 702367-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watt, D. K., H. Ono, and K. Hayashi. 1998. Agrobacterium tumefaciens beta-glucosidase is also an effective beta-xylosidase, and has a high transglycosylation activity in the presence of alcohols. Biochim. Biophys. Acta 138578-88. [DOI] [PubMed] [Google Scholar]
- 69.Whitehead, T. R. 1993. Analyses of the gene and amino acid sequence of the Prevotella (Bacteroides) ruminicola 23 xylanase reveals unexpected homology with endoglucanases from other genera of bacteria. Curr. Microbiol. 2727-33. [DOI] [PubMed] [Google Scholar]
- 70.Whitehead, T. R., and R. B. Hespell. 1989. Cloning and expression in Escherichia coli of a xylanase gene from Bacteroides ruminicola 23. Appl. Environ. Microbiol. 55893-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whitehead, T. R., and D. A. Lee. 1991. Cloning and comparison of xylanase genes from ruminal and colonic Bacteroides species. Curr. Microbiol. 2315-19. [Google Scholar]
- 72.Wilkie, K. C. B. 1983. Hemicellulose. Chemtech 13306-319. [Google Scholar]
- 73.Wulff-Strobel, C. R., and D. B. Wilson. 1995. Cloning, sequencing, and characterization of a membrane-associated Prevotella ruminicola B(1)4 beta-glucosidase with cellodextrinase and cyanoglycosidase activities. J. Bacteriol. 1775884-5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiong, J. S., M. Balland-Vanney, Z. P. Xie, M. Schultze, A. Kondorosi, E. Kondorosi, and C. Staehelin. 2007. Molecular cloning of a bifunctional beta-xylosidase/alpha-l-arabinosidase from alfalfa roots: heterologous expression in Medicago truncatula and substrate specificity of the purified enzyme. J. Exp. Bot. 582799-2810. [DOI] [PubMed] [Google Scholar]
- 75.Zupancic, M. L., M. Frieman, D. Smith, R. A. Alvarez, R. D. Cummings, and B. P. Cormack. 2008. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 68547-559. [DOI] [PubMed] [Google Scholar]