

Abstract
Clostridium botulinum cultures are classified into seven types, types A to G, based on the antigenicity of the neurotoxins produced. Of these seven types, only types C and D produce C2 toxin in addition to the neurotoxin. The C2 toxin consists of two components designated C2I and C2II. The genes encoding the C2 toxin components have been cloned, and it has been stated that they might be on the cell chromosome. The present study confirmed by using pulsed-field gel electrophoresis and subsequent Southern hybridization that these genes are on a large plasmid. The complete nucleotide sequence of this plasmid was determined by using a combination of inverse PCR and primer walking. The sequence was 106,981 bp long and contained 123 potential open reading frames, including the c2I and c2II genes. The 57 products of these open reading frames had sequences similar to those of well-known proteins. It was speculated that 9 these 57 gene products were related to DNA replication, 2 were responsible for the two-component regulatory system, and 3 were σ factors. In addition, a total of 20 genes encoding proteins related to diverse processes in purine catabolism were found in two regions. In these regions, there were 9 and 11 genes rarely found in plasmids, indicating that this plasmid plays an important role in purine catabolism, as well as in C2 toxin production.
Clostridium botulinum is a gram-positive, spore-forming, anaerobic bacterium. Cultures of this species produce poisonous botulinum neurotoxins (BoNTXs) that are lethal to humans and animals and are classified into seven types, types A to G, based on the antigenicity of the BoNTXs produced (actually, the cultures producing type G toxin were recently classified in a new species, Clostridium argentinens [40]).
C. botulinum type C and D cultures produce a binary C2 toxin in addition to C (or C1) and D BoNTXs; this additional toxin consists of two nonlinked proteins, C2I and C2II (28), that occur independently in the culture supernatant and are not chemically joined to each other. The C. botulinum C2 toxin used here is a representative of the family of binary actin-ADP-ribosylating toxins, which includes, in addition to C2 toxin, the Clostridium perfringens iota toxin, Clostridium difficile toxin, Clostridium spiroforme toxin, and the vegetative insecticidal proteins from Bacillus cereus (4).
The enzyme component of C2 toxin (C2I) ADP ribosylates G-actin at arginine 177 (1). This leads to depolymerization of actin filaments and finally to cell rounding. The proteolytically activated binding-translocation component (C2IIa) forms heptamers, which assemble with C2I and bind to the cellular receptor (5). Following receptor-mediated endocytosis, C2IIa forms pores in the membrane of acidic endosomes. Subsequently, C2I translocates across the membrane into the cytosol through these C2IIa pores.
The production of C1 and D BoNTXs is governed by bacteriophages (12, 13, 18, 19, 26), and both toxin genes have been cloned from the corresponding phage DNAs (16, 21). Recently, we determined the whole-genome sequence of a type C toxin-converting phage (c-st) genome (31). Eklund et al. reported that C2 toxin toxigenicity (mouse lethality) became clear when strains were cultured in fortified egg-meat medium and the culture supernatants were treated with trypsin (12). They also reported that C2 toxin production was not related to the BoNTX-converting phages; some non-BoNTX-producing cells produced C2 toxin. Fujii et al. (15) and Kimura et al. (22) determined the whole nucleotide sequences of the c2I and c2II genes and speculated that these genes might be located on the bacterial chromosome (15, 22).
In this study, we determined that these genes are present not on the cell chromosome but on a large plasmid; we first speculated that this was this was the case based on the results of both pulsed-field gel electrophoresis (PFGE) and Southern hybridization analysis and then confirmed it by determining the complete nucleotide sequence of the plasmid. Since the plasmid was extremely unstable, we could not purify the complete plasmid DNA; therefore, we determined the whole-genome sequence by using the inverse PCR method, which enables rapid determination of the flanking regions of unknown sequences and determination of unidentified sequences in the genome, and the primer-walking method. This is the first case in which an entire DNA sequence of a large plasmid was determined using only these two procedures. The process used to determine the whole-plasmid DNA sequence and several interesting features of the plasmid are described below.
MATERIALS AND METHODS
Bacterial strains and preparation of DNAs.
Five toxigenic type C and D strains, C-Stockholm (C-ST), C-203, C-468, D-1873, and D-4947, and two nontoxigenic strains, (C)-AO2 and (C)-203U28, that were obtained from toxigenic strains C-ST and C-203 by treatment with acridine orange and UV irradiation, respectively, were used (26, 27). C-ST, C-203, C-468, and D-1873 produced C2 toxin, but (C)-AO2 and D-4947 did not. The strains were cultivated overnight at 37°C in cooked meat medium (Becton Dickinson and Company, Sparks, MD), and the bacterial DNAs were prepared as described previously (31).
PFGE analysis.
Plugs were prepared as reported previously (31). PFGE was performed with a CHEF (clamped homogeneous electric fields) Mapper apparatus (Bio-Rad Laboratories K.K., Tokyo, Japan) by using 1% agarose gels (pulsed-field-certified agarose; Bio-Rad Laboratories) in 0.5× Tris-borate-EDTA run at 6 V/cm and 14°C. The pulse times were ramped from 6.75 to 15.54 s for 34.28 h, and there was a linear increase in pulse intervals.
Sequence analysis.
The sequence of the pC2C203U28 plasmid was determined by a combination of inverse PCR and primer-walking methods. Restriction enzymes that recognize ≥6-base sequences and “rare cutting” restriction enzymes were used for DNA digestion. Purified DNA (1 μg) was digested separately by incubation with the enzymes AclI, BglII, Bsp106I (BanIII, BspDI, ClaI), BspHI, BsrGI, EcoRI, MfeI, NsiI (AvaIII, EcoT22I), PciI, and SpeI overnight at the temperature recommended by the manufacturer. Digested DNA was extracted with a phenol-chloroform-isoamyl alcohol (25:24:1; pH 8.0) mixture by centrifugation. The DNA was precipitated with ethanol and collected by centrifugation. For circularization, the restriction fragments were dissolved in 10 mM Tris-HCl (pH 8.0). The ligation reaction was initiated by addition of T4 DNA ligase (Toyobo Co., Ltd., Osaka, Japan) and was allowed to proceed for 30 min at 16°C. The ligation samples were then treated with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1; pH 8.0), the aqueous phase was collected, and the DNA was precipitated with ethanol and collected by centrifugation, resulting in preparation of 10 libraries of circularized DNA from (C)-203U28 DNA. These libraries were then subjected to PCR analysis using specific oligonucleotide primers to anneal the ends of the known sequences as templates. The upstream and downstream flanking regions of unknown sequences were obtained by inverse PCR using the known sequence of the C2 toxin gene in (C)-203U28, and then the sequences were determined by the primer-walking method. These steps were repeated to determine the complete nucleotide sequence of the pC2C203U28 plasmid. PCR was performed manually with reaction mixtures containing circularized DNA obtained as described above. We prepared about 500 primers for the inverse PCR and primer-walking methods. We used 30 cycles of denaturation at 94°C for 1 min, primer annealing at 50°C for 1 min, and extension using an LA Taq PCR kit (Takara Bio Inc., Shiga, Japan) at 68°C for 5 min, followed by an additional extension at 68°C for 5 min. The PCR products were analyzed on 1% agarose gels, and their DNA sequences were determined by using BigDye Terminator chemistry with an ABI Prism 3130xl automated DNA sequencer (Applied Biosystems Inc., Foster City, CA). To close the remaining sequence gaps, PCR and primer-walking methods were applied to the PCR products.
Annotation and computer analysis.
Initially, potential protein-encoding regions (open reading frames [ORFs]) that were ≥150 bp long were identified by using the In Silico Molecular Cloning software package, Genomics Edition (In Silico Biology Inc., Yokohama, Japan), and each ORF was reviewed manually for the presence of a ribosomal binding sequence. Functional annotation was based on homology searches against the GenBank nonredundant protein sequence database by using the program BLASTP (2). The Sequencher DNA sequencing software (Gene Codes Corp, Ann Arbor, MI) was used for sequence assembly and multiple-sequence alignment.
PCR amplification.
PCR amplification was carried out by using an LA Taq PCR kit with the following settings: preheating for 96°C for 1 min, 30 cycles of denaturation at 96°C for 1 min, annealing at 50°C for 1 min, and extension at 68°C for various times, and an additional extension at 68°C for 5 min. The extension time was varied according to the expected product size (1 min/kb). PCR products were analyzed on 1% agarose gels. DNA sequences of PCR products were determined by the primer-walking method. The primers used for PCR scanning are shown in Table S1 in the supplemental material.
Southern hybridization analysis.
Bacterial DNAs in 1% agarose plugs (Bio-Rad Laboratories K.K.) were prepared as described previously (31). After cultivation of the strains overnight at 37°C in cooked meat medium, the cells were collected by centrifugation at 8,000 × g for 10 min at 4°C, washed twice with 10 mM Tris-HCl (pH 8.0), and embedded in 1% agarose plugs (CleanCut agarose; Bio-Rad Laboratories K.K.). The embedded cells were lysed with 1.2 mg/ml lysozyme (Sigma-Aldrich Japan K.K, Tokyo, Japan) and 0.5% N-lauroylsarcosine (Nacalai Tesque Inc., Kyoto, Japan) at 37°C for 16 h and then with 0.1% proteinase K (Roche Diagnostics K.K., Tokyo, Japan) and 0.5% N-lauroylsarcosine at 50°C for 48 h with gentle shaking. Each cellular DNA prepared by this procedure was subjected to PFGE, transferred onto nylon membranes (Hybond-N Plus; GE Healthcare Bio-Sciences K.K., Tokyo, Japan), and incubated with a specific probe. The hybridized probe was detected by using the ECL detection system (GE Healthcare Bio-Sciences K.K.) according to the manufacturer's instructions. The probe used for hybridization was prepared by performing PCR with (C)-203U28 DNA as a template and with forward primer 5′-AATAACTTTTATATAGCTTAAAATGTATC-3′ and reverse primer 5′-CGCAATCTAAATATAAAACGAG-3′, which amplified a fragment corresponding to the genes encoding C2I. The amplification reaction consisted of 30 cycles of denaturation at 94°C for 1 min, primer annealing at 50°C for 1 min, and extension at 68°C for 1 min. The PCR products were electrophoresed in a 1% agarose gel, eluted with a QIAquick gel extraction kit (Qiagen K.K., Tokyo, Japan), and labeled using the ECL direct nucleic acid labeling system (GE Healthcare Bio-Sciences K.K.).
Phylogenetic analysis of ECF σ factors.
The GENES database of the Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.ad.jp/kegg/) was searched for the genes encoding bacterial extracytoplasmic function (ECF) σ factors. Homology searches among three proteins as novel σ-like factors (C2P015, C2P030, and C2P052) encoded by their genes identified in pC2C203U28 were also performed using the NCBI BLAST databases (http://www.ncbi.nlm.nih.gov/). After this, based on the amino acid sequences, an alignment was used to build a neighbor-joining distance-based phylogenetic tree using the Kimura method with 1,000 replicates for statistical bootstrapping and the ClustalW algorithm, version 1.83 (http://clustalw.ddbj.nig.ac.jp/top-j.html) (42), which resulted in retrieval of many ECF σ factors of clostridia, RpoE of Escherichia coli, and SigY of Bacillus subtilis.
Nucleotide sequence accession numbers.
The nucleotide sequences reported here have been deposited in the GenBank database under accession numbers AP010934 (pC2C203U28 in Fig. 3) and AB465553, AB465554, and AB465552 (the upstream and downstream regions including the C2 toxin gene in C-ST, C-468, and D-1873, respectively) (Fig. 9).
FIG. 3.

Genome organization of the pC2C203U28 plasmid of (C)-203U28. The horizontal arrows represent ORFs identified for pC2C203U28, and the ORF numbers are indicated above the arrows. ORFs were placed into 10 groups according to their predicted functions. IS elements are indicated by yellow-green boxes. The results of a PCR scanning analysis of the large plasmid of C-ST, C-468, D-1873, (C)-AO2, and D-4947 are shown below the ORF map. The solid lines indicate the segments that yielded the PCR products, and the dashed lines indicate the segments from which no PCR products were obtained.
FIG. 9.

Genome structure of the C2 gene-flanking regions of type C and D strains. The DNA sequences of the upstream and downstream regions including the C2 genes of C-ST, C-468, and D-1873 were determined and compared with the analogous regions of (C)-203U28. The horizontal arrows represent the ORFs in the genome of each strain, and the ORFs are connected by gray shading. The levels of sequence identity (expressed as percentages) between homologous regions and deletions of c2p053 and c2p054 in C-ST and C-468 are indicated.
RESULTS
Southern hybridization test.
It has been assumed that the C2 toxin gene in C. botulinum types C and D is located on the chromosome. To determine whether the C2 toxin gene is indeed located on the chromosome or whether it might in fact be located on a plasmid, Southern hybridization analysis was performed by using a C2 probe. The intact DNAs from C-ST, C-468, and D-1873 not treated with restriction enzymes were subjected to PFGE and subsequently hybridized with a C2I probe (Fig. 1). Surprisingly, in the (C)-203U28 DNA, the C2I probe hybridized to a 110-kb fragment. In addition, the probe hybridized to 100-kb fragments of the C-ST, C-468, and D-1873 DNAs. The same result was obtained using a C2II probe (data not shown). These results indicate that the C2 toxin gene may be on a large plasmid in the type C and D strains.
FIG. 1.
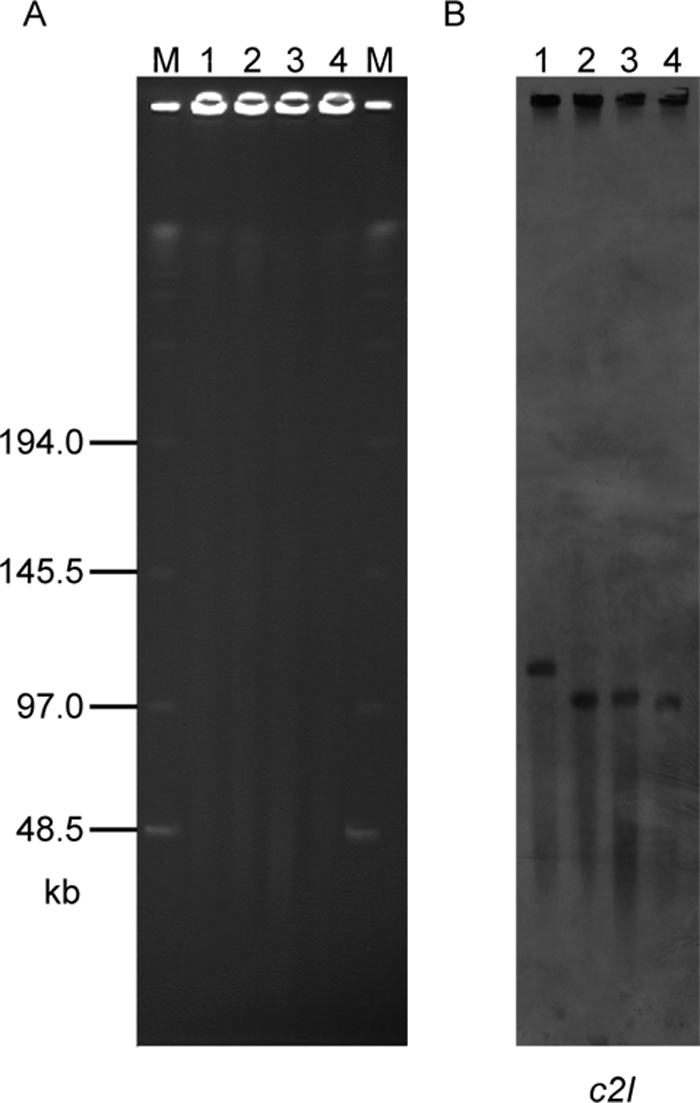
PFGE and Southern hybridization analyses with type C and D strains: PFGE performed with nondigested DNAs of strains (C)-203U28, C-ST, C-468 and D-1873 (A) and Southern hybridization performed with these DNAs and the c2I probe (B). Lanes M, size maker; lane 1, (C)-203U28; lane 2, C-ST; lane 3, C-468; lane 4, D-1873.
Determination of the pC2C203U28 sequence and its general features.
In order to confirm the conclusion described above, we tried to determine the whole sequence of the plasmid. Although we tried to purify intact whole plasmids from (C)-203U28, C-ST, C-468, and D-1873, we did not succeed because the plasmids were extremely unstable. Therefore, we employed a unique technique that consisted of a combination of inverse PCR and primer-walking methods as described in Materials and Methods, and we finally succeeded in determining the complete nucleotide sequence of the large plasmid designated pC2C203U28 from (C)-203U28.
The DNA of pC2C203U28 was circular 106,981-bp double-stranded DNA. The average G+C content was very low, 26.2% (Fig. 2), like the G+C contents of the chromosomal DNAs, plasmids, and bacteriophages of clostridia (6, 25, 31, 35-37, 39), including the chromosomal DNAs of C. botulinum type A (28.2%) and type B1 (28.3%), Clostridium tetani (28.6%), Clostridium acetobutylicum (30.9%), C. perfringens type A (28.6%), and C. difficile (29.1%); the plasmids of C. botulinum type A3 (25.6%), type B1 (25.4%), and type Ba4 (25.6%), and C. tetani (24.5%); and a bacteriophage of C. botulinum type C (26.2%).
FIG. 2.

Circular map of the pC2C203U28 plasmid of (C)-203U28. The numbers in the outermost circle indicate the scale (in kb). Position 1 is the putative origin of replication. Starting with the outermost circle with different colors, circles 1 and 2 show predicted coding sequences on the forward strand (circle 1) and reverse strand (circle 2). In the plasmid map the ORFs are color coded according to their putative functions. The genes encoding the C2 toxin are c2I and c2II (c2p056 and c2p057) and are indicated by red. The innermost circle (circle 3) shows the variation in the G+C content.
We identified 123 ORFs in the pC2C203U28 plasmid (c2p001 to c2p123) but no gene for tRNA (Fig. 3). Only 57 of the gene products of the 123 ORFs identified were functionally assigned based on sequence homology to known proteins, and 29 gene products shared no significant sequence homology with any protein in the databases. The remaining 37 gene products exhibited sequence similarity to proteins with unknown functions (see Table S2 in the supplemental material). They were categorized into 10 groups based on their predicted functions. There were several interesting points concerning the genes identified, as follows.
(i) DNA processing.
The pC2C203U28 genes encoded nine proteins (C2P001, C2P020, C2P031, C2P032, C2P034, C2P038, C2P058, C2P061, and C2P116) predicted to be involved in DNA processing (Fig. 3). Six of these proteins (C2P001, C2P020, C2P031, C2P032, C2P038, and C2P061) were thought to be related to various kinds of DNA-processing events (replication), two were involved in recombination (C2P058 and C2P116; the latter resembled XerC/D), and one was involved in partitioning of the plasmid (C2P034, which resembled ParA). Of the six gene products that were thought to play a role in replication, it seemed that only C2P001 was complete, while the other five proteins were nonfunctional. C2P031 and C2P032 were split, C2P020 and C2P038 were truncated in both the N- and C-terminal regions, and C2P061 was truncated only in the C-terminal region.
(ii) σ Factor-like proteins.
In clostridia, several σ factor-like proteins (Ctp10, Ctp11, CLC_1645, CLD_2938, BotR, TetR, UviA, and TxeR) have been described (9). BotR, TetR, UviA, and TxeR were identified as ECF σ factor proteins belonging to the σ70 family (10, 24, 29). In the present study, genes encoding three products (C2P015, C2P030, and C2P052) considered to be σ factor-like proteins were identified on pC2C203U28. C2P015, C2P030, and C2P052 consist of 146, 182, and 180 amino acids, respectively. C2P030 and C2P052 seemed to be complete, but C2P015 seemed to be truncated in the C-terminal region. Comparison of these three gene products showed that they were highly homologous (Fig. 4) and were very similar to Ctp10 (45.3% identity with 179 overlapping amino acids) encoded by a gene located on circular plasmid pE88 of C. tetani, CLC_1645 (46.6% identity with 176 overlapping amino acids) encoded by a gene located on the C. botulinum type A chromosome, CLD_2938 (47.2% identity with 176 overlapping amino acids) encoded by a gene located on a C. botulinum type B plasmid, and UviA (23.2% identity with 177 overlapping amino acids) encoded by a gene located on the C. perfringens plasmid but different from the BotR, TetR, and TxeR proteins. This was confirmed by constructing a phylogenetic tree by the neighbor-joining method; the three gene products (C2P015, C2P030, and C2P052) were close to Ctp10 and CLC_1645 but were distant from BotR, TetR, and TxeR (Fig. 5). However, it also became clear after construction of the phylogenetic tree that C2P015, C2P030, C2P052, Ctp10, Ctp11, CLC_1645, and CLD_2938 each could be grouped with the ECF σ factor proteins in the σ70 family, like BotR, TetR, UviA, TxeR, RpoE, and SigY.
FIG. 4.

Sequence alignment of the putative σ factor proteins, including the three σ factor-like proteins (C2P015, C2P030, and C2P052) encoded on the pC2C203U28 plasmid and Ctp10 encoded on the toxin plasmid of C. tetani. Residues with a black background are identical in >75% of the sequences, residues with a gray background are similar, and the dashes indicate gaps in the alignment.
FIG. 5.

Phylogenetic analysis of the σ70 family σ factors. The σ70 family σ factors were retrieved from the KEGG and NCBI GenBank databases. The phylogenetic tree was constructed based on the amino acid sequences of all 22 σ factors. The neighbor-joining method was applied to genetic distances between σ70 family σ factor protein sequences. The scale bar indicates a genetic distance of 0.1.
(iii) Two-component system.
Two-component systems have been reported for many bacteria, including C. perfringens (VirR/VirS), C. tetani, C. botulinum, C. acetobutylicum, C. difficile, B. subtilis, B. cereus, and Bacillus halodurans (6, 20, 23, 25, 35-39, 41). Here, we showed that two gene products, C2P002 and C2P003, were highly homologous to these two-component systems.
(iv) Transport systems.
Eight of the gene products (C2P005, C2P019, C2P042, C2P102, C2P107, C2P108, C2P109, and C2P120) were considered to be permease-, ATP binding-, and substrate-binding domains of an ATP-binding cassette (ABC) transporter. It seemed that c2p107 encoded the molybdate-binding protein, c2p102 and c2p108 encoded the permease domain, and the remaining five genes encoded the ATP-binding domain (Fig. 3). Three of the eight gene products, C2P120, C2P019, and C2P042, were predicted to be nonfunctional because C2P120 was split by frameshift mutation into two parts (C2P120a and C2P120b), and C2P019 and C2P042 were truncated in their C-terminal regions. The other five ORFs were presumed to be functional. In particular, it was thought that the products of the c2p107 to c2p109 genes might form a unit of the ABC transporter and play a role in the uptake of molybdenum into the cells (Fig. 6 and 7).
FIG. 6.

Possible purine catabolic pathway. This metabolic pathway is based on the B. subtilis motif (32). The genes of (C)-203U28 that appeared to participate in this pathway are indicated by their c2p numbers. The gene-enzyme relationships of the purine degradation pathway are shown in Fig. 3.
FIG. 7.

Putative model for the assembly of XDH. This model is based on the Moco biosynthesis pathway in E. coli and XDH assembly in R. capsulatus (30, 33, 34). Abbreviations: MPT-AMP, adenylated molybdopterin; bis-MGD, bis-molybdopterin guanine dinucleotide cofactor; MOS, Moco sulfurase; MOSC, Moco sulfurase C-terminal domain; FAD, flavin adenine dinucleotide.
(v) Purine catabolism.
Many genes involved in purine catabolism were identified (Fig. 3). The metabolism of hypoxantine or guanine to NH3 and CO2 is summarized in Fig. 6. In this metabolism, xanthine dehydrogenase (XDH) and molybdenum cofactor (Moco) are the important enzymes. The assembly of these enzymes is shown in Fig. 7. We identified 20 functional ORFs (c2p068, c2p70 to c2p077, c2p100 to c2p102, c2p105 to c2p109, and c2p111 to c2p113) encoding enzymes or proteins for this pathway. These genes were located in two regions: an 11-kb region (map positions 57 to 68) and a 12-kb region (map positions 90 to 102) downstream of the C2 toxin gene. All of the products of the 20 ORFs (C2P068, C2P070 to C2P077, C2P100 to C2P102, C2P105 to C2P109, and C2P111 to C2P113) exhibited some similarities to the proteins participating in purine catabolism in E. coli, B. subtilis, and Rhodobacter capsulatus (30, 32-34). It seemed that C2P070 to C2P072 and C2P074 to C2P077 were related to assembly of XDH and that C2P101, C2P106 to C2P109, and C2P111 to C2P113 participated in Moco biosynthesis; C2P107 to C2P109 were identified as a molybdate ABC transporter as described above. In addition, C2P112 and C2P113 catalyzed the conversion of GTP to precursor Z, and C2P106 catalyzed the conversion of precursor Z to molybdopterin (MPT). C2P111 and C2P073 catalyzed the conversion of MPT to adenylated molybdopterin and the conversion of Moco to bis-molybdopterin guanine dinucleotide cofactor, respectively. C2P101 was homologous to the Moco sulfurase C-terminal domain (sulfur carrier domain) of molybdenum cofactor sulfurase that transfers sulfur to Mo, forming the sulfido version of Moco (3). Active XDH is composed of three subunits encoded by the xdhA, xdhB, and xdhC genes in R. capsulatus (these genes correspond to pucC/E, pucD, and pucA, respectively, in B. subtilis). The products of the c2p070 and c2p076, c2p072 and c2p074, and c2p077 genes were homologous to PucC/E, PucD, and PucA, respectively. The C2P068 and C2P100 proteins and the C2P105 protein were homologous to the enzymes that catalyze the conversion of guanine to xanthine and the conversion of carbamoylphosphate to NH3 and CO2 in the last step, respectively. However, in E. coli, B. subtilis, and R. capsulatus, none of the expected genes associated with purine catabolism were identified on the chromosomes. The pC2C203U28 plasmid also lacked some genes needed for purine catabolism (Fig. 6 and Fig. 7).
(vi) IS elements.
Four insertion sequence (IS) copies were identified (Fig. 8). Two copies (c2p086-c2p087 and c2p117-c2p118) seemed to be functional, but the other two copies (c2p028 and c2p047) seemed to be nonfunctional; c2p047 was truncated, and c2p028 was degenerate. In the two functional IS elements (c2p086-c2p087 and c2p117-c2p118), some similarities were detected; both elements had an IS target site with the same sequence (TTTAA), and both lacked terminal inverted repeats. The level of homology of the total amino acid sequences encoded by these IS elements, however, was low. Therefore, the regions including c2p086-c2p087 and c2p117-c2p118 were designated ISCbt5 (or ISCbt5-a) and ISCbt6 (or ISCbt6-a), respectively. ISCbt5-a and ISCbt6-a were similar to the IS3/911 and IS200/605 families, respectively. The C2P087 protein encoded by ISCbt5-a showed a high level of amino acid sequence homology to the product of the cst045 gene in the c-st phage genome that we previously identified as forming an IS element (31). Thus, it was concluded that ISCbt5-a in the pC2C203U28 plasmid and the IS element (designated ISCbt5-c) including cst045 in the c-st phage genome could be grouped with ISCbt5, although ISCbt5-c lacked 282 bp in its 5′ region. When the total DNAs of the C-ST, C-203, C-468, and D-1873 toxigenic strains were analyzed to determine whether they had IS elements like ISCbt5-a or ISCbt6-a, only D-1873 possessed both of these IS elements (designated ISCbt5-b and ISCbt6-b), while all of the other strains had only the IS target site described above. Therefore, ISCbt5 and ISCbt6 seemed to be inserted specifically into the TTTAA sequence without target site duplication.
FIG. 8.

Structures of three IS elements. A total four copies of IS elements were identified on the pC2C203U28 plasmid, but only two copies (c2p086-c2p087 and c2p117-c2p118) seemed to be functional. The regions including c2p086-c2p087 and c2p117-c2p118 were designated ISCbt5 (or ISCbt5-a) and ISCbt6 (or ISCbt6-a), respectively. An IS element similar to ISCbt5-a was found in the c-st phage genome, and it was designated ISCbt5-c; C2P087 of ISCbt5-a was similar to CST045 of ISCbt5-c, although ISCbt5-c had a deletion in its 5′-terminal region. These IS elements had the special target sites (TTTAA). The total DNA of the D-1873 strain contained both these IS elements (designated ISCbt5-b and ISCbt6-b) with the TTTAA target site, but the DNAs of C-ST, C-203, and C-468 strains contained only the IS target site.
(vii) PCR scanning analysis.
In order to determine whether the gene features observed in the (C)-203U28 strain were conserved in other C. botulinum type C and D strains, we employed C2 toxin-producing strains C-ST, C-468, and D-1873 and C2 toxin-negative strains (C)-AO2 and D-4947. DNAs were extracted from the cells and analyzed by PCR scanning using a set of PCR primers which were prepared based on the pC2C203U28 sequence in order to cover all of pC2C203U28 (Fig. 3; see Table S3 in the supplemental material). As expected, many of these primers yielded PCR products with C-ST, C-468, and D-1873 DNAs as templates, indicating that pC2C203U28 was significantly conserved in them, whereas none of the PCR products were generated from (C)-AO2 and D-4947 DNAs. In the three C2 toxin-producing strains, the genes coding for the putative replication protein (C2P001), resolvase (C2P058), XerC/D (C2P116), and three σ factors (C2P015, C2P030, and C2P052) and seven genes involved in purine catabolism (encoding C2P072, C2P074, C2P077, C2P100, C2P102, C2P106, and C2P112) were also conserved along with the C2 toxin gene. However, the strains differed with respect to the presence of the two-component system genes (c2p002 and c2p003) and the parA gene (c2p034); these genes were conserved in D-1873 but not in C-ST and C-468.
Upstream and downstream regions including the C2 toxin gene.
We sequenced 5 kb of each of the upstream and downstream regions including the C2 toxin gene in C-ST, C-468, and D-1873 and compared them with the pC2C203U28 sequences (Fig. 9). The data showed that, although C2P053 and C2P054 were not present in C-ST and C-468, both regions including the C2 toxin gene were highly conserved (99 to 100% nucleotide sequence identity) in the type C and D strains examined.
DISCUSSION
Recently, the whole nucleotide sequences of many clostridial toxin genes have been determined, and their locations on cell chromosomes, plasmids, or bacteriophages have been elucidated. It was confirmed that C. botulinum type C and D toxin genes are carried by specific bacteriophages (12, 13, 18, 19, 26) and that the genes for C. botulinum type G toxin and tetanus toxin are on 114-kb and 74-kb plasmids, respectively (6, 11). For C. botulinum types A, B, E, and F, it has been postulated that the genes are on the chromosome. However, it has also become clear that type A and B toxin genes are located on a plasmid in some type A and B strains. Recently, C. botulinum type A and B strains have been classified as A1 to A4, B1 to B3, bivalent B, and bivalent (Ab, Ba, and Bf) (17). Smith et al. reported that in an A3 strain (Loch Maree) and a B1 strain (Okra) the toxin genes are on 267-kb and 149-kb plasmids, respectively (39). They also found that a bivalent Ba4 strain (657) possessed a 270-kb plasmid containing both type A (A4) and type B (bivalent B) toxin genes.
The complete nucleotide sequences of the c2I and c2II genes encoding C. botulinum C2 toxin were reported by Fujii et al. (15) and Kimura et al. (22). Although these authors speculated that these genes might be on the chromosome, this was uncertain, because they cloned each c2I and c2II gene fragment from the total DNA of (C)-203U28 cells. In the present study, we demonstrated that the C2 toxin gene is located on a large plasmid. We first found that the c2I and c2II genes were in a 100-kb band in a Southern hybridization analysis performed with specific probes for these genes and then confirmed that the 100-kb fragment is a large plasmid by determining the entire nucleotide sequence by both inverse PCR and primer-walking procedures. It also became clear that the c2 genes are on a similar large plasmid in other C. botulinum type C and D strains (C-ST, C-468, and D-1873), as well as in (C)-203U28, and these genes were designated pC2C203U28.
The pC2C203U28 plasmid of (C)-203U28 was a 106,981-bp double-stranded DNA and encoded 123 proteins. The most characteristic features of this plasmid compared with other plasmids were the presence of many genes involved in DNA processing (replication, recombination, and plasmid partition), regulation of gene expression (σ factor-like proteins and two-component system proteins), IS elements, and purine catabolism. The pC2C203U28 plasmid contained six replication genes (c2p001, c2p020, c2p031, c2p032, c2p038, and c2p061) (although five of them seemed to be nonfunctional), two recombination genes (c2p058 and c2p116), and one partition gene (c2p034). The products of the c2p034 and c2p116 genes resembled ParA and XerC/D family proteins, respectively, indicating that the plasmid may be stably maintained as a low-copy-number plasmid. Of the four IS elements, two were structurally intact (designated ISCbt5 and ISCbt6). Although ISCbt5 and ISCbt6 had different structures, they had the same target sites (TTTAA). When we analyzed the presence of these IS elements and their target sites in the DNAs of several type C and D strains and c-st phage, it became clear that they are widespread; both ISCbt5 and ISCbt6 were detected in D-1873, an ISCbt5-like element was found in c-st phage, and a TTTAA target sequence was detected in all of the samples examined. Therefore, it was estimated that these IS elements play an important role in transfer of the genes (C2 toxin genes) into type C and D strains and that the pC2C203U28 plasmid might be formed by fusion with several small replicons.
In clostridia, many RNA polymerase σ factor proteins, including ECF σ-like proteins belonging to the σ70 family, have been reported (10, 24, 29). In the present work, we found that many RNA polymerase σ factor proteins, such as CLC_1645 (C. botulinum type A), CLD_2938 (C. botulinum type B), and Ctp10 and Ctp11 (C. tetani), can be classified as ECF σ-like proteins belonging to the σ70 family, just like BotR, TetR, TxeR, and UviA. BotR, TetR, TxeR, and UviA regulate the synthesis of the C. botulinum neurotoxin, C. tetani neurotoxin, C. difficile toxins (ToxA and ToxB), and a bacteriocin of C. perfringens, respectively. It has been shown that the ctp10 and ctp11 genes are located on the same plasmid as the tetR gene, and it has been speculated that Ctp10 might be involved in the regulation of tetanus toxin production as well as TetR or in the synthesis of collagenase (7). In the present study, three ECF σ factor-like proteins (C2P015, C2P030, and C2P052) were identified. These three gene products were very similar to Ctp10, CLC_1645, CLD_2938, and UviA but not to BotR, TetR, and TxeR. It seemed that c2p030 and c2p052 were complete genes, and c2p052 was about 3 kb upstream of the C2 toxin gene in the same orientation. Thus, it is speculated that C2P052 might be involved in regulation of C2 toxin formation.
A total of 20 genes that were predicted to be involved in purine catabolism were identified in the pC2C203U28 plasmid. All gene products were predicted to participate in the formation of XDH and Moco, and the products of the remaining genes, the c2p068 and c2p100 genes and the c2p105 gene, were the enzymes guanine deaminase and carbamate kinase, respectively. The other genes required to catabolize the purine were not identified. This is similar to the results reported for E. coli, B. subtilis, and R. capsulatus. Recently, the whole-genome sequences of C. botulinum type A and B were reported (35, 39). In C. botulinum chromosomal DNA, the genes for XDH formation and for carbamate kinase were also identified. The 20 genes identified in the pC2C203U28 plasmid seemed to be functional based on the gene sizes. Therefore, we speculated that the products of these 20 genes might be used for purine catabolism. The remaining enzymes or proteins needed for purine catabolism might be products of genes on the chromosome. The final products in this pathway are NH3 and CO2, which can be used as sources of nitrogen and carbon for growth. In an attempt to confirm that the growth of the (C)-203U28 strain is influenced by pC2C203U28, we tried to cure pC2C203U28 by incubating the cells at 48°C (the maximum temperature for growth) for 1 to 4 days, but we were not successful. We are now trying to knock out some of the 19 genes and/or regulation genes (especially c2p052) in order to clarify their functions.
We compared homologies between 123 ORFs identified on the pC2C203U28 plasmid and the ORFs in C. botulinum type A3 (pCLK), type B1 (pCLD), and type Ba4 (pCLJ) plasmids harboring neurotoxin genes (39). Although the three latter plasmids (pCLK, pCLD, and pCLJ) share significant sequence identity, all of the ORFs on the pC2C203U28 plasmid exhibited similarity to ORFs on them, indicating that pC2C203U28 is really different from them.
The possibility that the pC2C203U28 plasmid harboring C2 toxin genes is transferred between strains is quite interesting. In our genome analysis, we found no gene involved in transmission of a plasmid like a self-transmissible plasmid through conjugation, as reported elsewhere (8, 14). Further investigation is needed to resolve this issue.
Supplementary Material
Acknowledgments
This work was supported by grants-in-aid for scientific research 19790321 and 19390126 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Published ahead of print on 6 March 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Aktories, K., M. Barmann, I. Ohishi, S. Tsuyama, K. H. Jakobs, and E. Habermann. 1986. Botulinum C2 toxin ADP-ribosylates actin. Nature 322390-392. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anantharaman, V., and L. Aravind. 2002. MOSC domains: ancient, predicted sulfur-carrier domains, present in diverse metal-sulfur cluster biosynthesis proteins including molybdenum cofactor sulfurases. FEMS Microbiol. Lett. 20755-61. [DOI] [PubMed] [Google Scholar]
- 4.Barth, H., K. Aktories, M. R. Popoff, and B. G. Stiles. 2004. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 68373-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth, H., D. Blocker, J. Behlke, W. Bergsma-Schutter, A. Brisson, R. Benz, and K. Aktories. 2000. Cellular uptake of Clostridium botulinum C2 toxin requires oligomerization and acidification. J. Biol. Chem. 27518704-18711. [DOI] [PubMed] [Google Scholar]
- 6.Bruggemann, H., S. Baumer, W. F. Fricke, A. Wiezer, H. Liesegang, I. Decker, C. Herzberg, R. Martinez-Arias, R. Merkl, A. Henne, and G. Gottschalk. 2003. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc. Natl. Acad. Sci. USA 1001316-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruggemann, H., and G. Gottschalk. 2004. Insights in metabolism and toxin production from the complete genome sequence of Clostridium tetani. Anaerobe 1053-68. [DOI] [PubMed] [Google Scholar]
- 8.Brynestad, S., M. R. Sarker, B. A. McClane, P. E. Granum, and J. I. Rood. 2001. Enterotoxin plasmid from Clostridium perfringens is conjugative. Infect. Immun. 693483-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupuy, B., and S. Matamouros. 2006. Regulation of toxin and bacteriocin synthesis in Clostridium species by a new subgroup of RNA polymerase sigma-factors. Res. Microbiol. 157201-205. [DOI] [PubMed] [Google Scholar]
- 10.Dupuy, B., S. Raffestin, S. Matamouros, N. Mani, M. R. Popoff, and A. L. Sonenshein. 2006. Regulation of toxin and bacteriocin gene expression in Clostridium by interchangeable RNA polymerase sigma factors. Mol. Microbiol. 601044-1057. [DOI] [PubMed] [Google Scholar]
- 11.Eklund, M. W., F. T. Poysky, L. M. Mseitif, and M. S. Strom. 1988. Evidence for plasmid-mediated toxin and bacteriocin production in Clostridium botulinum type G. Appl. Environ. Microbiol. 541405-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eklund, M. W., F. T. Poysky, and S. M. Reed. 1972. Bacteriophage and the toxigenicity of Clostridium botulinum type D. Nat. New Biol. 23516-17. [DOI] [PubMed] [Google Scholar]
- 13.Eklund, M. W., F. T. Poysky, S. M. Reed, and C. A. Smith. 1971. Bacteriophage and the toxigenicity of Clostridium botulinum type C. Science 172480-482. [DOI] [PubMed] [Google Scholar]
- 14.Francia, M. V., A. Varsaki, M. P. Garcillan-Barcia, A. Latorre, C. Drainas, and F. de la Cruz. 2004. A classification scheme for mobilization regions of bacterial plasmids. FEMS Microbiol. Rev. 2879-100. [DOI] [PubMed] [Google Scholar]
- 15.Fujii, N., T. Kubota, S. Shirakawa, K. Kimura, I. Ohishi, K. Moriishi, E. Isogai, and H. Isogai. 1996. Characterization of component-I gene of botulinum C2 toxin and PCR detection of its gene in clostridial species. Biochem. Biophys. Res. Commun. 220353-359. [DOI] [PubMed] [Google Scholar]
- 16.Fujii, N., K. Oguma, N. Yokosawa, K. Kimura, and K. Tsuzuki. 1988. Characterization of bacteriophage nucleic acids obtained from Clostridium botulinum types C and D. Appl. Environ. Microbiol. 5469-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill, K. K., T. J. Smith, C. H. Helma, L. O. Ticknor, B. T. Foley, R. T. Svensson, J. L. Brown, E. A. Johnson, L. A. Smith, R. T. Okinaka, P. J. Jackson, and J. D. Marks. 2007. Genetic diversity among botulinum neurotoxin-producing clostridial strains. J. Bacteriol. 189818-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue, K., and H. Iida. 1970. Conversion of toxigenicity in Clostridium botulinum type C. Jpn. J. Microbiol. 1487-89. [DOI] [PubMed] [Google Scholar]
- 19.Inoue, K., and H. Iida. 1971. Phage-conversion of toxigenicity in Clostridium botulinum types C and D. Jpn. J. Med. Sci. Biol. 2453-56. [PubMed] [Google Scholar]
- 20.Ivanova, N., A. Sorokin, I. Anderson, N. Galleron, B. Candelon, V. Kapatral, A. Bhattacharyya, G. Reznik, N. Mikhailova, A. Lapidus, L. Chu, M. Mazur, E. Goltsman, N. Larsen, M. D'Souza, T. Walunas, Y. Grechkin, G. Pusch, R. Haselkorn, M. Fonstein, S. D. Ehrlich, R. Overbeek, and N. Kyrpides. 2003. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 42387-91. [DOI] [PubMed] [Google Scholar]
- 21.Kimura, K., N. Fujii, K. Tsuzuki, T. Murakami, T. Indoh, N. Yokosawa, K. Takeshi, B. Syuto, and K. Oguma. 1990. The complete nucleotide sequence of the gene coding for botulinum type C1 toxin in the C-ST phage genome. Biochem. Biophys. Res. Commun. 1711304-1311. [DOI] [PubMed] [Google Scholar]
- 22.Kimura, K., T. Kubota, I. Ohishi, E. Isogai, H. Isogai, and N. Fujii. 1998. The gene for component-II of botulinum C2 toxin. Vet. Microbiol. 6227-34. [DOI] [PubMed] [Google Scholar]
- 23.Kunst, F., N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessieres, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S. K. Choi, J. J. Codani, I. F. Connerton, A. Danchin, et al. 1997. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390249-256. [DOI] [PubMed] [Google Scholar]
- 24.Mani, N., and B. Dupuy. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc. Natl. Acad. Sci. USA 985844-5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nolling, J., G. Breton, M. V. Omelchenko, K. S. Makarova, Q. Zeng, R. Gibson, H. M. Lee, J. Dubois, D. Qiu, J. Hitti, Y. I. Wolf, R. L. Tatusov, F. Sabathe, L. Doucette-Stamm, P. Soucaille, M. J. Daly, G. N. Bennett, E. V. Koonin, and D. R. Smith. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 1834823-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oguma, K., H. Iida, and K. Inoue. 1973. Bacteriophage and toxigenicity in Clostridium botulinum: an additional evidence for phage conversion. Jpn. J. Microbiol. 17425-426. [DOI] [PubMed] [Google Scholar]
- 27.Oguma, K., H. Iida, and M. Shiozaki. 1976. Phage conversion to hemagglutinin production in Clostridium botulinum types C and D. Infect. Immun. 14597-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohishi, I., M. Iwasaki, and G. Sakaguchi. 1980. Purification and characterization of two components of botulinum C2 toxin. Infect. Immun. 30668-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raffestin, S., B. Dupuy, J. C. Marvaud, and M. R. Popoff. 2005. BotR/A and TetR are alternative RNA polymerase sigma factors controlling the expression of the neurotoxin and associated protein genes in Clostridium botulinum type A and Clostridium tetani. Mol. Microbiol. 55235-249. [DOI] [PubMed] [Google Scholar]
- 30.Regulski, E. E., R. H. Moy, Z. Weinberg, J. E. Barrick, Z. Yao, W. L. Ruzzo, and R. R. Breaker. 2008. A widespread riboswitch candidate that controls bacterial genes involved in molybdenum cofactor and tungsten cofactor metabolism. Mol. Microbiol. 68918-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaguchi, Y., T. Hayashi, K. Kurokawa, K. Nakayama, K. Oshima, Y. Fujinaga, M. Ohnishi, E. Ohtsubo, M. Hattori, and K. Oguma. 2005. The genome sequence of Clostridium botulinum type C neurotoxin-converting phage and the molecular mechanisms of unstable lysogeny. Proc. Natl. Acad. Sci. USA 10217472-17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz, A. C., P. Nygaard, and H. H. Saxild. 2001. Functional analysis of 14 genes that constitute the purine catabolic pathway in Bacillus subtilis and evidence for a novel regulon controlled by the PucR transcription activator. J. Bacteriol. 1833293-3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schumann, S., M. Saggu, N. Moller, S. D. Anker, F. Lendzian, P. Hildebrandt, and S. Leimkuhler. 2008. The mechanism of assembly and cofactor insertion into Rhodobacter capsulatus xanthine dehydrogenase. J. Biol. Chem. 28316602-16611. [DOI] [PubMed] [Google Scholar]
- 34.Schwarz, G., and R. R. Mendel. 2006. Molybdenum cofactor biosynthesis and molybdenum enzymes. Annu. Rev. Plant Biol. 57623-647. [DOI] [PubMed] [Google Scholar]
- 35.Sebaihia, M., M. W. Peck, N. P. Minton, N. R. Thomson, M. T. Holden, W. J. Mitchell, A. T. Carter, S. D. Bentley, D. R. Mason, L. Crossman, C. J. Paul, A. Ivens, M. H. Wells-Bennik, I. J. Davis, A. M. Cerdeno-Tarraga, C. Churcher, M. A. Quail, T. Chillingworth, T. Feltwell, A. Fraser, I. Goodhead, Z. Hance, K. Jagels, N. Larke, M. Maddison, S. Moule, K. Mungall, H. Norbertczak, E. Rabbinowitsch, M. Sanders, M. Simmonds, B. White, S. Whithead, and J. Parkhill. 2007. Genome sequence of a proteolytic (group I) Clostridium botulinum strain Hall A and comparative analysis of the clostridial genomes. Genome Res. 171082-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sebaihia, M., B. W. Wren, P. Mullany, N. F. Fairweather, N. Minton, R. Stabler, N. R. Thomson, A. P. Roberts, A. M. Cerdeno-Tarraga, H. Wang, M. T. Holden, A. Wright, C. Churcher, M. A. Quail, S. Baker, N. Bason, K. Brooks, T. Chillingworth, A. Cronin, P. Davis, L. Dowd, A. Fraser, T. Feltwell, Z. Hance, S. Holroyd, K. Jagels, S. Moule, K. Mungall, C. Price, E. Rabbinowitsch, S. Sharp, M. Simmonds, K. Stevens, L. Unwin, S. Whithead, B. Dupuy, G. Dougan, B. Barrell, and J. Parkhill. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38779-786. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu, T., K. Ohtani, H. Hirakawa, K. Ohshima, A. Yamashita, T. Shiba, N. Ogasawara, M. Hattori, S. Kuhara, and H. Hayashi. 2002. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. USA 99996-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu, T., H. Yaguchi, K. Ohtani, S. Banu, and H. Hayashi. 2002. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol. Microbiol. 43257-265. [DOI] [PubMed] [Google Scholar]
- 39.Smith, T. J., K. K. Hill, B. T. Foley, J. C. Detter, A. C. Munk, D. C. Bruce, N. A. Doggett, L. A. Smith, J. D. Marks, G. Xie, and T. S. Brettin. 2007. Analysis of the neurotoxin complex Genes in Clostridium botulinum A1-A4 and B1 strains: BoNT/A3, /Ba4 and /B1 clusters are located within plasmids. PLoS ONE 2e1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suen, J. C., C. L. Hatheway, A. G. Steigerwalt, and D. J. Brenner. 1988. Clostridiurn argentinense sp. nov.: a genetically homogeneous group composed of all strains of Clostridium botulinum toxin type G and some nontoxigenic strains previously identified as Clostridium subterminale or Clostridium hastiforme. Int. J. Syst. Bacteriol. 38375-381. [Google Scholar]
- 41.Takami, H., K. Nakasone, Y. Takaki, G. Maeno, R. Sasaki, N. Masui, F. Fuji, C. Hirama, Y. Nakamura, N. Ogasawara, S. Kuhara, and K. Horikoshi. 2000. Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res. 284317-4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.