

Abstract
The human immunodeficiency virus type 1 (HIV-1) gp41 coiled-coil domain is an important target for fusion inhibitors, including the peptide T20, which has been approved as a drug against HIV-1. Research into nonpeptide fusion inhibitors has focused primarily on a hydrophobic pocket located within the coiled coil and has so far yielded compounds with relatively weak fusion inhibitory activity. Here, we describe metal ion-assisted stabilization of an extended 39-residue construct of gp41, which includes residues of the hydrophobic pocket and also of an extended groove N terminal to the hydrophobic pocket. We show that the presence of a metal ion and the high-affinity interaction between the receptor construct and cognate C-peptides result in a simple and highly selective assay for fusion inhibitors that may be used to scan large compound libraries. The long construct presents multiple potential binding sites along the extended coiled-coil groove. We demonstrate the modular use of assay probes to detect whether compounds bind in the hydrophobic pocket or elsewhere along the groove. Rapid detection and quantitation of hits can lead to the discovery of compounds binding to different sites along the groove and provide structure-activity relationship data for optimization. Compounds binding to adjacent sites could be linked to form more potent fusion inhibitors.
Fusion inhibitors are a promising new class of human immunodeficiency virus type 1 (HIV-1) therapeutics, with currently only one FDA-approved drug, T20 (Fuzeon) (17). T20 is a 36-residue peptide subject to the limitations of a peptide drug, i.e., high cost, limited half-life, and the requirement for parenteral or subcutaneous administration. It is derived from the C-heptad repeat (CHR) region of the HIV-1 transmembrane glycoprotein gp41. It is believed to act in a dominant negative manner, preventing the formation of the gp41 trimer of hairpins by binding to the N-heptad repeat (NHR) coiled-coil domain and the cell membrane (5, 12, 26). Many CHR peptides have been investigated as fusion inhibitors, including various derivatives of T20 (27) and of the peptide C34 (16), which partially overlaps T20 but includes residues that bind in a known hydrophobic pocket on the coiled coil. The long protein-protein interaction surface results in nanomolar binding between the coiled coil and its cognate CHR peptide. Fusion inhibition in vitro appears to correlate with the peptide binding affinity (4).
Low-molecular-weight compounds would be an attractive alternative to peptides as anti-HIV fusion inhibitors. Small molecules with high binding affinities have, however, proven to be difficult to develop (14). Most small-molecule inhibition studies have targeted the hydrophobic pocket, long considered a hot spot for inhibiting the protein-protein interaction (3). It appears likely that the extension of small-molecule inhibitors beyond the pocket will be necessary to obtain higher potency. The development of such inhibitors requires the accessibility of a long segment of the coiled-coil groove, as well as detailed knowledge of the binding locations of small molecules along the groove.
Many of the biochemical detection schemes for peptide and/or small-molecule binding have involved the use of a GCN4-gp41 fusion construct. The soluble trimeric GCN4 segment stabilizes and solubilizes the hydrophobic gp41 trimer, providing access to the coiled-coil grooves. gp41 segment lengths from 17 to 50 residues have been tested using this concept. A 17-residue segment encompassing the residues of the hydrophobic pocket forms a discreet well-behaved trimer which has been used in multiple crystal studies of peptide binding (6, 23). However, longer segments of gp41 tend to display less optimal characteristics. GCN4-gp41 fusion proteins with 36 and 50 residues have shown limited stability or aggregation in the absence of the accompanying C-peptide (22, 24). Subsequently, several protein complexes containing a mixture of NHR and CHR segments were designed (15, 21). The most useful of these is a complex called 5-helix, which consists of alternating NHR and CHR segments interspersed with short loops (21). The protein folds into a five-helix bundle in which one groove of the NHR coiled coil is accessible for binding. 5-Helix was constructed with a 40-residue NHR, although recently, a 53-residue form of 5-helix was constructed to better evaluate the T20 binding site (2). 5-Helix has been used in polarization assays to detect small-molecule binding (7). The peptide N36 has been stabilized as a mannose binding protein fusion product for use in polarization assays (18) and employed along with C34 in an enzyme-linked immunosorbent assay in which antibodies detect the six-helix bundle that spontaneously forms between the two peptides (11). Small molecules which interfere with the formation of the six-helix bundle can be detected by these methods.
In this report, we describe the construction and evaluation of an extended stabilized coiled-coil segment of gp41 from a 39-residue NHR peptide, using metal ion coordination to N-terminal bipyridine groups. We have used this method previously to demonstrate metal ion-induced self-assembly of a 26-residue segment of the NHR, called env2.0 (1). Here, we show that bipyridine-metal coordination has the ability to stabilize longer segments of the gp41 coiled coil, leaving exposed grooves of the coiled coil for binding studies. There are only six nonnative residues in the construct, at the N- and C-terminal ends. Importantly, the metal coordination complex provides an intrinsic probe of binding, in addition to its effect in stabilizing the structure. We describe fluorescence experiments similar to those described earlier for env2.0, in which the binding of a fluorescently labeled probe peptide can be monitored quantitatively through fluorescence quenching. While env2.0 bound to its cognate C-peptide C18-Aib with 1 μM affinity, we show that probes with mid- to low-nanomolar affinities can be selected for the longer complex, permitting the quantitative analysis of high-affinity inhibitors by using very small amounts of a sample and enabling the targeting of more than one binding site along the length of the groove.
The ease of the fluorescence measurement lends itself to a high-throughput-ready, inexpensive assay for fusion inhibitors. The high binding affinity between the probe and the receptor enables the new assay to quantitatively detect inhibitors with nanomolar potencies, permitting a range of detection similar to that for mannose binding protein-N36 and 5-helix. These large protein constructs and antibody detection systems can be cumbersome or expensive for high-throughput screening, typically requiring significant amounts of material and/or complex plate preparation. They also can neither identify nor target the specific location of inhibitor binding. We will show that the modular nature of our receptor and probe construction enables us to develop screens selective for both compound affinity and binding location.
MATERIALS AND METHODS
Materials.
Peptides were prepared by standard solid-phase synthesis (Biosynthesis, Inc.). NHR peptides were capped with 5-carboxy-2,2′-bipyridine while on the resin. The coiled-coil receptor was prepared by the addition of a mixture of freshly prepared ferrous ammonium sulfate and NHR peptide at a stoichiometry of 1/3 in 25 mM Tris-acetate buffer, pH 7.0. The stock concentration was determined using an ɛ291 of 17,200 M−1 cm−1. CHR peptide probes were prepared with a C-terminal cysteine, which was labeled with 5-iodoacetamino-fluorescein (FL; Sigma-Aldrich) or Lucifer yellow iodoacetamide (LY; Invitrogen) by a reaction with CHR peptide in 100 mM phosphate buffer, pH 7.0, at room temperature. The C-peptide was subsequently purified using a PD10 column (GE Healthcare), eluted with water, and lyophilized. Designations of C-peptides labeled with a fluorophore include LY or FL to indicate an LY or fluorescein label, respectively. Concentrations were determined using an ɛ425 of 10,800 M−1 cm−1 in 6 M guanidine for LY-labeled peptides (Molecular Probes) or an ɛ493 of 35,000 M−1 cm−1 in buffer at pH 7 for fluorescein-labeled peptides (25).
CD measurements.
Circular dichroism (CD) studies were performed on a DSM20 CD spectrophotometer from On-Line Instruments Systems, Inc., Bogart, GA, using 20 μM solutions of peptides in 15 mM Tris-acetate buffer, pH 7.0, at 25°C.
Fluorescence measurements.
Fluorescence measurements were performed on a Molecular Devices M5 plate reader using Greiner Bio-One 384-well small-volume clear-bottom plates. Each well contained 10 μl of sample solution in 25 mM Tris-acetate buffer, with 4% dimethyl sulfoxide and 0.01% Tween 20, pH 6.5 to 7. Measurements were recorded within 30 min after the mixing of assay components. Direct binding experiments were conducted using 15 to 150 nM fluorescently labeled CHR peptide as a probe. The dissociation constant (Kd) of the interaction was obtaining by fitting the data to a 1:1 binding model given by the following equation:
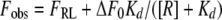 |
where Fobs is the observed fluorescence, [R] is the receptor concentration, equal to {Rt − Lt − Kd + [(Rt − Lt − Kd)2 + 4KdRt]1/2}, Lt is the total probe peptide concentration, and Rt is equal to 3[Fe(env5.0)3]. env5.0 is a 45-residue peptide containing 39 residues of the wild-type gp41 NHR, and Fe(env5.0)3 is the Fe2+-stabilized env5.0 trimer. The difference between the fluorescence of the free probe (Fmax) and the fluorescence of the receptor-probe complex (FRL) is expressed by the following equation: ΔF0 = Fmax − FRL. Data were fit using Kaleidagraph.
In inhibition experiments, samples were typically prepared by the sequential addition of the inhibitor, probe, and receptor and measured using the bottom read mode of the plate reader, with excitation and emission wavelengths set at 485 and 538 nm, respectively, for fluorescein and 425 and 538 nm, respectively, for LY. A control measurement was made under identical conditions in the absence of a receptor to check whether a spurious interaction between the probe and inhibitor occurred. The data are presented as fractional fluorescence levels relative to that for the control and were used to determine Ki by the numerical solution of 1:1 binding equations using Mathcad (Mathsoft, Cambridge, MA) (1).
RESULTS
Peptide design.
Fig. 1 depicts the peptides used in this study that were carved out of the full-length coiled-coil domain of gp41. The 45-residue peptide env5.0 contains 39 residues of the wild-type gp41 NHR and includes all of the residues of N36, plus an additional three wild-type gp41 residues at the C terminus. The remaining six residues include a bipyridine linker sequence (GQAV) and the two C-terminal residues, which were replaced with lysine. The positively charged lysine residues and the amidation at the C terminus assist in stabilizing the helical structure. env5.0 was matched to probe peptides C32-e5.0 and C39-e5.0, 32- and 39-residue segments of the gp41 CHR, each with a C-terminal cysteine for fluorophore labeling. The CHR-derived peptides were modified on the face exposed to the solvent according to strategies described previously for increasing solubility and stabilizing the helix by salt bridge and alanine substitutions (4, 19, 20).
FIG. 1.

Peptides used in assay development. (A) Schematic representation of the gp41 hairpin structure showing the boundaries of NHR and CHR helices. (B) Helical representation of the construct Fe(env5.0)3, shown in gray with bipyridine groups and ferrous ion in magenta; associated peptide probes are shown in green. C28-e5.0 overlaps with C39-e5.0 and is highlighted in yellow-green. Distances between the Cβ proton of the C-terminal cysteine and the ferrous ion are indicated by dashed lines. (C) Sequences of the peptides discussed in the text. The locations of peptides N36, C34, and T20, described in the literature, are given for comparison. Ac, acetyl; bpy, 5-carboxy-2,2′-bipyridine.
CD spectroscopy.
CD analyses of env5.0 in the absence and presence of Fe2+ and upon the addition of C32-e5.0, presented in Fig. 2, clearly demonstrated both the stabilization of the helical structure of env5.0 with Fe2+ and the interaction of Fe(env5.0)3 with its cognate C-peptide. A 20 μM solution of env5.0 in pH 7 buffer displayed a helical content of 57%, which increased to 70% upon the addition of Fe2+ in a 1/3 stoichiometry. No precipitate was detected in solutions of Fe(env5.0)3 at pH 7 with concentrations of up to 1 mM, and the receptor was stable at −80°C for an extended period. C32-e5.0 at 20 μM in solution was 69% helical. An equimolar mixture of the two peptides exhibited a total helical content of 82% for the system, representing a significant increase over the sum of component spectra. This result indicated an interaction between the NHR and CHR peptides, leading to enhanced helical structures of component N- and C-peptides, as is expected for six-helix bundle formation.
FIG. 2.

CD spectra of env5.0 and C32-e5.0 in Tris-acetate buffer, pH 7.0: ⋄, C32-e5.0; ○, env5.0; •, Fe(env5.0)3; ♦, mixture of Fe(env5.0)3 and C32-e5.0 at a ratio of 1:3; +, sum of the C32-e5.0 and Fe(env5.0)3 spectra (the expected spectrum of the mixture if the peptides did not interact). All spectra were obtained at 20 μM peptide and 25°C.
Fluorescence spectroscopy.
The Fe2+-stabilized trimer is a useful receptor in fluorescence experiments with C-peptide, since binding between the peptides can be monitored through changes in fluorescence intensity. Fluorescence quenching is expected when C-peptide is labeled with a fluorophore that emits at 540 nm, the wavelength of absorbance of Fe(5-carboxy-2,2′-bipyridine)3, provided the fluorophore comes into close proximity to the metal center upon binding (8). The selection of fluorescein for labeling ensured a signal intensity an order of magnitude higher than that of LY, the dye selected for studies of the micromolar interaction between env2.0 and C18-Aib (1). This approach permitted the use of much lower concentrations of probe peptides, suitable for the study of the higher-affinity interaction expected in this case, than those used for the env2.0-C18-Aib assay. Fluorescein was attached at position b of the heptad repeat through a cysteine side chain ∼18 or 13.6 Å away from the metal center in C32-e5.0 or C39-e5.0, respectively (Fig. 1). The fluorophore points away from the NHR-CHR interaction surface.
Figure 3 demonstrates the quenching of 150 nM C32-e5.0 or C39-e5.0 fluorescence that occurred upon the binding of Fe(env5.0)3. There is clearly flexibility in the allowed positions of the fluorophore, since the fluorescence intensities of both the 32- and 39-residue peptides were significantly quenched. The observed Kds were calculated to be 4.2 ± 1.8 nM and 9.7 ± 3.5 nM for C32-e5.0 and C39-e5.0, respectively, by fitting the data to equation 1, with residual fluorescence levels for the receptor-probe complexes (FRL in equation 1) of 6 and 9%. C32-e5.0 and C39-e5.0 also demonstrated low-nanomolar fusion inhibitory activities in a cell-cell fusion assay, yielding 50% inhibitory concentrations of 2 and 6 nM, respectively. The fluorescence intensity was reduced to 20% of the free-peptide value at a 125 nM receptor concentration, as shown in Fig. 3, presenting a large dynamic range for a potential competitive inhibition assay. At lower probe peptide concentrations, the amount of the receptor required to quench the fluorescence to the 20% level was reduced accordingly. The results of Z′ factor (28) determination for 15 nM probe peptide and 50 nM Fe(env5.0)3 are shown in Fig. 4. A Z′ factor of 0.88 and a signal-to-noise ratio of 32.6 were obtained. The assay is therefore highly sensitive and robust for inhibitor detection by high-throughput screening.
FIG. 3.

Fluorescence quenching upon the interaction of fluorescein-labeled C32-e5.0 (○) or C39-e5.0 (•) with Fe(env5.0)3. The concentration of metallopeptide indicated refers to the concentration of component peptide env5.0. The C-peptide concentration was constant at 150 nM, and the fluorescence intensity is reported relative to that of the C-peptide in the absence of Fe(env5.0)3. Data points represent mean values, and error bars show the standard deviations for four measurements.
FIG. 4.

Forty-eight repeat measurements of the upper and lower bounds of the assay using 15 nM C39-e5.0 for the upper bound and a mixture of C39-e5.0 at 15 nM and Fe(env5.0)3 at 50 nM (given as concentration of component peptides) for the lower bound. The y axis is the fluorescence intensity in arbitrary units.
Competitive inhibition.
Inhibitors of the Fe(env5.0)3-C32-e5.0-FL or Fe(env5.0)3-C39-e5.0-FL interaction in a ternary mixture with the assay components are expected to cause an increase in fluorescence. Inhibitors with Kis in the 1 to 100 nM range should be easily ranked, based on the Kd and the Z′ factor, by applying the single-point assay using a 0.2 μM inhibitor concentration. Kis and the stoichiometries of binding can be effectively determined from dose-response curves. High-affinity inhibitors (Ki < 1 nM) will readily be detected with ∼100% fluorescence signal recovery, although their relative potencies cannot be distinguished. These tight-binding inhibitors will also display overlapping dose-response curves, with 50% inhibitory concentrations close to the receptor concentration ([R] − Kd). Weaker binding inhibitors (Ki > 100 nM) may give dose-response curves if a high inhibitor concentration is accessible, but the binding stoichiometries and, hence, the mechanisms for these weaker inhibitors may not be ascertained, because all the dose-response curves will have similar curve shapes.
The ability of the assay system to quantify fusion inhibitors was tested by competitive inhibition of unlabeled peptides C32-e5.0 and C39-e5.0. The result is shown in Fig. 5A. Observed Kis for these two peptides were equal to the Kds (6.4 ± 1.4 nM for C32-e5.0 and 9.9 ± 1.8 nM for C39-e5.0). The assay was robust, with minimal aggregation and no precipitation of assay peptides, despite their lengths and levels of hydrophobicity. There was evidence of slight aggregation of Fe(env5.0)3 in the competitive inhibition experiment (Fig. 5A). Aggregation caused a 27% reduction in available binding sites but had no measurable effect on the quality of the assay. This finding contrasts favorably with the aggregation observed for unmodified NHR peptides, which showed 70% reduced effective concentrations (10). C39-e5.0-FL also showed a tendency to aggregate, as exhibited by a failure to consistently observe 100% fluorescence recovery with this probe peptide (Fig. 5A). Assays using C32-e5.0-FL may be preferable because they did not show this effect.
FIG. 5.

(A) Dose-response curves for peptide inhibitors C32-e5.0 (○) and C39-e5.0 (•), as determined using their corresponding fluorescently labeled probes at 25 nM and Fe(env5.0)3 at 75 nM (the concentration of component peptide env5.0). Fluorescence was measured relative to that of the control, a mixture containing labeled C-peptide and the same concentration of the inhibitor as the experimental assay mixture. Experiments were repeated in quadruplicate; data points show the mean values, and error bars show the standard deviations. Data were fit with 73% of binding sites available (see the text). (B) Effects of the order of addition of assay components on the Fe(env5.0)3-C39-e5.0 competitive inhibition assay response with 200 nM Fe(env5.0)3 and 150 nM C39-e5.0. Fluorescence intensities were measured at 0 h (□), 1 h (⊞), and 2 h (▪) after the addition of the receptor to the inhibitor, followed by the addition of the probe; 0 h (○), 1 h (⊙), and 2 h (•) after the addition of the probe to the inhibitor, followed by the addition of the receptor; and 0 h (▵), 1 h (⋄), and 2 h (♦) after the mixing of the receptor and the probe, followed by the addition of the inhibitor. Inhibition curves were drawn through the data points represented by ○, ⊙, •, and ▪.
Due to the high affinity of the peptide-peptide interaction, we expected and found a significant effect of the order of the addition of assay components on the behavior of the assay mixture. Shown in Fig. 5B is a time course plot of the assay responses with varied orders of addition of the inhibitor, probe, and receptor. An optimal outcome occurred when the probe and inhibitor were mixed prior to the addition of the receptor. Equilibrium was rapidly obtained, and the observed dose-response curve was time independent. However, prior mixing of the receptor with either the probe or the inhibitor (which in this case resembles the probe) led to time-sensitive data, with 2 h or more required to attain equilibrium values. This result was attributed to the formation of a very tight complex of the probe or inhibitor with the extended receptor. Under certain conditions, the time course could provide information on the kinetics of helix bundle formation. The signal amplitude was most sensitive for a mixture of the inhibitor and the receptor close to the binding transition (Fig. 5B). Protocols described in the literature (7, 11) use preincubation of the inhibitor and the receptor for several hours to overnight, followed by the addition of the probe or C-peptide. No reason was given for the order of addition, but it likely avoids the formation of a six-helix bundle structure which cannot be reversed by the inhibitor (13) (Fig. 5B).
Using our established protocol, with probe and receptor concentrations at 15 and 50 nM, we obtained excellent agreement between the observed signal recovery levels and the Kis for several nonpeptide inhibitors that were calibrated using our env2.0-C18-Aib assay (unpublished results). A correlation coefficient of 0.9 was obtained by testing seven inhibitors at 20 μM, with a range of Kis from 0.3 to 6.7 μM.
Identification and targeting of the inhibitor binding site.
The long groove presented by Fe(env5.0)3 includes both the hydrophobic pocket and a proximal groove that is the target of the N-terminal 60% of T20. This N-terminal segment is considered to play an important role in the interaction of T20 with the NHR (2, 26). Small molecules targeting the proximal groove may conceivably be linked to hydrophobic pocket binders to create a highly potent inhibitor. We therefore prepared the probe C28-e5.0, a 28-residue peptide which spans the T20 binding site on Fe(env5.0)3 but does not overlap with the hydrophobic pocket (Fig. 1). C28-e5.0-LY bound to Fe(env5.0)3 with a Kd of 100 ± 20 nM (Fig. 6A), confirming the importance of the N-terminal residues of T20 in six-helix bundle stability. A comparison of the results of competitive inhibition experiments utilizing env5.0-C28-e5.0-LY with those obtained using either env2.0-C18-Aib-LY or env5.0-C32-e5.0-FL would enable us to distinguish between small molecules that bind in the hydrophobic pocket and those that bind along the proximal groove. The findings of such an analysis, in which a small-molecule hydrophobic pocket binding inhibitor discovered using the env2.0-C18-Aib-LY assay failed to elicit a response in the env5.0-C28-e5.0-LY assay, are shown in Fig. 6B. This comparison confirmed that the inhibitor bound in the hydrophobic pocket. An inhibitor binding to the proximal groove, in this case the unlabeled peptide C28-e5.0, demonstrated the expected dose-response curve in the env5.0-C28-e5.0-LY assay (data not shown). In this way, we can discriminate between inhibitors according to their binding locations, potentially leading to the rational design of nonpeptide high-affinity HIV-1 gp41 binders by using linkers to connect inhibitors which are located in adjacent sites (9).
FIG. 6.

Selection of coiled-coil binding site. (A) Titration of receptor Fe(env5.0)3 into 0.2 μM truncated probe C28-e5.0-LY, fit to a 1:1 binding curve assuming 71% available binding sites (see the text). (B) Results of competitive inhibition experiments with a low-molecular-weight hydrophobic pocket binder using the assay systems 7.2 μM env2.0-1 μM C18-Aib-LY (○) and 3 μM env5.0-0.2 μM C28-e5.0-LY (•).
DISCUSSION
We have demonstrated that Fe(env5.0)3 is a stable and soluble form of an extended gp41 coiled coil in solution. It encompasses all of the residues of the N36 peptide plus three additional residues and includes a metal complex which facilitates the detection of inhibitor binding. The high affinity and high sensitivity of the Fe(env5.0)3-based assay lend themselves well to the use of the assay for the discovery of gp41 inhibitors from a library. The C-peptide probe C32-e5.0-FL performed slightly better than C39-e5.0-FL in the assay. The high binding affinity between the probe and the receptor will enable quantitative measurement of inhibitors with high potencies for lead optimization. The low concentrations of peptides required, 15 nM probe and 50 nM receptor, result in the need for less than 0.3 mg of each peptide to run the assay for 100,000 compounds or to perform 14,000 dose-response measurements. The assay is therefore suitable for high-throughput screening because of the low cost and mix-and-measure features.
The present assay using Fe(env5.0)3 employs a readout similar to that used by our recently developed assay focusing on the hydrophobic pocket (1) but broadens both the range of detection and the size of the target. In the previous assay, peptides Fe(env2.0)3 and C18-Aib-LY (Fig. 1) were used at concentrations of 7.2 and 1 μM, respectively, to obtain a 20% residual signal for the lower bound. This approach resulted in an assay that is highly sensitive to inhibitors in the mid- to submicromolar range and selective for hydrophobic pocket binders. The range of the assay for the quantification of high-affinity binders is restricted by the relatively weak interaction between the probe and receptor, resulting in a minimum quantifiable Ki of ∼0.2 μM, ∼1/5 of the receptor-probe Kd. Dose-response curves for tighter interactions become difficult to distinguish using the Fe(env2.0)3-C18-Aib assay system, because the curves become very steep. This cutoff occurs at ∼0.3 nM for Fe(env5.0)3-C32-e5.0 or Fe(env5.0)3-C39-e5.0, a natural outcome of the Kd and the peptide concentrations used in the assay. The modular assay system described here and in our previous publication (1) can be used to quantitatively detect gp41 coiled-coil inhibitors with nanomolar to millimolar affinities, covering the entire range from lead generation to final structure and activity optimization. Furthermore, the higher-affinity assay can be used to detect inhibitors of grooves other than the hydrophobic pocket. This feature may be of value in discovering compounds that bind to adjacent sites that may be linked to form potent fusion inhibitors.
Acknowledgments
This work was supported by NIH grant NS059403 to M.G.
We thank Dong Wu for preparation of bipyridine carboxylate and the small-molecule inhibitor 3-p-fluorobenzylbenzoic acid and Richard Shafer, UCSF, for the use of the CD machine.
Footnotes
Published ahead of print on 13 April 2009.
REFERENCES
- 1.Cai, L., and M. Gochin. 2007. A novel fluorescence intensity screening assay identifies new low-molecular-weight inhibitors of the gp41 coiled-coil domain of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 51:2388-2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Champagne, K., A. Shishido, and M. J. Root. 2009. Interactions of HIV-1 inhibitory peptide T20 with the gp41 N-HR coiled coil. J. Biol. Chem. 284:3619-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan, D. C., C. T. Chutkowski, and P. S. Kim. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 95:15613-15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dwyer, J. J., K. L. Wilson, D. K. Davison, S. A. Freel, J. E. Seedorff, S. A. Wring, N. A. Tvermoes, T. J. Matthews, M. L. Greenberg, and M. K. Delmedico. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 104:12772-12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777-810. [DOI] [PubMed] [Google Scholar]
- 6.Eckert, D. M., V. N. Malashkevich, L. H. Hong, P. A. Carr, and P. S. Kim. 1999. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103-115. [DOI] [PubMed] [Google Scholar]
- 7.Frey, G., S. Rits-Volloch, X. Q. Zhang, R. T. Schooley, B. Chen, and S. C. Harrison. 2006. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc. Natl. Acad. Sci. USA 103:13938-13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gochin, M., R. K. Guy, and M. A. Case. 2003. A metallopeptide assembly of the HIV-1 gp41 coiled coil is an ideal receptor in fluorescence detection of ligand binding. Angew. Chem. (international ed.) 42:5325-5328. [DOI] [PubMed] [Google Scholar]
- 9.Hajduk, P. J. 2006. SAR by NMR: putting the pieces together. Mol. Interv. 6:266-272. [DOI] [PubMed] [Google Scholar]
- 10.He, Y., S. Liu, W. Jing, H. Lu, D. Cai, D. J. Chin, A. K. Debnath, F. Kirchhoff, and S. Jiang. 2007. Conserved residue Lys574 in the cavity of HIV-1 Gp41 coiled-coil domain is critical for six-helix bundle stability and virus entry. J. Biol. Chem. 282:25631-25639. [DOI] [PubMed] [Google Scholar]
- 11.Jiang, S., K. Lin, L. Zhang, and A. K. Debnath. 1999. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J. Virol. Methods 80:85-96. [DOI] [PubMed] [Google Scholar]
- 12.Kilgore, N. R., K. Salzwedel, M. Reddick, G. P. Allaway, and C. T. Wild. 2003. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J. Virol. 77:7669-7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kliger, Y., and Y. Shai. 2000. Inhibition of HIV-1 entry before gp41 folds into its fusion-active conformation. J. Mol. Biol. 295:163-168. [DOI] [PubMed] [Google Scholar]
- 14.Liu, S., S. Wu, and S. Jiang. 2007. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr. Pharm. Des. 13:143-162. [DOI] [PubMed] [Google Scholar]
- 15.Louis, J. M., C. A. Bewley, and G. M. Clore. 2001. Design and properties of NCCG-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J. Biol. Chem. 276:29485-29489. [DOI] [PubMed] [Google Scholar]
- 16.Lu, M., and P. S. Kim. 1997. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dyn. 15:465-471. [DOI] [PubMed] [Google Scholar]
- 17.Matthews, T., M. Salgo, M. Greenberg, J. Chung, R. DeMasi, and D. Bolognesi. 2004. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 3:215-225. [DOI] [PubMed] [Google Scholar]
- 18.Mo, H., A. K. Konstantinidis, K. Stewart, T. Dekhtyar, T. Ng, K. Swift, E. Matayoshi, W. Kati, W. Kohlbrenner, and A. Molla. 2004. Conserved residues in the coiled-coil pocket of human immunodeficiency virus type 1 gp41 are essential for viral replication and interhelical interaction. Virology 329:319-327. [DOI] [PubMed] [Google Scholar]
- 19.Oishi, S., S. Ito, H. Nishikawa, K. Watanabe, M. Tanaka, H. Ohno, K. Izumi, Y. Sakagami, E. Kodama, M. Matsuoka, and N. Fujii. 2008. Design of a novel HIV-1 fusion inhibitor that displays a minimal interface for binding affinity. J. Med. Chem. 51:388-391. [DOI] [PubMed] [Google Scholar]
- 20.Otaka, A., M. Nakamura, N. Daisuke, E. Kodama, S. Uchiyama, H. Nakamura, Y. Kobayashi, M. Matsuoka, and N. Fujii. 2002. Remodelling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew. Chem. (international ed.) 41:2938-2939. [DOI] [PubMed] [Google Scholar]
- 21.Root, M. J., M. S. Kay, and P. S. Kim. 2001. Protein design of an HIV-1 entry inhibitor. Science 291:884-888. [DOI] [PubMed] [Google Scholar]
- 22.Shu, W., H. Ji, and M. Lu. 1999. Trimerization specificity in HIV-1 gp41: analysis with a GCN4 leucine zipper model. Biochemistry 38:5378-5385. [DOI] [PubMed] [Google Scholar]
- 23.Sia, S. K., P. A. Carr, A. G. Cochran, V. N. Malashkevich, and P. S. Kim. 2002. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. USA 99:14664-14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissenhorn, W., L. J. Calder, A. Dessen, T. Laue, J. J. Skehel, and D. C. Wiley. 1997. Assembly of a rod-shaped chimera of a trimeric GCN4 zipper and the HIV-1 gp41 ectodomain expressed in Escherichia coli. Proc. Natl. Acad. Sci. USA 94:6065-6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wendt, H., R. M. Thomas, and T. Ellenberger. 1998. DNA-mediated folding and assembly of MyoD-E47 heterodimers. J. Biol. Chem. 273:5735-5743. [DOI] [PubMed] [Google Scholar]
- 26.Wexler-Cohen, Y., B. T. Johnson, A. Puri, R. Blumenthal, and Y. Shai. 2006. Structurally altered peptides reveal an important role for N-terminal heptad repeat binding and stability in the inhibitory action of HIV-1 peptide DP178. J. Biol. Chem. 281:9005-9010. [DOI] [PubMed] [Google Scholar]
- 27.Wild, C. T., D. C. Shugars, T. K. Greenwell, C. B. McDanal, and T. J. Matthews. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91:9770-9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, J., T. Chung, and K. Oldenburg. 1999. A simple statistical parameter for use in evaluation and validation of high throughout screening assays. J. Biomol. Screen. 4:67-73. [DOI] [PubMed] [Google Scholar]