

Abstract
PF-00868554 is a nonnucleoside inhibitor of the hepatitis C virus (HCV) RNA polymerase, which exerts its inhibitory effect by binding to the thumb base domain of the protein. It is a potent and selective inhibitor, with a mean 50% inhibitory concentration of 0.019 μM against genotype 1 polymerases and a mean 50% effective concentration (EC50) of 0.075 μM against the genotype 1b-Con1 replicon. To determine the in vitro antiviral activity of PF-00868554 against various HCV strains, a panel of chimeric replicons was generated, in which polymerase sequences derived from genotype 1a and 1b clinical isolates were cloned into the 1b-Con1 subgenomic reporter replicon. Our results indicate that PF-00868554 has potent in vitro antiviral activity against a majority (95.8%) of genotype 1a and 1b replicons, with an overall mean EC50 of 0.059 μM. PF-00868554 showed no cytotoxic effect in several human cell lines, up to the highest concentration evaluated (320 μM). Furthermore, the antiviral activity of PF-00868554 was retained in the presence of human serum proteins. An in vitro resistance study of PF-00868554 identified M423T as the predominant resistance mutation, resulting in a 761-fold reduction in susceptibility to PF-00868554 but no change in susceptibility to alpha interferon and a polymerase inhibitor that binds to a different region. PF-00868554 also showed good pharmacokinetic properties in preclinical animal species. Our results demonstrate that PF-00868554 has potent and broad-spectrum antiviral activity against genotype 1 HCV strains, supporting its use as an oral antiviral agent in HCV-infected patients.
The CDC estimates that 4.5 million individuals in the United States and more than 170 million worldwide are infected with the hepatitis C virus (HCV), with about 25,000 new infections occurring each year. Approximately 80% of hepatitis C patients become persistently infected, among which 10% to 20% develop progressive liver disease during their lifetime, including cirrhosis, hepatocellular carcinoma, and liver failure, thus making hepatitis C the leading indication for liver transplantation (38, 44). HCV was primarily transmitted via a contaminated blood supply before the discovery of HCV in 1989 and the development of sensitive detection methods in 1992. Since then, intravenous drug use has become the major route of HCV transmission and the incidence of HCV infection has decreased rapidly. However, the large number of patients infected before 1992 are now in the later stages of the disease, in which treatment becomes necessary to prevent liver damage, transplantation, or death.
The current standard of care for HCV is pegylated alpha interferon (IFN-α) in combination with ribavirin. The success rate of this therapy with genotype 1 infection, which is the most prevalent in the United States and worldwide, is only about 40% to 50% (9, 11, 32). In addition, IFN-α therapy is associated with significant side effects, including fatigue, headache, myalgia, fever, nausea, and insomnia in >30% of patients (9, 11, 32). Ribavirin also causes hemolytic anemia in 10% to 20% of patients (28, 40). Due to the tolerability and safety concerns of current therapies, as well as the slow progression and uncertain course of this disease, physicians generally withhold treatment from patients until symptoms manifest or liver function begins to be impaired. Currently, fewer than 10% of chronically infected patients in the United States are receiving HCV treatment. Thus, there remains a significant unmet need for more effective, more tolerable, and safer HCV therapies.
The HCV genome is a single-stranded, positive-sense RNA of approximately 9.6 kb (7). The genomic RNA encodes a polyprotein that is processed by host and viral proteases into at least 10 structural and nonstructural (NS) proteins (10, 14, 15). Most of the HCV NS proteins are required for viral RNA replication (1). The NS5B protein, encoding the viral RNA-dependent RNA polymerase (RdRp), is an essential component of the HCV RNA replication complex (20). Due to its apparent sequence and structural differences from human DNA and RNA polymerases, the HCV RNA polymerase is considered an attractive target for antiviral drug discovery. In addition to nucleoside analogs (4) and pyrophosphate mimics (41) that target the active site, a number of structurally diverse nonnucleoside polymerase inhibitors have been reported (19). They were shown to interact with at least four distinct allosteric sites by a combination of crystallographic analysis and in vitro resistance studies (18, 19).
We have reported the discovery, using high-throughput screening and structure-based drug design, of a novel and potent class of nonnucleoside HCV polymerase inhibitors characterized by a dihydropyrone core (23, 24). These molecules bind within the thumb base portion of the polymerase (pocket II) (2) and are noncompetitive inhibitors with respect to nucleotide. In this report, we characterize the preclinical activity and pharmacokinetic profile of PF-00868554 (25), a molecule that retains optimal biochemical potency and selectivity and yet has improved pharmacokinetic and physicochemical properties compared to its predecessors (23, 24). In a phase I clinical trial with genotype 1 HCV-infected, treatment-naïve subjects, PF-00868554 potently inhibited viral replication, with mean maximum reductions in HCV RNA ranging from 0.97 to 2.13 logs (12). It is currently in phase II clinical trials. The antiviral activity of PF-00868554 against different HCV genotype 1 strains was determined by generating chimeric replicons containing polymerase sequences derived from a panel of genotype 1 clinical isolates in the genotype 1b (Con1 strain) backbone. The pharmacokinetic properties of the molecule were profiled, revealing promising oral bioavailability in rodent and nonrodent species. In addition, an in vitro resistance study was carried out to identify the resistance changes associated with exposure to PF-00868554.
MATERIALS AND METHODS
Compounds.
PF-00868554 (25) was synthesized at Pfizer Global Research and Development (La Jolla, CA) (Fig. 1). The benzimidazole compound C (21) and benzothiadiazine SB-750330 (5) were prepared according to published protocols (Fig. 1). IFN-α was purchased from Sigma Aldrich (St. Louis, MO). Individual compounds, except IFN-α, were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 mM or 100 mM and diluted to appropriate concentrations in buffer or growth medium. IFN-α was dissolved in phosphate-buffered saline to a stock concentration of 105 IU/ml and diluted to appropriate concentrations in growth medium.
FIG. 1.

Structures of HCV polymerase inhibitors.
Reagents, enzymes, cells, and plasmids.
Recombinant constructs of HCV NS5B polymerase 1a H77, 1b BK, 1b Con1, 1b Con1 M423T, 2b, and 4a enzymes each bore a 21-amino-acid C-terminal deletion of the membrane insertion fragment (Δ21) and a C-terminal six-His tag and were expressed in Escherichia coli and purified as previously described (29). Recombinant His-tagged NS5B polymerase genotypes (Δ21) 1b J1 and (Δ21) 3a were expressed in insect cells using a baculovirus expression system and were similarly purified. Human DNA polymerase alpha and delta were cloned, expressed, and purified according to published procedures (8, 36, 42). Human DNA polymerase gamma was purchased from Replizyme Ltd. (Heslington, York, United Kingdom). Recombinant human immunodeficiency virus (HIV) reverse transcriptase and HIV Q7K protease were purified from E. coli, as described previously (3, 16). Huh7 and Huh7.5 cells, obtained from Apath LLC (St. Louis, MO), were propagated in Dulbecco's modified Eagle medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; HyClone, Logan, UT), 0.1 mM nonessential amino acid (Invitrogen), 100 units/ml of penicillin (Invitrogen), and 100 μg/ml of streptomycin sulfate (Invitrogen). HEK-293, HeLa, and HepG2 cells were propagated in Dulbecco's modified Eagle medium containing 10% FBS, 100 IU/ml of penicillin, and 100 mg/ml of streptomycin sulfate.
The dicistronic selectable replicon BB7, containing the HCV genes NS3 to NS5B derived from the Con1 strain of genotype 1b, was licensed from Apath LLC. The construction of the HCV subgenomic reporter replicon, BB7M4hRLuc, and the generation of the stable cell lines, dicistronic single reporter (DSR) and dicistronic dual reporter (DDR), were described previously (13, 39). DSR is a stable cell line that harbors the BB7M4hRLuc reporter replicon, whereas DDR also contains the CMV-firefly luciferase (FLuc), allowing for simultaneous monitoring of cytotoxicity. The monocistronic selectable replicon RB10, containing the same HCV sequences under the control of the HCV internal ribosome entry site, was licensed from ReBLikon GmbH (Mainz, Germany). The dicistronic selectable subgenomic replicon H77-SG, containing the HCV genes NS3 to NS5B (derived from the H77 strain of genotype 1a) under the control of the encephalomyocarditis virus internal ribosome entry site, was licensed from Apath LLC.
Biochemical assays.
Recombinant HCV NS5B genotypes 1b BK, 1b J1, 1b Con1, 1b Con1 M423T, and 2b were assayed by measuring the incorporation of [α-33P]GMP into dG12:poly(rC)350 primer-template substrate [dG12 DNA primer and poly(rC)350 RNA template preannealed in a 12.5:1 primer-to-template molar ratio]. NS5B genotypes 1a H77, 3a, and 4a were assayed using either the GMP incorporation or UMP incorporation format; the latter measured the incorporation of [α-33P]UMP into dT17:poly(rA)350 primer-template [dT17 DNA primer and poly(rA)350 RNA template preannealed in a 12:1 molar ratio]. Assays were performed in 96- or 384-well microplates, with final reaction volumes of 100 μl or 75 μl, respectively. The reaction mixtures of GMP incorporation assays contained 20 mM Tris-Cl (pH 7.6), 10 mM MgCl2, 20 mM NaCl, 0.05% Tween 20, 1 mM dithiothreitol (DTT), 30 nM polymerase enzyme, 2% (vol/vol) dimethyl sulfoxide, 62.5 nM dG12 primer, 5 nM poly(rC)350 template, 1 μM GTP, and 1 nCi/μl [α-33P]GTP. Reaction mixtures for UMP incorporation assays were similar, except that 60 nM dT17 primer, 5 nM poly(rA)350 template, 2.5 uM UTP, and 10 nCi/μl [α-33P]UTP were substituted for primer, template, and nucleotide. Reactions were routinely initiated by enzyme addition and incubated for 30 min at room temperature for GMP assays or 60 min at 30°C for UMP assays. Separate experiments demonstrated that product formation was linear with respect to time, and substrate conversion was ≪10%, conforming to initial rate kinetics under these assay conditions. Reactions were stopped by addition of 59 mM EDTA, and the nucleotide product was detected by phosphorimaging after filtration onto Biodyne B nylon 6,6 membranes (Thermo Fisher Scientific, Rochester, NY). Noncompetitive, tight-binding 50% inhibitory concentration (IC50) constants were measured by the fitting of the dose-response data to the following equation (51):
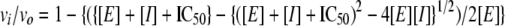 |
(1) |
where vi is the inhibited velocity, vo is the uninhibited velocity, [E] is the enzyme concentration, and [I] is the inhibitor concentration. Unless indicated otherwise, all reported inhibition data represent averages of at least two measurements in duplicate.
Human DNA polymerase alpha (20 nM) was assayed in the presence of 20 mM Tris (pH 7.5), 1 uM dTTP (plus 100 nCi/well of [α-33P]dTTP), 10 mM MgCl2, 0.1 mg/ml bovine serum albumin, 1 mM DTT, 8 nM dA-dT primer-template, and 2% dimethyl sulfoxide. Human DNA polymerase delta (5 nM) was assayed in the presence of 19 mM Tris (pH 7.6), 1.9 mM MnCl2, 0.85 μM dATP, 0.43 uM dTTP (plus 200 nCi/well of [α-33P]dTTP), 1.3 mM DTT, 4.2% (vol/vol) glycerol, 2% (vol/vol) DMSO, and 8.5 ng/μl poly(dA-dT)/poly(dA-dT). Reactions were initiated with enzyme, and the incorporation of [α-32P]dTMP into high-molecular-weight polynucleotide was measured in the presence of an oligo(dT)/poly(dA) primer-template. The elongated primer-template was detected by phosphorimaging after microfiltration through Biodyne B membranes (Thermo Fisher Scientific). HIV reverse transcriptase (1 nM) was assayed similarly in the presence of 50 mM Tris-Cl (pH 8.0), 60 mM KCl, 1 mM DTT, 20 nM template, 160 nM primer (preannealed), 8 mM MgCl2, and 20 μM [α-32P]dTTP for 30 min before microfiltration and phosphorimaging. HeLa cell nuclear extracts were used as a source of human nuclear RNA polymerases I, II, and III. Activity was assayed using calf thymus DNA as template, and incorporation of [α-32P]GMP into high-molecular-weight RNA was measured by microfiltration onto Biodyne B membranes in the presence of 50 mM Tris-Cl (pH 8.0), 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 50 mM (NH4)2SO4, 5 μg/well calf thymus DNA, 1% (vol/vol) DMSO, 100 μM ATP, 100 μM UTP, 100 μM CTP, 5 μM GTP, 0.2 μCi/well [α-32P]GTP, and 5 μl/well nuclear extract. RNA polymerase I activity was assessed in the presence of 230 μg/ml α-amanitin, RNA polymerase (I plus II) activity was assessed in the presence of 0.77 μg/ml α-amanitin, and RNA polymerase III activity was assessed differentially in the absence of α-amanitin. The HIV protease Q7K was assayed according to published methods (3).
Generation of patient-derived NS5B chimeric replicons.
To eliminate the necessity of ScaI-mediated linearization prior to in vitro transcription and to allow transcript production from patient sequences containing ScaI sites, an 84-nucleotide hepatitis delta virus ribozyme sequence was introduced into BB7M4hRLuc immediately downstream of the HCV sequence so that the replicon RNA transcript with an exact 3′ terminus of the 3′ untranscribed region (UTR) could be generated by self-cleavage. The incorporation of ribozyme was accomplished by two rounds of PCRs, as described previously (43), using Herculase DNA polymerase and PCR primer pairs Ribo1 and Ribo2 for the first and second rounds of PCR, respectively (see the supplemental material). Both rounds of amplification were carried out for 35 cycles consisting of 95°C for 1 min, 55°C for 90 s, and 72°C for 90 s. The first-round PCR generated the fusion between a sequence from a unique NcoI site in NS5B and the end of the 3′ UTR and the 5′-end sequence of the ribozyme. The resulting fragment was used as a template for the second-round PCR to fuse the entire ribozyme sequence to the HCV 3′ UTR. The final product was cloned into BB7M4hRLuc at the unique NcoI and SpeI sites to create BB7M4hRLuc.ribo. The SbfI and PacI restriction sites were introduced into BB7M4hRLuc.ribo in the NS5A region and 3′ UTR, respectively, by site-directed mutagenesis using QuikChange mutagenesis (Stratagene, La Jolla, CA), according to the manufacturer's protocol, and PCR primer pairs SbfI and PacI (see the supplemental material). Amplification was carried out for 16 cycles of 94°C for 30 s, 55°C for 1 min, and 68°C for 26 min. All mutations were confirmed by sequencing.
Plasma samples from HCV-infected patients were obtained from Teragenix (Fort Lauderdale, FL) and Cliniqa (San Diego, CA). Samples with a viral load over 105 IU/ml were randomly selected for RNA isolation using Qiagen QIAamp viral RNA mini isolation kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. Purified RNA was reverse transcribed using Thermo-X reverse transcriptase (Invitrogen) and a mixture of two poly(A) primers of 20 and 34 nucleotides in length, according to the manufacturer's protocol.
Amplification of the HCV NS5B gene was achieved by two rounds of PCR using AccuPrime high-fidelity Taq polymerase (Invitrogen). For both rounds, the PCRs were carried out for 40 cycles consisting of 94°C for 30 s and 68°C for 3 min. The first-round primers (1a1 and 1b1; see the supplemental material) amplified the full-length NS5B gene, while the second-round primers (1a2 and 1b2) incorporated the SbfI and PacI restriction sites at the 5′ and 3′ ends of the products, respectively, to allow for cloning. Amplified full-length NS5B products were cloned into the BB7M4hRLuc.ribo vector at the engineered SbfI and PacI sites to generate chimeric replicons containing patient NS5B sequences in the 1b Con1 backbone.
Luciferase reporter assay.
The wild-type replicon cells, DSR or DDR, or resistant replicon cells were seeded at a density of 2 × 104 cells per well in 96-well plates in the absence of selection antibiotics. In some experiments, medium was supplemented with 50% human serum (HS) or a combination of 45 mg/ml HS albumin (HSA) and 1 mg/ml α1-acid glycoprotein (AAG), all of which were obtained from Sigma. Compounds were tested at half-log serial dilutions over a range of concentrations with appropriate solvent controls (compound free). Cells were incubated with compounds for 3 days at 37°C. HCV RNA replication was monitored by humanized Renilla luciferase (hRLuc) reporter activity. In DDR cells, cytotoxicity was measured simultaneously by the FLuc reporter activity. For DSR, hRLuc activity was determined using the RLuc assay system (Promega, Pittsburgh, PA), according to the manufacturer's instructions, in a Perkin Elmer MicroBeta 1450 jet (Perkin Elmer, Wellesley, MA). For DDR, reporter activities were determined using the dual luciferase reporter kit (Promega) according to the manufacturer's instructions. The 50% effective concentration (EC50) was calculated as the concentration of compound that effected a decrease in viral RNA replication (as measured by hRLuc activity or quantitative PCR) in compound-treated cells to 50% of that produced in compound-free cells. The 50% cytotoxicity concentration was calculated as the concentration of compound that decreased host cell viability (as measured by FLuc activity) in compound-treated cells to 50% of that produced in compound-free cells. The values were determined by nonlinear regression analyses.
The XTT cytotoxicity assay.
Cells were seeded at a density of 2 × 104 per well in 96-well plates. Compounds were tested at half-log serial dilutions over a range of concentrations with appropriate solvent controls (compound free). After incubation with compounds for 3 days, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT; Sigma) and phenazine methosulfate (Sigma) were added to the cells at final concentrations of 200 μg/ml and 1 mM, respectively. After an additional 4-h incubation at 37°C, the amount of formazan produced was quantified at a test reference of 450 nm and a reference wavelength of 650 nm in a spectrophotometer (Molecular Devices, Sunnyvale, CA).
In vitro resistance selection at fixed concentrations.
DSR cells were seeded at densities of 5 × 104, 1 × 105, and 2.5 × 105 in 10-cm plates in medium containing either 3×, 6×, or 12× EC90 (0.96, 1.9, or 3.8 μM, respectively) of PF-00868554, with weekly changes of selection medium. As a positive control, DSR cells were cultured in the presence of 3× EC90 (15 μM) of a benzimidazole polymerase inhibitor, compound C (Fig. 1) (21). As a negative control, DSR cells were cultured in medium without the presence of inhibitor. Colonies were isolated after 21 days of selection and expanded to cell lines in larger culture vessels. Ten individual resistant cell lines were randomly chosen for genotypic and phenotypic characterizations at each selection concentration for PF-00868554 and compound C. Construction of the M423T mutant replicon and enzyme was described previously (39).
Population sequence analysis of the NS5B gene.
Replicon RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Reverse transcription (RT)-PCR of the NS5B region was completed using primers 5′-AAGAATTCTTCACAGAAGTGGATGGG-3′ and 5′-ATTGGCCTGGAGTGTTTAGGTCC-3′ and SuperScript one-step RT-PCR with platinum Taq (Invitrogen) under the following conditions: 45°C for 30 min and 94°C for 2 min. PCR was performed for a total of 30 cycles in a GeneAmp 9700 PCR system (PE Applied Biosystems, Foster City, CA) under the following conditions: 94°C for 30 s, 60°C for 1.5 min, and 72°C for 3 min, followed by a single cycle of 10 min at 72°C. The amplified products were purified and subsequently subjected to population sequencing.
Transient replication assay.
In vitro transcripts were generated from replicon constructs using the MEGAscript T7 kit (Ambion, Austin, TX) according to the manufacturer's protocol and stored at −80°C before use. Ten micrograms of replicon transcripts were electroporated into 8 × 106 Huh7.5 cells in a 0.4-cm cuvette using a Bio-Rad Gene Pulser II electroporator (Bio-Rad, Hercules, CA) at 220 V and 950 μF. Electroporated cells were resuspended in growth medium, plated on 96-well black plates with clear bottoms at 6 × 104 cells per well, and allowed to adhere for 4 h at 37°C. Compounds were tested at half-log serial dilutions over a range of concentrations, with appropriate DMSO controls (compound free). After incubation for 3 days at 37°C, hRLuc activity was determined using the RLuc assay system, according to the manufacturer's protocol, in the Perkin Elmer MicroBeta 1450 jet.
Pharmacokinetic assessment with rats, dogs, and monkeys.
Male Sprague-Dawley rats were housed in individual cages with food and water provided ad libitum. For the intravenous (i.v.) study, a single i.v. dose of 2 mg/kg PF-00868554 in a solution of 50% polyethylene glycol (PEG) 200, 10% ethanol, and 40% water at a dosing volume of 2 ml/kg was prepared. Oral evaluation with solution for PF-00868554 was at 2 mg/kg in a solution of 50% PEG 200, 10% ethanol, and 40% water at a dosing volume of 2 ml/kg. Oral dosing of PF-00868554 was also prepared using a 0.5% methylcellulose suspension at a dosing volume of 2 ml/kg. Both the solution and suspension oral gavage doses were followed by a 10-ml tap water flush of the gavage tube. Blood samples (∼0.25 ml) were collected from the jugular vein of each rat for determination of PF-00868554 plasma concentrations. Blood samples for the i.v. arm of these studies were collected at predose; 2, 5, 10, 15, and 30 min; and 1, 2, 4, 6, 8, and 24 h postdose. For the oral arm of these studies, blood samples were collected at 10, 20, and 30 min and at 1, 2, 3, 4, 6, 8, and 24 h postdose.
Male beagle dogs were housed in individual cages with water provided ad libitum. The dogs were fasted overnight prior to dosing and during the first 4 h of blood sampling. In the i.v. arm of the study, three dogs received an i.v. dose of 1 mg/kg PF-00868554 in a solution of 50% PEG 200, 10% ethanol, and 40% water at a dosing volume of 0.5 ml/kg. For the oral arm of the study, three dogs received a single oral dose of 2 mg/kg PF-00868554 in a solution of 50% PEG 200, 10% ethanol, and 40% water and two dogs received a single oral dose of PF-00868554 in a 0.5% methylcellulose suspension, both at a dosing volume of 1 ml/kg. The oral gavage doses were followed by a 10-ml tap water flush of the gavage tube. Blood samples (1 ml) were collected from the jugular vein of each dog for determination of PF-00868554 plasma concentrations. Blood samples for the i.v. portion of the study were collected at predose; 2, 5, 10, 20, 30, and 45 min; and 1, 2, 4, 6, 8, 12, and 24 h postdose. For the oral portion of the study, blood samples were collected at 10, 20, 30, and 45 min and at 1, 2, 3, 4, 6, 8, 12, and 24 h postdose.
Male cynomolgus monkeys were housed in individual cages with water provided ad libitum. The animals were fasted overnight, and food was also withheld during the first 4 h of blood sampling. Monkeys received a single i.v. dose of 2-mg/kg PF-00868554 solution at a dosing volume of 1 ml/kg. Blood samples (1 ml) were collected via femoral venipuncture for each monkey for determination of PF-00868554 plasma concentrations. Blood samples were collected at predose; 2, 5, 10, 20, 30, and 45 min; and 1, 2, 4, 6, 8, and 12 h postdose.
For all studies, each blood sample was placed in a tube containing heparin and centrifuged to obtain plasma. The plasma was frozen immediately and stored at −80°C until samples were analyzed by liquid chromatography-tandem mass spectrometry.
Pharmacokinetic parameters were estimated using the noncompartmental analysis by WinNonlin 3.1 software (Pharsight, Mountain View, CA). Graphing was accomplished by Microsoft Excel 5.0. Estimated parameters reported here include the values of the area under the curve from time zero to infinity (AUC0-∞), the maximum mean plasma concentration (Cmax), the time-to-maximum-plasma concentration (Tmax), terminal half-life (t1/2), clearance (CL), and volume of distribution at steady state (Vss). Oral bioavailability (Foral) was calculated using the following equation:
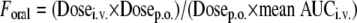 |
where p.o. indicates orally.
RESULTS
Biochemical potency and selectivity of PF-00868554 in vitro.
Previous data showed that members of the dihydropyrone class of inhibitors are potent, noncompetitive inhibitors of HCV polymerase 1b, binding approximately 30 Å from the active site at the region between the thumb and finger domains of the enzyme. Optimization of this class of compounds using combinatorial efforts and structure-based design has led to the discovery of PF-00868554 (25) (Fig. 1). The IC50s against polymerases of the 1b Con1 and BK strains were 0.015 ± 0.010 and 0.0066 ± 0.0018 μM, respectively (Table 1). Inhibitory potency was clearly the greatest against the genotype 1 polymerases tested, with IC50s in the nanomolar range, whereas the IC50s were approximately 1 μM against non-genotype 1 HCV polymerases. PF-00868554 displayed at least 1,600- to 7,500-fold selectivity for the genotype 1 viral polymerases, compared to the human DNA and RNA polymerases assayed (Table 1).
TABLE 1.
Spectrum of biochemical potency and selectivity of PF-00868554
| Enzyme | % Inhibition (at 50 μM) | IC50 (μM) |
|---|---|---|
| HCV NS5B polymerases | ||
| 1b Con1 | 0.015 ± 0.010 | |
| 1b BK | 0.0066 ± 0.0018a | |
| 1b J1 | 0.030 ± 0.005 | |
| 1a H77 | 0.021 ± 0.009b | |
| 2b | 1.1 ± 0.3 | |
| 3a | 1.0 ± 0.2 | |
| 4a | 1.7 ± 0.3 | |
| 1b M423T | 11 ± 3 | |
| Human DNA polymerases | ||
| Alpha | 0 | >50 |
| Delta | 0 | >50 |
| Gamma | 0c | >200 |
| Human RNA polymerases | ||
| I | 0 | >50 |
| II | 0 | >50 |
| III | 0 | >50 |
| HIV reverse transcriptase | 0 | >50 |
| HIV protease Q7K | 2.18 |
n = 9.
n = 8.
% inhibition at 200 μM.
Because an early predecessor of the dihydropyrone series originated from an HIV protease inhibitor program (29), the inhibitory potency of PF-00868554 was tested against a recombinant HIV protease and several human proteases of physiological relevance. While the molecule remains a weak inhibitor of HIV protease, with an IC50 of 2.18 μM, it displayed no significant inhibition of a wide variety of human aspartyl and serine proteases, including plasma thrombin, neutrophil elastase, liver cathepsin B, human caspase-2, and pancreatic chymotrypsin, at a concentration of 10 μM (data not shown).
In vitro antiviral activity and cytotoxicity of PF-00868554.
The ability of PF-00868554 to inhibit HCV RNA replication was evaluated against subgenomic replicons derived from the 1b Con1 and 1a H77 strains in replicon assays using the luciferase reporter end point. PF-00868554 demonstrated strong antiviral activity against the 1b Con1 replicon, with a mean EC50 of 0.075 μM, and reduced activity against the 1a H77 replicon, with a mean EC50 of 0.39 μM (Table 2). The cytotoxicity of PF-00868554 was determined by the XTT assay with various human cell lines, including Huh7, HepG2, HeLa, and HEK293 cells, after 3 days of incubation. PF-00868554 demonstrated little or no cytopathic effects in multiple cell lines, up to the highest concentration of compound evaluated, with 50% cytotoxicity concentration values of >320 μM.
TABLE 2.
In vitro antiviral activity of PF-00868554 against replicons containing genotype 1 NS5B sequencesa
| Genotype of NS5B | Strain of NS5Bb | PF-00868554 EC50 (μM) | Benzothiadiazine SB-750330 EC50 (μM) |
|---|---|---|---|
| 1b | Con1 | 0.075 ± 0.059 | 0.0037 ± 0.0017 |
| BK | 0.011 | 0.051 | |
| 33778 | 0.027 ± 0.018 | 0.062 ± 0.043 | |
| 45969 | 0.040, 0.024 | 0.18 ± 0.04 | |
| 45967 | 0.022 ± 0.009 | 0.024 ± 0.014 | |
| 45970 | 0.022 ± 0.013 | 0.045 ± 0.042 | |
| 46002 | 0.0088 ± 0.0039 | 0.021 ± 0.006 | |
| 51499 | 0.045 ± 0.034 | 0.0029 ± 0.0007 | |
| 5903 | 0.023 ± 0.013 | 0.049 ± 0.045 | |
| 66193 | 0.075 ± 0.036 | <0.0010d | |
| 7601 | 0.025 ± 0.023 | 0.095 ± 0.067 | |
| 1a | H77c | 0.39 ± 0.42 | 0.013 ± 0.023 |
| 33772 | 0.057 ± 0.045 | 0.26 ± 0.14 | |
| 33776 | 0.049 ± 0.036 | 0.061 ± 0.020 | |
| 33779 | 0.040 ± 0.008 | 0.45 ± 0.09 | |
| 33781 | 0.028 ± 0.032 | 1.1 ± 1.2 | |
| 33783 | 0.059 ± 0.043 | 0.70 ± 0.17 | |
| 34161 | 0.042 ± 0.016 | 0.93 ± 0.81 | |
| 36941 | 0.087 ± 0.062 | 0.18 ± 0.02 | |
| 42134 | 0.050 ± 0.024 | 0.15 ± 0.03 | |
| 45958 | 0.051, 0.065 | 0.91, 0.71 | |
| 45992 | 0.079 ± 0.012 | 2.1 ± 1.4 | |
| 45996 | 0.042 ± 0.019 | 0.97 ± 0.56 | |
| 5904 | 0.036 ± 0.011 | 0.50 ± 0.18 |
Results represent the means ± standard deviations (of three to five experiments) or individual values (of one or two experiments).
Except where it is indicated, the NS3-to-NS5A sequence in the replicon is derived from 1b Con1.
The NS3-to-NS5A sequence is derived from either 1a H77 or 1b Con1.
n = 3.
In vitro antiviral activity of PF-00868554 against chimeric replicons containing NS5B sequences from clinical isolates.
To evaluate the activity of PF-00868554 against different HCV strains within genotype 1, chimeric replicons containing polymerase sequences derived from a panel of genotype 1 clinical isolates in the genotype 1b Con1 backbone were generated. The replication fitness of these chimeric replicons was first determined in the transient replication assay with Huh7.5 cells by use of the luciferase reporter end point (Fig. 2). Only the replicons that displayed luciferase activities of at least 2 × 103 relative light units (RLU) (about 20 times above the background) were included in the subsequent antiviral assays so that the EC50s could be accurately determined. PF-00868554 demonstrated potent in vitro antiviral activity against HCV replicons containing polymerase sequences from 23 out of 24 (95.8%) strains of genotype 1a and 1b, with a mean EC50 of 0.043 ± 0.022 μM and a range of 0.0088 to 0.087 μM (Table 2). The mean antiviral activity against 11 of 11 1b strains (EC50 = 0.033 ± 0.023 μM) was comparable to that demonstrated against 12 of 13 1a strains (EC50 = 0.052 ± 0.017 μM). PF-00868554 showed reduced activity against the H77 strain of genotype 1a, with a mean EC50 of 0.39 ± 0.42 μM. Because PF-00868554 has similar activities against all genotype 1 polymerases tested, including Con1, BK, J1 and H77 (Table 1), the molecular basis for the reduction in activity of PF-00868554 against the H77 replicon is not known. It is possible that other NS proteins in the replication complex may have an effect on the replicon's sensitivity to polymerase inhibitors. However, even with the inclusion of the H77 replicon, the overall mean and median EC50s of PF-00868554 against all genotype 1 replicons were 0.059 and 0.042 μM, respectively, with a range of 0.0088 to 0.39 μM. In contrast, a benzothiadiazine polymerase inhibitor (Fig. 1) displayed EC50s varying from <0.001 to 2.1 μM, corresponding to a larger than 2,000-fold difference in activity levels (Table 2). As expected, all of the chimeric replicons showed similar sensitivities to an NS3 protease inhibitor (data not shown).
FIG. 2.

Replication fitness of chimeric replicons containing patient-derived NS5B sequences. The NS5B sequences from genotype 1a (open bars) and 1b (solid bars) HCV-infected patients were cloned into the Con1 subgenomic replicons. The NS5B sequence from the BK strain was also cloned in the Con1 replicon to produce the BK/Con1 chimeric replicon. Huh7.5 cells were electroporated with in vitro transcripts of chimeric replicons, seeded in 96-well plates at 6 × 104/well, and cultured for 3 days before hRLuc activity was determined. The replication fitness of the parental Con1 replicon is designated 100%. Results represent the average of triplicate values, with error bars showing standard deviations.
The in vitro antiviral activity of PF-00868554 was also evaluated against the genotype 2a JFH1 replicon and chimeric replicons, in which polymerase sequences from genotype 2b virus isolates were substituted for the corresponding sequence in the 1b replicon. PF-00868554 demonstrated antiviral activity against the replicons that contain non-genotype 1 polymerase sequences, with EC50s ranging from 11 to 17 μM (data not shown). No apparent structural correlations between sensitivity to PF-00868554 and polymerase sequences were identified.
The effects of protein binding on the in vitro antiviral activity of PF-00868554.
The effect of protein binding on the in vitro antiviral activity of PF-00868554 was evaluated in the luciferase reporter assay with DSR cells. Cell culture medium that contained 10% FBS was supplemented with either 50% HS or a combination of 1 mg/ml AAG and 45 mg/ml HSA in these experiments. In the presence of 50% HS, no statistically significant change in the antiviral activity of PF-00868554 was observed (3.5-fold change from 0.053 to 0.19 μM, P ≥ 0.05). In the presence of 45 mg/ml HSA and 1 mg/ml AAG, PF-00868554 demonstrated antiviral activity with a mean EC50 of 0.69 μM, which represented a 13-fold increase compared with that observed in medium containing 10% FBS alone (P < 0.05).
Phenotypic and genotypic determination of PF-00868554-resistant cell lines.
The PF-00868554-resistant cell lines generated by exposing the DSR cell line to fixed concentrations of the inhibitor were subjected to replication fitness analysis using the luciferase reporter assay. The mutant replicons displayed luciferase activities ranging from 4.9 × 104 to 1.15 × 106 RLU, which were well above the background, to allow for accurate EC50 determinations (Table 3). Phenotypic analysis of the 30 PF-00868554-resistant cell lines showed an 8.9- to >2,202-fold reduction in susceptibility to PF-00868554 compared to the wild-type cells (Table 3). After confirmation of resistance, replicon RNA was isolated from PF-00868554-resistant cell lines, amplified by RT-PCR, and subjected to population sequencing to determine the genotypic changes in the NS5B region of the replicon. The replicons in these cell lines were shown to harbor one to four mutations that resulted in amino acid changes in the polymerase (Table 3). Every resistant cell line contained at least one change at an amino acid that was shown to be part of the inhibitor-binding pocket, which includes M423, M426, and I482 (29). The methionine-to-threonine change at residue 423 (M423T) in the NS5B region was identified as the most prevalent amino acid substitution, occurring alone in 14 of 30 cell lines (47%) and in combination with other mutations in 8 of 30 cell lines (27%) that were selected across different concentrations of PF-00868554 (Table 3). Overall, M423T was observed at a frequency of 73% (22 of 30 cell lines), and changes at amino acid M423, including M423V and M423I, were observed in 87% (26 out of 30) of PF-00868554-resistant cell lines (Table 3). Changes at other amino acid residues, M426T and I482T, existed at a much lower frequency (Table 3). Substitution of proline by alanine or serine at residue 496 (P496A or P496S) and substitution of valine by alanine at residue 499 (V499A) were observed in cell lines resistant to a benzimidazole polymerase inhibitor, compound C (Fig. 1), consistent with the in vitro resistance changes previously reported for this compound (21).
TABLE 3.
Genotypic and phenotypic changes of PF-00868554-resistant cell lines
| Amino acid change(s)a | Selection pressureb
|
Frequency (%) | RLuc activity (103 RLU) | Fold change in PF-00868554 EC50c | ||
|---|---|---|---|---|---|---|
| 3× EC90 | 6× EC90 | 12× EC90 | ||||
| M423T | 3 | 7 | 4 | 47 | 160-1,151 | 715->2,202 |
| N110N/S, M423T | 1 | 3 | 652 | 2,198 | ||
| I134I/V, M423T | 1 | 3 | 256 | >2,202 | ||
| M423T, K535N | 1 | 1 | 7 | 704, 185 | 1,107, 731 | |
| M423T, K535N, N590S | 1 | 3 | 678 | 880 | ||
| M423T, I585M | 1 | 3 | 313 | 2,180 | ||
| M423T, F572L, I585M | 1 | 3 | 122 | 1,057 | ||
| M423T, F572F/V, M573T, W574R | 1 | 3 | 457 | 897 | ||
| M423V, I585M | 1 | 3 | 277 | 1,495 | ||
| M423I | 1 | 3 | 483 | 1,000 | ||
| M423I, I432T | 1 | 3 | 49 | 912 | ||
| K81N, M423I | 1 | 3 | 189 | 706 | ||
| M426T, I432V | 1 | 3 | 848 | 20 | ||
| M426T, I432T, Y586C | 1 | 3 | 554 | 8.9 | ||
| A300V, I482T | 1 | 3 | 582 | 49 | ||
RNA was extracted from stable cell lines obtained at 3×, 6×, and 12× the EC90s of PF-00868554 and subjected to RT-PCR amplification and population sequencing of the NS5B region. M423T in combination with one or more amino acid changes occurred at a frequency of 27% (n = 8).
Results represent the number of PF-00868554-resistant cell lines established at each concentration (μM) that contain the observed resistance mutations.
Results represent the average of duplicate values from one experiment. The ranges of fold changes in the EC50s for benzimidazole compound C and IFN-α are 0.6 to 4.5 and 0.1 to 6, respectively.
All of the amino acid substitutions found to be associated with resistance to PF-00868554, including M423T, M423I, M423V, M426T, and I482T, were also observed in the in vitro resistance study of a structurally related compound, AG-021541 (39). The 22 resistant cell lines that contain the M423T change alone or in combination with other amino acid changes demonstrated a 715- to >2,202-fold increase in the EC50 when tested for their susceptibility to PF-00868554. Consistent with our previous findings, the M426T and I482T substitutions resulted in a lower level of resistance than the changes from M to T, V, or I at amino acid 423 of the polymerase (8.9- to 49-fold versus 706- to >2,202-fold), confirming that M423 is a key residue involved in the interaction between dihydropyrone compounds and the polymerase (Table 3). M426T and I482T were present only in replicon cell lines generated at lower concentrations of PF-00868554 (Table 3). Regardless of their replication fitness or level of resistance to PF-00868554, all of the resistant replicons remained sensitive to IFN-α, with changes in the EC50 ranging from 0.1 to 6. Furthermore, the resistance changes at the thumb base site did not significantly affect the activity of compound C (Fig. 1), known to interact with a different allosteric site of the polymerase protein, with changes in the EC50 ranging from 0.6 to 4.5.
The predominant resistance change, M423T, was introduced back into the wild-type (wt) replicon, from which a stable cell line was generated for the determination of its resistance phenotype. M423T conferred 761-fold resistance to PF-00868554, but no change in susceptibility to IFN-α or compound C was observed (Table 4). The replication fitness of the M423T replicon was reduced to 1.8 × 105 RLU, which was less than 50% of that of the wt replicon (5.2 × 105 RLU), as indicated by the hRLuc reporter activity. However, the colony-forming efficiency of the M423T replicon was comparable to that of the wt replicon in the colony formation assay (data not shown), suggesting that the mutant replicon replicates at a lower level but is still sufficient to support colony formation. Similarly, the purified recombinant 1b Con1 polymerase enzyme that contained the M423T substitution also displayed 733-fold resistance to PF-00868554 but no change in susceptibility to compound C in the enzymatic assay in vitro (Table 4).
TABLE 4.
Susceptibility of the M423T mutant replicon and polymerase to PF-00868554a
| Compound | Replicon EC50 (μM)c
|
FC in EC50 | Biochemical IC50 (μM)
|
FC in IC50 | ||
|---|---|---|---|---|---|---|
| wt | M423T | NS5BΔ21 wt | NS5BΔ21M423T | |||
| PF-00868554 | 0.035 ± 0.031 | 27 ± 9 | 761 | 0.015 ± 0.010 | 11 ± 3 | 733 |
| Benzimidazole compound C | 1.6 ± 0.9 | 2.4 ± 0.7 | 1.4 | 0.20 ± 0.01b | 0.38 ± 0.03 | 1.9 |
| IFN-α | 0.97 ± 0.09 | 1.3 ± 0.2 | 1.4 | ND | ND | ND |
The susceptibility of the 1b Con1 wt and M423T replicon cell line to inhibitors was evaluated in the reporter replicon assay. Cells were exposed to compounds for 3 days before RLuc activity was determined. The susceptibility of the 1b Con1 wt and M423T mutant polymerase to inhibitors was evaluated in the HCV polymerase biochemical assay as described in Materials and Methods. Results represent the means ± standard deviations (three experiments) or individual values (one to two experiments of duplicates). FC, fold change; ND, not determined.
The result was generated against the 1b BK polymerase.
The replicon EC50s for IFN-α are given as IU/ml.
Pharmacokinetic properties of PF-00868554 in preclinical species.
In both rats and monkeys, PF-0868554 exhibited moderate plasma CL and V (Table 5). In dogs, however, it exhibited both low plasma CL (4.4 ± 1.1 ml/min/kg) and V (0.25 liter/kg). The t1/2 of PF-00868554 in rats, monkeys, and dogs were between 2 and 4.4 h, but the calculated effective half-life (0.693 × V/CL) was substantially shorter (0.5 h in both rats and monkeys and 3 h in dogs). The preclinical results suggest that PF-00868554 would be amenable for a twice-daily dosing regimen. PF-00868554 was rapidly absorbed after an oral dose in both species (with a Tmax of 0.94 ± 0.9 h and 1 h for rats and dogs, respectively) with acceptable F (75% and 49% for rats and dogs, respectively) (Table 5). In summary, the absorption of PF-00868554 in preclinical species was rapid, resulting in a high predicted human absorption rate constant (Ka) of 1.1 h−1.
TABLE 5.
In vivo pharmacokinetics of PF-00868554 in rats, dogs, and monkeysa
| Pharmacokinetic parameterb | Rat
|
Dog
|
Monkey
|
||||
|---|---|---|---|---|---|---|---|
| i.v. | Oral solution | Oral suspension | i.v. | Oral solution | Oral suspension | i.v. | |
| Dose (mg/kg) | 2 | 2 | 2 | 1 | 2 | 2 | 2 |
| No. of animals | 5 | 3 | 3 | 3 | 3 | 2 | 3 |
| CLblood (ml/min/kg) | 21 ± 4.8 | 0.93 ± 0.1 | 21 ± 5 | ||||
| Vss (liters/kg) | 1 ± 0.4 | 0.25 | 0.8 ± 0.05 | ||||
| AUC0-∞ (μg·h/ml) | 2.9 ± 0.7 | 0.93 ± 0.2 | 0.93 ± 0.2 | 18 ± 2.3 | 17.7 ± 1.8 | 14.1 | 2.6 ± 1.0 |
| t1/2 (h) | 2.5 ± 1 | 2.8 ± 1.2 | 2.85 ± 0.2 | 4.4 ± 1.1 | 3.6 ± 0.8 | 3.1 | 2.1 ± 0.4 |
| Cmax (μg/ml) | 0.64 ± 0.28 | 0.7 ± 0.1 | 4.7 ± 1.1 | 4.3 | |||
| Tmax (h) | 0.94 ± 0.9 | 0.2 | 1 | 0.7 | |||
| F (%) | 75 | 31 | 49 | 39 | |||
i.v. and oral solution formulation was as follows: 50% PEG 200, 10% ethanol, 40% water. Oral suspension formulation was as follows: 0.5% methylcellulose.
CLblood, blood clearance; Vss, volume of distribution at steady state.
DISCUSSION
Current therapy for HCV is poorly tolerated and of limited efficacy, such that patients infected with the 1a and 1b genotypes have more severe liver disease and lower response rates to current IFN therapy (54). In the United States, infection by the 1a and 1b genotypes is the most prevalent, whereas in Europe and Japan, the prevalence of infection by the 1b genotype significantly exceeds that of any other HCV genotypes (53). Thus, there remains a significant unmet need for targeted, efficacious chemotherapy against HCV genotypes 1a and 1b.
We have demonstrated that PF-00868554 displays both potency and selectivity as a promising inhibitor of the 1a and 1b genotypes of HCV RNA-dependent RNA polymerase. With low nanomolar inhibitory potency against several recombinant genotype 1 NS5B constructs in vitro, the molecule displays no detectable inhibition of several human polymerases and no detectable inhibition of several human proteases, in spite of its weak inhibitory activity against recombinant HIV protease. This biochemical selectivity likely translates into cellular selectivity and a favorable therapeutic index, in that no detectable cytotoxicity was detected with several immortalized human cell lines at concentrations more than 5,400-fold greater than the potent activity against the genotype 1 HCV replicons.
One obstacle to developing effective HCV therapies is the heterogeneity of the viral genomes present in infections. The RNA-dependent RNA polymerase that synthesizes the viral genome lacks a proof-reading mechanism. The error rate of the polymerase is estimated at 10−4 (35), which results in significant genetic diversity among the viral population within each patient. This heterogeneity has been associated with reductions in activity of HCV polymerase inhibitors up to 25-fold when tested in a chimeric replicon system containing NS5B sequences derived from genotype 1a and 1b patients (31, 48). Therefore, an important part of preclinical development for HCV inhibitors is the assessment of the antiviral activity against a variety of clinical isolates. PF-00868554 demonstrated potent antiviral activity against polymerases derived from a majority (95.8%) of HCV strains, including primary clinical isolates and laboratory strains. The overall mean and median EC50s of PF-00868554 against all genotype 1 replicons were 0.059 and 0.042 μM, respectively, with a range of 0.0088 to 0.39 μM. The presence of 50% HS only modestly attenuated the antiviral activity of PF-00868554. These studies demonstrate that PF-00868554 has potent in vitro antiviral activity in the HCV replicon system against a variety of genotype 1 polymerases and support its potential use as an antiviral agent with patients infected with genotype 1 HCV.
The emergence of drug-resistance variants has been a major factor limiting the efficacy of virus-specific inhibitors against retroviruses and many other RNA viruses. In vitro resistance studies of various HCV inhibitors, including NS3 protease (26, 27, 30, 47, 52) and NS5B polymerase inhibitors (17, 21, 22, 33, 34, 45, 46, 50), identified resistance mutations in the corresponding viral target regions, some of which have also been observed in subsequent clinical studies. A recent report indicated that resistance mutations observed in vitro were also developed in vivo after a 14-day monotherapy treatment with an NS3 protease inhibitor, VX-950, and correlated strongly with clinical outcome (37). A nonnucleoside polymerase inhibitor from Viropharma, HCV-796, achieved a peak reduction in viral load of >1 log on day 4, but the reduction dropped to approximately 0.7 log on day 14 (6) as a result of the emergence of resistance (49). These results highlight the importance of conducting in vitro resistance studies, which could provide important insights into resistance development in future clinical trials.
Our in vitro resistance study of PF-00868554 in the HCV subgenomic replicon cells identified amino acid changes at the thumb base allosteric site of the polymerase, including M423T/V/I, M426T, and I482T, all of which were observed in our previous study of a structurally related compound, AG-021541 (39). Similar to our previous findings, amino acid substitutions at M423 resulted in a significantly higher level of resistance than those of substitutions at M426 and I482 (Table 3). More importantly, there was no cross-resistance of replicons containing these resistance changes to IFN-α and a number of other polymerase inhibitors (39), supporting the use of PF-00868554 in combination therapies with other inhibitors targeting different regions of the polymerase for the treatment of HCV.
In conclusion, we have presented a summary of preclinical in vitro and in vivo data demonstrating that PF-00868554 has potent and broad-spectrum activity against HCV polymerase 1a and 1b viral strains, while displaying a selectivity profile that is consistent with a significant margin of safety. Furthermore, the pharmacokinetic properties of PF-00868554 were determined by in vivo studies to be favorable in preclinical species, suggesting that a twice-daily dosing regimen for human patients will be feasible.
Supplementary Material
Acknowledgments
We acknowledge all members of the HCV polymerase project team at Pfizer Global Research and Development, La Jolla Laboratories, and Kalamazoo Laboratories (formerly Pharmacia and Upjohn, Inc.). In addition, we thank Karen Maegley and Laura Lingardo for conducting protease selectivity assays and Robert Hunter for conducting the in vivo rat pharmacokinetic studies.
Footnotes
Published ahead of print on 23 March 2009.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Bartenschlager, R., M. Frese, and T. Pietschmann. 2004. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63:71-180. [DOI] [PubMed] [Google Scholar]
- 2.Beaulieu, P. L. 2007. Non-nucleoside inhibitors of the HCV NS5B polymerase: progress in the discovery and development of novel agents for the treatment of HCV infections. Curr. Opin. Investig. Drugs 8:614-634. [PubMed] [Google Scholar]
- 3.Canon-Koch, S. S., T. N. Alexander, M. Barvian, G. Bolton, F. E. Boyer, B. J. Burke, T. Holler, T. M. Jewell, J. V. Prasad, D. J. Kucera, J. Machak, L. J. Mitchell, S. T. Murphy, S. H. Reich, D. J. Skalitzky, J. H. Tatlock, M. D. Varney, S. C. Virgil, S. T. Worland, M. Melnick, M. A. Linton, and S. E. Webber. December 2002. Preparation of amino acid amides as HIV protease inhibitors. U.S. patent 02/100845.
- 4.Carroll, S. S., and D. B. Olsen. 2006. Nucleoside analog inhibitors of hepatitis C virus replication. Infect. Disord. Drug Targets 6:17-29. [DOI] [PubMed] [Google Scholar]
- 5.Chai, D., M. G. Darcy, D. Dhanak, K. J. Duffy, G. A. Erickson, D. M. Fitch, A. T. Gates, V. K. Johnston, T. Sariskyrobert, M. J. Sharp, A. N. Shaw, R. Tedesco, K. J. Wiggall, and M. N. Zimmerman. December 2002. Quinolinylthiadiazine dioxides as antiviral agents for treating hepatitis C. U.S. patent WO 2002098424 A1 20021212.
- 6.Chandra, P., D. Raible, D. Harper, J. Speth, S. Villano, and G. Bichier. 2006. Antiviral activity of the non-nucleoside polymerase inhibitor, HCV-796, in patients with chronic hepatitis C virus: preliminary results from a randomized, double-blind, placebo-controlled, ascending multiple dose study. Abstr. Dig. Dis. Week, abstr.1.
- 7.Choo, Q. L., K. H. Richman, J. H. Han, K. Berger, C. Lee, C. Dong, C. Gallegos, D. Coit, R. Medina-Selby, P. J. Barr, et al. 1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 88:2451-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Copeland, W. C., and T. S. Wang. 1991. Catalytic subunit of human DNA polymerase alpha overproduced from baculovirus-infected insect cells. Structural and enzymological characterization. J. Biol. Chem. 266:22739-22748. [PubMed] [Google Scholar]
- 9.Fried, M. W., M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Goncales, Jr., D. Haussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman, and J. Yu. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975-982. [DOI] [PubMed] [Google Scholar]
- 10.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice. 1993. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. USA 90:10583-10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadziyannis, S. J., H. Sette, Jr., T. R. Morgan, V. Balan, M. Diago, P. Marcellin, G. Ramadori, H. Bodenheimer, Jr., D. Bernstein, M. Rizzetto, S. Zeuzem, P. J. Pockros, A. Lin, and A. M. Ackrill. 2004. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 140:346-355. [DOI] [PubMed] [Google Scholar]
- 12.Hammond, J. L., M. C. Rosario, F. Wagner, D. Mazur, C. Kantaridis, V. S. Purohit, L. K. Durham, S. Jagannatha, and M. F. DeBruin. 2008. Antiviral activity of the HCV polymerase inhibitor PF-00868554 administered as monotherapy in HCV genotype 1 infected subjects. Hepatology 48:LB11. [Google Scholar]
- 13.Hao, W., K. J. Herlihy, N. J. Zhang, S. A. Fuhrman, C. Doan, A. K. Patick, and R. Duggal. 2007. Development of a novel dicistronic reporter-selectable hepatitis C virus replicon suitable for high-throughput inhibitor screening. Antimicrob. Agents Chemother. 51:95-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hijikata, M., N. Kato, Y. Ootsuyama, M. Nakagawa, and K. Shimotohno. 1991. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. USA 88:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hijikata, M., H. Mizushima, T. Akagi, S. Mori, N. Kakiuchi, N. Kato, T. Tanaka, K. Kimura, and K. Shimotohno. 1993. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J. Virol. 67:4665-4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hostomsky, Z., Z. Hostomska, G. O. Hudson, E. W. Moomaw, and B. R. Nodes. 1991. Reconstitution in vitro of RNase H activity by using purified N-terminal and C-terminal domains of HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 88:1148-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howe, A. Y., H. Cheng, I. Thompson, S. K. Chunduru, S. Herrmann, J. O'Connell, A. Agarwal, R. Chopra, and A. M. Del Vecchio. 2006. Molecular mechanism of a thumb domain hepatitis C virus nonnucleoside RNA-dependent RNA polymerase inhibitor. Antimicrob. Agents Chemother. 50:4103-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howe, A. Y. M., H. Cheng, S. Johann, S. Mullen, S. K. Chunduru, D. C. Young, J. Bard, and R. Chopra. 2006. Identification and characterization of HCV replicon variants with reduced susceptibility to HCV-796. Abstr. 13th Int. Meet. Hepat. C Virus Relat. Viruses, abstr. 717.
- 19.Koch, U., and F. Narjes. 2006. Allosteric inhibition of the hepatitis C virus NS5B RNA dependent RNA polymerase. Infect. Disord. Drug Targets 6:31-41. [DOI] [PubMed] [Google Scholar]
- 20.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kukolj, G., G. A. McGibbon, G. McKercher, M. Marquis, S. Lefebvre, L. Thauvette, J. Gauthier, S. Goulet, M. A. Poupart, and P. L. Beaulieu. 2005. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 280:39260-39267. [DOI] [PubMed] [Google Scholar]
- 22.Le Pogam, S., H. Kang, S. F. Harris, V. Leveque, A. M. Giannetti, S. Ali, W. R. Jiang, S. Rajyaguru, G. Tavares, C. Oshiro, T. Hendricks, K. Klumpp, J. Symons, M. F. Browner, N. Cammack, and I. Najera. 2006. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 80:6146-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, H., A. Linton, J. Tatlock, J. Gonzalez, A. Borchardt, M. Abreo, T. Jewell, L. Patel, M. Drowns, S. Ludlum, M. Goble, M. Yang, J. Blazel, R. Rahavendran, H. Skor, S. Shi, C. Lewis, and S. Fuhrman. 2007. Allosteric inhibitors of hepatitis C polymerase: discovery of potent and orally bioavailable carbon-linked dihydropyrones. J. Med. Chem. 50:3969-3972. [DOI] [PubMed] [Google Scholar]
- 24.Li, H., J. Tatlock, A. Linton, J. Gonzalez, A. Borchardt, P. Dragovich, T. Jewell, T. Prins, R. Zhou, J. Blazel, H. Parge, R. Love, M. Hickey, C. Doan, S. Shi, R. Duggal, C. Lewis, and S. Fuhrman. 2006. Identification and structure-based optimization of novel dihydropyrones as potent HCV RNA polymerase inhibitors. Bioorg. Med. Chem. Lett. 16:4834-4838. [DOI] [PubMed] [Google Scholar]
- 25.Li, H., J. Tatlock, A. Linton, J. Gonzalez, T. Jewell, L. Patel, S. Ludlum, M. Drowns, S. V. Rahavendran, H. Skor, R. Hunter, S. T. Shi, K. J. Herlihy, H. Parge, M. Hickey, X. Yu, F. Chau, J. Nonomiya, and C. Lewis. 2009. Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554) as a potent and orally available hepatitis C virus polymerase inhibitor. J. Med. Chem. 52:1255-1258. [DOI] [PubMed] [Google Scholar]
- 26.Lin, C., C. A. Gates, B. G. Rao, D. L. Brennan, J. R. Fulghum, Y. P. Luong, J. D. Frantz, K. Lin, S. Ma, Y. Y. Wei, R. B. Perni, and A. D. Kwong. 2005. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 280:36784-36791. [DOI] [PubMed] [Google Scholar]
- 27.Lin, C., K. Lin, Y. P. Luong, B. G. Rao, Y. Y. Wei, D. L. Brennan, J. R. Fulghum, H. M. Hsiao, S. Ma, J. P. Maxwell, K. M. Cottrell, R. B. Perni, C. A. Gates, and A. D. Kwong. 2004. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 279:17508-17514. [DOI] [PubMed] [Google Scholar]
- 28.Lindahl, K., L. Stahle, A. Bruchfeld, and R. Schvarcz. 2005. High-dose ribavirin in combination with standard dose peginterferon for treatment of patients with chronic hepatitis C. Hepatology 41:275-279. [DOI] [PubMed] [Google Scholar]
- 29.Love, R. A., H. E. Parge, X. Yu, M. J. Hickey, W. Diehl, J. Gao, H. Wriggers, A. Ekker, L. Wang, J. A. Thomson, P. S. Dragovich, and S. A. Fuhrman. 2003. Crystallographic identification of a noncompetitive inhibitor binding site on the hepatitis C virus NS5B RNA polymerase enzyme. J. Virol. 77:7575-7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu, L., T. J. Pilot-Matias, K. D. Stewart, J. T. Randolph, R. Pithawalla, W. He, P. P. Huang, L. L. Klein, H. Mo, and A. Molla. 2004. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 48:2260-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludmerer, S. W., D. J. Graham, E. Boots, E. M. Murray, A. Simcoe, E. J. Markel, J. A. Grobler, O. A. Flores, D. B. Olsen, D. J. Hazuda, and R. L. LaFemina. 2005. Replication fitness and NS5B drug sensitivity of diverse hepatitis C virus isolates characterized by using a transient replication assay. Antimicrob. Agents Chemother. 49:2059-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 33.Mo, H., L. Lu, T. Pilot-Matias, R. Pithawalla, R. Mondal, S. Masse, T. Dekhtyar, T. Ng, G. Koev, V. Stoll, K. D. Stewart, J. Pratt, P. Donner, T. Rockway, C. Maring, and A. Molla. 2005. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 49:4305-4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen, T. T., A. T. Gates, L. L. Gutshall, V. K. Johnston, B. Gu, K. J. Duffy, and R. T. Sarisky. 2003. Resistance profile of a hepatitis C virus RNA-dependent RNA polymerase benzothiadiazine inhibitor. Antimicrob. Agents Chemother. 47:3525-3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogata, N., H. J. Alter, R. H. Miller, and R. H. Purcell. 1991. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA 88:3392-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oien, N. L., R. J. Brideau, T. A. Hopkins, J. L. Wieber, M. L. Knechtel, J. A. Shelly, R. A. Anstadt, P. A. Wells, R. A. Poorman, A. Huang, V. A. Vaillancourt, T. L. Clayton, J. A. Tucker, and M. W. Wathen. 2002. Broad-spectrum antiherpes activities of 4-hydroxyquinoline carboxamides, a novel class of herpesvirus polymerase inhibitors. Antimicrob. Agents Chemother. 46:724-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarrazin, C., T. L. Kieffer, D. Bartels, B. Hanzelka, U. Muh, M. Welker, D. Wincheringer, Y. Zhou, H. M. Chu, C. Lin, C. Weegink, H. Reesink, S. Zeuzem, and A. D. Kwong. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767-1777. [DOI] [PubMed] [Google Scholar]
- 38.Seeff, L. B. 1995. Natural history of viral hepatitis, type C. Semin. Gastrointest. Dis. 6:20-27. [PubMed] [Google Scholar]
- 39.Shi, S. T., K. J. Herlihy, J. P. Graham, S. A. Fuhrman, C. Doan, H. Parge, M. Hickey, J. Gao, X. Yu, F. Chau, J. Gonzalez, H. Li, C. Lewis, A. K. Patick, and R. Duggal. 2008. In vitro resistance study of AG-021541, a novel nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 52:675-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sulkowski, M. S. 2003. Anemia in the treatment of hepatitis C virus infection. Clin. Infect. Dis. 37(Suppl. 4):S315-S322. [DOI] [PubMed] [Google Scholar]
- 41.Summa, V., A. Petrocchi, V. G. Matassa, M. Taliani, R. Laufer, R. De Francesco, S. Altamura, and P. Pace. 2004. HCV NS5b RNA-dependent RNA polymerase inhibitors: from alpha,gamma-diketoacids to 4,5-dihydroxypyrimidine- or 3-methyl-5-hydroxypyrimidinonecarboxylic acids. Design and synthesis. J. Med. Chem. 47:5336-5339. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka, S., S. Z. Hu, T. S. Wang, and D. Korn. 1982. Preparation and preliminary characterization of monoclonal antibodies against human DNA polymerase alpha. J. Biol. Chem. 257:8386-8390. [PubMed] [Google Scholar]
- 43.Thill, G., M. Blumenfeld, F. Lescure, and M. Vasseur. 1991. Self-cleavage of a 71 nucleotide-long ribozyme derived from hepatitis delta virus genomic RNA. Nucleic Acids Res. 19:6519-6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas, D. L., and L. B. Seeff. 2005. Natural history of hepatitis C. Clin. Liver Dis. 9:383-398. [DOI] [PubMed] [Google Scholar]
- 45.Tomei, L., S. Altamura, L. Bartholomew, A. Biroccio, A. Ceccacci, L. Pacini, F. Narjes, N. Gennari, M. Bisbocci, I. Incitti, L. Orsatti, S. Harper, I. Stansfield, M. Rowley, R. De Francesco, and G. Migliaccio. 2003. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77:13225-13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomei, L., S. Altamura, L. Bartholomew, M. Bisbocci, C. Bailey, M. Bosserman, A. Cellucci, E. Forte, I. Incitti, L. Orsatti, U. Koch, R. De Francesco, D. B. Olsen, S. S. Carroll, and G. Migliaccio. 2004. Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 78:938-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tong, X., R. Chase, A. Skelton, T. Chen, J. Wright-Minogue, and B. A. Malcolm. 2006. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antivir. Res. 70:28-38. [DOI] [PubMed] [Google Scholar]
- 48.Tripathi, R. L., P. Krishnan, Y. He, T. Middleton, T. Pilot-Matias, C. M. Chen, D. T. Lau, S. M. Lemon, H. Mo, W. Kati, and A. Molla. 2007. Replication efficiency of chimeric replicon containing NS5A-5B genes derived from HCV-infected patient sera. Antivir. Res. 73:40-49. [DOI] [PubMed] [Google Scholar]
- 49.Villano, S., A. Howe, D. Raible, D. Harper, J. Speth, and G. Bichier. 2006. Analysis of HCV NS5B genetic variants following monotherapy with HCV-796, a non-nucleoside polymerase inhibitor, in treatment-naïve HCV-infected patients. Hepatology 44:607A-608A. [Google Scholar]
- 50.Wang, M., K. K. Ng, M. M. Cherney, L. Chan, C. G. Yannopoulos, J. Bedard, N. Morin, N. Nguyen-Ba, M. H. Alaoui-Ismaili, R. C. Bethell, and M. N. James. 2003. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 278:9489-9495. [DOI] [PubMed] [Google Scholar]
- 51.Williams, J. W., and J. F. Morrison. 1979. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 63:437-467. [DOI] [PubMed] [Google Scholar]
- 52.Yi, M., X. Tong, A. Skelton, R. Chase, T. Chen, A. Prongay, S. L. Bogen, A. K. Saksena, F. G. Njoroge, R. L. Veselenak, R. B. Pyles, N. Bourne, B. A. Malcolm, and S. M. Lemon. 2006. Mutations conferring resistance to SCH6, a novel hepatitis C virus NS3/4A protease inhibitor. Reduced RNA replication fitness and partial rescue by second-site mutations. J. Biol. Chem. 281:8205-8215. [DOI] [PubMed] [Google Scholar]
- 53.Zein, N. N. 2000. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 13:223-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zein, N. N., J. Rakela, E. L. Krawitt, K. R. Reddy, T. Tominaga, D. H. Persing, et al. 1996. Hepatitis C virus genotypes in the United States: epidemiology, pathogenicity, and response to interferon therapy. Ann. Intern. Med. 125:634-639. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.