

Abstract
3,3′-Diindolylmethane (DIM), a novel poison targeting Leishmania donovani topoisomerase I (LdTOP1LS), induces programmed cell death in Leishmania parasites. The development of resistant parasites by adaptation with increasing concentrations of DIM generates random mutations in LdTOP1LS. Single-nucleotide mutations result in the amino acid substitutions F270L and K430N in the large subunit and N184S in the small subunit of the enzyme. DIM failed to inhibit the catalytic activity of the recombinant mutant enzyme (LdTOP1DRLS). Transfection studies of the mutant genes showed that the mutated topoisomerase I confers DIM resistance on wild-type Leishmania parasites. Site-directed mutagenesis studies revealed that a substantial level of resistance is conferred by the F270L mutation alone; however, all three mutations (F270L, K430N, and N184S) together are required to reach a higher-resistance phenotype. DIM fails to stabilize the topoisomerase I-DNA covalent complexes in the F270 mutant. Moreover, DIM cannot interfere with the religation step in the catalytic cycle of the recombinant F270L mutant enzyme. Taken together, these findings identify novel mutations in topoisomerase I that hinder its interaction with DNA, thereby modulating enzyme catalysis and conferring resistance to DIM. These studies advance our understanding of the mechanism of cell poisoning by DIM and suggest a specific modification of the drug that may improve its efficacy.
Eukaryotic DNA topoisomerase I is an essential enzyme that alters the topological changes of DNA that accompany DNA replication, transcription, recombination, and chromosomal segregation during mitotic cell division (3, 21). Topoisomerase I induces a transient single-stranded break of the DNA duplex and results in a reversible topoisomerase I-DNA covalent complex (2). Most eukaryotic type IB topoisomerases are monomeric enzymes, including human topoisomerase I, which comprises 765 amino acids (91 kDa). But interestingly, DNA topoisomerase I of the kinetoplast protozoan parasite Leishmania donovani is an unusual bisubunit enzyme, consisting of a large subunit (73 kDa) and a small subunit (29 kDa) (6). Recently, we demonstrated for the first time that in vitro reconstitution of the two recombinant proteins, the large subunit and the small subunit of L. donovani topoisomerase I (LdTOP1L and LdTOP1S, respectively), shows active enzyme. This active enzyme (LdTOP1LS) is located in both the nucleus and the kinetoplast of the parasite (6). Because topoisomerase I poisoning has been recognized as a promising pharmacological target for the development of therapeutic agents, a number of candidates have been identified. The potent antitumor compound 3,3′-diindolylmethane (DIM) is a well-characterized topoisomerase I inhibitor that stabilizes the topoisomerase I-DNA covalent complex. In addition to its antitumor effects, DIM has also been recognized to specifically target L. donovani topoisomerase I (16). Moreover, we recently demonstrated that DIM induces programmed cell death through the inhibition of mitochondrial FoF1 ATP synthase in the unicellular protozoan parasite L. donovani and reduces the parasitic loads in the livers and spleens of infected hamsters (17). Thus, DIM could be of interest in the treatment of human leishmaniasis. Therefore, precise determination of the mechanism of DIM resistance in Leishmania parasites could be an important strategic tool for acquiring a better understanding of its mechanism of molecular action.
Topoisomerase I is a target for anticancer as well as antiparasitic drugs, one important class of which is topoisomerase poison. These drugs act by stabilizing an intermediate stage of a reaction, where the enzyme is complexed with the cleaved gate helix. Resistance to topoisomerase I poison drugs is a clinical problem and may arise through a variety of mechanisms. Mammalian cell lines selected for resistance to camptothecin (CPT) exhibit decreased levels of topoisomerase I expression. In resistant murine P388 leukemia cells, CPT treatment is associated with a lower frequency of DNA single-strand breaks than that in wild-type (WT) cells and decreased expression of topoisomerase I at the mRNA and protein levels (8). Similar findings have been obtained with a human lung cancer cell line resistant to a CPT analog (CPT-11) (11) and with a CPT-resistant tumor cell line (19). The levels of expression of topoisomerase I mRNA and protein in CPT-resistant human nasopharyngeal carcinoma cells (CPT30) were 30% and 40% lower, respectively, than those in the parent human nasopharyngeal carcinoma cell line (HONE-1) (4). Decreases in topoisomerase I expression have been associated with rearrangement and hypermethylation of the topoisomerase I gene (20). Thus, decreased expression and/or alteration of the topoisomerase I gene can confer resistance to CPT. Mutations affecting the way the drug interacts with the enzyme can cause resistance. Mutations that confer resistance to CPT have been identified in several regions of human topoisomerase I (Phe361Ser, Arg364His, Glu418Lys, Gly503Ser, Asp533Gly, Ala653Pro, and Asn722Ser) (5). A form of human topoisomerase I containing an Ala653Pro mutation has been shown to exhibit enhanced DNA strand religation, preventing the binding of CPT to the topoisomerase I-DNA complex (9). A point mutation resulting in the replacement of threonine with alanine at residue 729 has been found in a CPT-11-resistant human lung cancer cell line (12). Another mutation in topoisomerase I, at nucleotide residue 1463 of the human nasopharyngeal carcinoma cell line HONE-1, confers resistance to CPT. This mutation (a G-to-A transition) results in a glutamate-to-lysine mutation at amino acid residue 418 (E418) in the CPT30 cell line (4). In L. donovani, mutations for resistance to CPT have been identified in two regions of the large subunit of LdTOP1LS, resulting from two single-nucleotide mutations (Gly185Arg and Asp325Glu). Moreover, by alignment of the sequence of L. donovani topoisomerase I with that of the human enzyme, it was observed that the Gly185 and Asp325 residues in LdTOP1LS are both conserved in human topoisomerase I (Gly359 and Asp500) (13).
CPT-resistant mutations in human topoisomerase I present structural evidence explaining their possible impact on drug binding. Several topoisomerase I mutations, located either near or far from the active site, have been shown to render the enzyme resistant to the drug (14, 18). The 3-dimensional structures of the ternary complexes with the native enzyme and with some specific mutants have clarified the molecular basis for the resistance conferred by most of the mutations located near the active site but have still left unexplained the effects of those located far from it, such as the Ala653Pro, Glu418Lys, and Thr729Ala mutations (4, 9, 12). The single mutation Glu418Lys in human topoisomerase I shows a different cleavage substrate specificity, pointing out the important role of core subdomain I in substrate recognition and confirming the strict requirement of a T base at the −1 position of the scissile strand for CPT binding (10).
To investigate the mechanism of action of resistance to DIM in Leishmania parasites, we have developed parasites resistant to DIM by stepwise exposure to the drug. We further determined the resistance mechanisms by which the parasites bypassed the cytotoxic effects of DIM. In the present study, we report for the first time that DIM resistance in protozoan parasites arises via topoisomerase I gene mutation. Understanding the mechanisms of resistance to DIM is crucial for the treatment of human leishmaniasis and for the design of new drugs.
MATERIALS AND METHODS
Chemicals.
The bioactive compound DIM was synthesized chemically from indole and urotropine by the addition of InCl2 (10 mol%) and isopropanol as described previously (16). CPT was purchased from Sigma Chemicals (St. Louis, MO). All drugs were dissolved in 100% dimethyl sulfoxide (DMSO) at 20 mM and were stored at −20°C.
Development of a DIM-resistant parasite strain.
A highly resistant L. donovani strain (AG83) called LdDR50 was developed by stepwise exposure of the parasites to DIM at 1, 2, 5, 10, 20, 40, and 50 μM concentrations. When a 50 μM concentration was reached, the culture was diluted to ∼10 cells/ml and separately cultured to generate a clonal population of parasites. The resistant parasites were maintained in drug-free medium for 72 h before use.
Cell viability testing by an MTT assay.
The effect of the drug on the viability of the WT and LdDR50 cell lines was determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (17). The cells were collected at the exponential phase and transferred to a 24-well plate (2 × 106 cells per well). The cells were then incubated for 12 h in the presence of various concentrations of DIM (1, 5, 10, 15, and 20 μM). After incubation, the cells were centrifuged, and the cell pellet was washed twice with phosphate-buffered saline (PBS) (1×) and finally suspended in 100 μl of PBS (1×) in a 96-well plate. Ten microliters (10 μg/ml) of MTT solution was added to each sample in the 96-well plates and incubated for 4 h. After incubation, 100 μl of stop solution (stock; 4,963 μl isopropanol and 17 μl concentrated HCl) was added to each sample and kept for 20 min at room temperature. The optical density was measured as the A570 on an enzyme-linked immunosorbent assay reader (Multiskan EX; Thermo Fisher Scientific, Waltham, MA).
Measurement of DIM accumulation in cells.
WT and DIM-resistant Leishmania parasites grown to logarithmic phase were incubated at a density of 3 × 107 cells per ml in culture medium containing 20 μM DIM at 22° C. At 1-, 5-, 10-, 20-, and 30-min intervals, 1-ml aliquots were taken, washed three times with ice-cold PBS (1.2 mM KH2PO4, 8.1 mM NaH2PO4, 130 mM NaCl, and 2.6 mM KCl, adjusted to pH 7.4) containing 0.01 N HCl, and suspended in the same buffer. After sonication, the cell lysate was used to measure the accumulated amount of DIM by equilibrium dialysis as described previously (16). Each experiment was repeated three times with duplicate samples. Error bars were calculated from the means ± standard errors.
Real-time RT-PCR.
Total RNA was prepared from WT and DIM-resistant promastigotes using the Total RNA isolation kit (Roche Biochemicals). cDNA was synthesized from 60 ng of total RNA using Superscript II RNaseH− reverse transcriptase (Invitrogen) and oligo(dT)12-18 primers (Invitrogen) according to the manufacturer's instructions. Semiquantitative PCR was performed in a 25-μl volume using 50 pmol each of sense and antisense primers corresponding to LdTOP1DRL, LdTOP1DRS, and L. donovani glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by using the following profile: initial denaturation at 95°C for 5 min; 25 cycles with denaturation at 95°C for 1 min, annealing at 55°C for 40 s, and extension at 68°C for 40 s; and a final extension of 3 min. The primers were designed such that each set amplified a 300-bp fragment. Three separate reactions were carried out using three different RNA preparations in a 25-μl volume using SYBR Green SuperMix (Applied Biosystems) and the same primer sets in a 7300 real-time PCR system (Applied Biosystems). Reactions were carried out with the following profile: initial denaturation at 95°C for 5 min, followed by 35 cycles with denaturation at 95°C for 40 s, annealing at 60°C for 40 s, and extension at 65°C for 40 s. The PCR was followed by a melt curve analysis to ascertain that the expected products were amplified. The relative amounts of PCR products generated were obtained from the threshold cycle (CT), and amplification efficiencies were normalized by dividing the values by the relative amount of the GAPDH gene, used as a control. The level of expression was calculated as 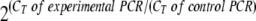 .
.
Western blotting.
The DIM-resistant Leishmania strain LdDR50 and WT L. donovani cells (3 × 107) were cultured for 12 h at 22°C with or without drugs. Nuclear fractions were isolated as described previously (16). Briefly, cells were suspended in hypotonic buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 0.1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine hydrochloride, and 5 mM dithiothreitol) and homogenized. The homogenate was centrifuged for 10 min at 10,000 × g. The pellet was washed and used as the source of the nuclear fraction. The nuclear fractions, after lysis with 1% sodium dodecyl sulfate (SDS), were subjected to SDS-polyacrylamide gel electrophoresis (12%), and the proteins were electrophoretically transferred to nitrocellulose membranes. Immobilized proteins were immunoblotted using rabbit antibodies raised against LdTOP1L (73 kDa) and LdTOP1S (29 kDa). Both polyclonal antibodies were diluted 1,000 times, and a horseradish peroxidase-conjugated secondary antibody was used to visualize the reactive band by a diaminobenzidine color reaction.
Cloning and construction of recombinant plasmids.
The full-length large-subunit gene (LdTOP1L) and small-subunit gene (LdTOP1S) of L. donovani topoisomerase IB were cloned into bacterial expression vector pET16b and transformed into Escherichia coli BL21(DE3) pLysS as described previously (6). Total cellular RNA was isolated from DIM-resistant L. donovani AG83 promastigotes (strain LdDR50) by using an RNA isolation kit (Roche Applied Science) according to the manufacturer's protocol. Reverse transcription-PCR (RT-PCR) was performed with gene-specific primers corresponding to a single open reading frame (ORF) of 1,905 bp (LdTOP1DRL) using sense primer 5′-GGAATTCCATATGATGAAGGTGGAGAATAGCAAG-3′, containing an NdeI site created at the initiation codon of the ORF, and antisense primer 5′-CGGGATCCCTACACCCTCAAAGCTGCAAG-3′, with a BamHI site immediately downstream from the termination codon. (Restriction endonuclease sites are underlined.) RT-PCR was performed with another set of gene-specific primers corresponding to a single ORF consisting of 786 bp (LdTOP1DRS): sense primer 5′-GGAATTCCATATGATGCAGCCTGTTCAAAGTCCT-3′, containing an NdeI site created at the initiation codon of the ORF, and antisense primer 5′-CGGGATCCTCAAAAATCGAAGTTCTCGGC-3′, with a BamHI site immediately downstream from the termination codon. The fragment amplified by RT-PCR was cloned into the NdeI/BamHI site of bacterial expression vector pET16b (with a six-His tag). The resultant constructs, pET16bLdTOP1DRL and pET16bLdTOP1DRS, were transformed into E. coli BL21(DE3) pLysS.
Overexpression and purification of recombinant proteins.
The recombinant constructs pET16bLdTOP1L, pET16bLdTOP1S, pET16bLdTOP1DRL, and pET16bLdTOP1DRS were separately transformed into Escherichia coli BL21(DE3) pLysS cells. The cells were grown in LB medium containing 0.4 M sorbitol to an A600 of 0.6 and were induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 22°C for 12 h. Cells harvested from 1 liter of culture were separately suspended in lysis buffer containing 1 mg/ml lysozyme and sonicated, and the proteins were purified using a Ni2+-nitrilotriacetic acid agarose column (Qiagen). Proteins were dialyzed and purified using a phosphocellulose column (P11; Whatman) as described previously (6). Finally, the purified proteins LdTOP1L, LdTOP1S, LdTOP1DRL, and LdTOP1DRS were stored at −70°C.
In vitro reconstitution of recombinant proteins.
Purified LdTOP1L was mixed with purified LdTOP1S and LdTOP1DRS, while LdTOP1S was mixed with purified LdTOP1DRL, and purified LdTOP1DRL was mixed with LdTOP1DRS, in a 1:1 molar ratio according to a standard protocol (6). The total protein concentration was 0.5 mg/ml in the standard reconstitution buffer containing 50 mM potassium phosphate (pH 7.5), 0.5 mM dithiothreitol, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, and 10% glycerol. The mixture was dialyzed at 4°C for 12 h, and the dialyzed proteins were used subsequently in all the assays.
DNA sequencing.
The LdTOP1DRL and LdTOP1DRS genes were sequenced with the primers described above by using an ABI Prism DNA sequencer (Perkin-Elmer, Norwalk, CT).
Site-directed mutagenesis.
Single mutations were introduced into the Leishmania heterodimeric topoisomerase I at positions Phy270 and Lys430 of the large subunit and Asp184 of the small subunit. Mutagenesis was performed by using the QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol. To carry out the desired mutations, pET16bLdTOP1L and pET16bLdTOP1S were used as templates for all mutagenesis experiments. For each mutation, the WT nucleotide was replaced using a specific pair of mutagenic primers. The following sense primers, along with their antisense counterparts (with codons in boldface and substitutions underlined), were used: for LdTOP1LF270L, 5′-CCGACATGGTGAAGTTGGAGAAGGCACGCAAGC-3′ and 5′-GCTTGCGTGCCTTCTC CAACTTCACCATGTCGG-3′; for LdTOP1LK430N, 5′-CCGGTCGACCCCAATTGGTCCACCGCGG-3′ and 5′-CCGCGGTGGACCAATT GGGGTCGACCGG-3′; and for LdTOP1SN184S, 5′-GGTGCGATCTTTCCCCAGTGACATCGGCAAGGC-3′ and 5′-GCCTTGCCGATGTCACTGGGGAAAGATCGCACC-3′. Mutations were confirmed through DNA sequencing in an ABI Prism DNA sequencer (Perkin-Elmer, Norwalk, CT).
Cloning of transfection constructs and transfection into Leishmania parasites.
LdTOP1DRL and LdTOP1DRS were PCR amplified using sense primers 5′-TCCCCCGGGATGAAGGTGGAGAATAGCAAG-3′, containing a SmaI site created at the initiation codon of the ORF, and 5′-ATAAGAATGCGGCCGCATGCAGCCTGTTCAAAGTCCT-3′, containing a NotI site created at the initiation codon of the ORF, respectively, along with antisense primers 5′-CGGGATCCCTACACCCTCAAAGCTGCAAG-3′ for LdTOP1DRL and 5′-CGGGATCCTCAAAAATCGAAGTTCTCGGC-3′ for LdTOP1DRS, each containing a BamHI site immediately downstream from the termination codon. (Restriction endonuclease sites are underlined.) The amplified fragments were cloned into the SmaI/BamHI site of the pXG-HYG vector (BD Biosciences) and the NotI/BamHI site of the pXG-GFP2+\ vector (a kind gift from S. M. Beverley) containing the green fluorescent protein (GFP) fragment. The transfection constructs pXG-HYG-LdTOP1DRL and pXG-GFP2+\LdTOP1DRS were electroporated according to the protocol in reference 15, and the transfected clones were obtained by selection in increasing concentrations of hygromycin B (50 to 300 mg/ml) for pXG-HYG-LdTOP1DRL and G418 (50 to 300 mg/ml) for pXG-GFP2+\LdTOP1DRS.
Plasmid relaxation assay.
The type I DNA topoisomerase was assayed by the decreased mobility of the relaxed isomers of supercoiled pBluescript SK(+) DNA in an agarose gel as described previously (16). The fluorescence of the supercoiled monomer DNA band after staining with ethidium bromide (0.5 μg/ml) was quantitated by integration using Gel Doc 2000 under UV illumination (Quantity One software; Bio-Rad).
Equilibrium cleavage assay.
The 25-mer oligonucleotide 1 (5′-GAAAAAAGACTT↓AGAAAAATTTTTA-3′) (with the downward-pointing arrow marking a cleavage site) was 5′ end labeled with [γ-32P]ATP and annealed with oligonucleotide 2 (5′-TAAAAATTTTTCTAAGTCTTTTTTC-3′) to make a duplex oligonucleotide containing a topoisomerase I binding motif as described previously (16). Cleavage was carried out using a 20-fold molar excess of the WT (LdTOP1LS) or mutant (LdTOP1LF270L/S, LdTOP1LK430N/S, or LdTOP1L/SN184S) enzyme over the duplex 25-mer DNA (enzymes, 0.2 μM; DNA, 10 nM). The reactions were carried out in the presence or absence of DIM at 37°C for 30 min. The samples were analyzed on a 12% denaturing polyacrylamide gel, followed by autoradiography as described previously (16).
Single-turnover cleavage and religation assay.
A 14-mer oligonucleotide containing a topoisomerase IB-specific cleavage site (5′-GAAAAAAGACTT↓AG-3′) was 5′ end labeled with 32P and annealed to a 25-mer oligonucleotide (3′-CTTTTTTCTGAATCTTTTTAAAAAT P-5′) as described previously (16). The suicidal cleavage reaction was carried out with 5 nM DNA substrate and 0.15 μM WT (LdTOP1LS) or mutant (LdTOP1LF270L/S or LdTOP1L/SN184S) enzyme in 20-μl reaction mixtures under standard assay conditions at 23°C for 4 h in the absence or presence of DIM as described previously (16). For religation experiments, covalent complexes generated by incubating the suicidal DNA substrate with the WT or mutant enzyme in the absence or presence of DIM (20 μM) were transferred to 30°C and preincubated for 2 min. The religation reaction was initiated by the addition of a 300-fold molar excess of the 11-mer religation acceptor oligonucleotide (5′-OH-AGAAAAATTTT-3′) to the same reaction mixture and incubation for the indicated times. Finally, all the reactions were stopped by the addition of SDS, and DNAs were subsequently precipitated by ethanol. Samples were digested with 5 μl of 1-mg/ml trypsin, electrophoresed in 12% denaturing polyacrylamide gels, and autoradiographed.
Equilibrium dialysis.
The equilibrium dialysis experiment was performed as described previously (16). The binding solution, as well as the buffer, used for equilibrium dialysis was 20 mM Tris-HCl (pH 7.5)-50 mM NaCl-10 mM MgCl2. Four sets of reactions were performed in 500 μl of binding solution containing 200 μM enzyme (LdTOP1DRLS, LdTOP1DRLF270L-K430N, LdTOP1DRLF270L, or LdTOP1DRSN184S, respectively) in each set and increasing concentrations of DIM (1, 5, 10, 15, and 20 μM) at 37°C for 30 min. The reaction mixture was loaded into the dialysis bag with a 12- to 14-kDa cutoff and placed in a 5-ml buffer solution. Corresponding “blanks” containing only the buffer solution or only the buffer-protein solution without any drug were run. The solutions were equilibrated for 24 h at 25°C with shaking at 100 rpm for the entire period. At the end of the equilibration, the concentration of DIM outside solutions was estimated by measuring the absorbance of the solution and using a molar extinction coefficient of 6.6 × 104 M−1 for DIM. Inside solutions could not be used, since protein interfered with the estimation. All the absorbance measurements were made at 25°C using a Bio-Rad SmartSpec 3000 UV spectrophotometer in a 10-mm-path-length cell.
RESULTS
Development of DIM-resistant parasites and accumulation of DIM in the cells of WT and resistant parasites.
In order to determine the DIM resistance mechanisms of Leishmania parasites, we selected the parental L. donovani parasites (AG83) for resistance to DIM in a step-by-step manner until they reached a resistance level of 42-fold over their WT counterparts. More specifically, a highly resistant L. donovani strain was selected by stepwise exposure to increasing concentrations of DIM (1, 2, 5, 10, 20, 40, and 50 μM) over a period of 1 year. The parasites were grown stably in the medium in the presence of 50 μM DIM and were designated LdDR50. Then LdDR50 parasites were cloned, and DIM was found to have similar 50% inhibitory concentrations for the single-cell clones. A representative clone was used for all subsequent assays. To compare their susceptibilities to DIM, we exposed freshly harvested parasites to increasing concentrations of the drug. The growth of LdDR50 parasites was hardly affected by DIM, even at the highest concentration used, whereas the growth of WT parasites was inhibited in a dose-dependent manner, with a 50% inhibitory concentration of 1.25 μM (Fig. 1A). This result clearly established the development of resistance mechanisms in LdDR50 to circumvent the cytotoxic effect of DIM. Because the resistance to topoisomerase I inhibitors is usually multifactorial in mammalian cells, we performed further experiments at different cellular levels to establish the potential mechanisms of resistance to DIM in L. donovani.
FIG. 1.

Effects of DIM on the growth of WT and LdDR50 parasites, and accumulation of DIM in both strains. (A) The growth of parasites (WT and strain LdDR50) was monitored in the presence of increasing concentrations (1, 5, 10, 15, and 20 μM) of DIM at 24 h. The level of proliferation of the WT Leishmania strain in the absence of any treatment was considered the maximal-growth control. Parasite growth was expressed as a percentage of the optical density of the control. Each result (percentage of the control value ± standard error) is representative of three experiments. (B) The accumulation of DIM was measured at given time intervals by spectrophotometric analysis at absorbance maxima of 280 nm as described in Materials and Methods. Data are means ± standard errors from three independent experiments.
One of the possible potential mechanisms of drug resistance in parasites is increased efflux or decreased influx. So for Leishmania parasites, one possible way to develop resistance to DIM could be to block the uptake or increase the efflux of DIM. Since DIM has an absorbance maximum at 280 nm, we tested these possibilities by measuring its accumulation inside the parasites. Moreover, spectrophotometric analysis of the drug kinetics was performed to determine whether the WT strain was more permeable to DIM than resistant parasites. As reported in Fig. 1B, no significant differences in permeability were detected between the WT strain and LdDR50, even at a high concentration of DIM (20 μM), suggesting no alteration in the accumulation of the drug in the resistant strain. Since DIM accumulation appears to remain same in the WT and resistant parasites, we further investigated the molecular aspects of topoisomerase I and its interaction with DIM.
ABC transporters are not involved in the resistance of parasites to DIM.
In CPT-resistant parasites, CPT accumulation decreased primarily through the ABC transporters present in the cell membrane. The ABCG6 gene is threefold overexpressed in that phenotype (1). Therefore, we investigated the involvement of ABC transporters in DIM resistance in parasites. Real-time RT-PCR quantitation revealed that the expression levels of ABCC3, ABCG4, and ABCG6 in DIM-resistant parasites were similar to those in WT parasites. Parasites resistant to CPT, sodium antimony gluconate (SAG), and miltefosine were used as positive controls. The ABCG6, ABCC3, and ABCG4 transporters were approximately threefold, fourfold, and threefold overexpressed in CPT-, SAG-, and miltefosine-resistant parasites, respectively (Fig. 2; see also Fig. S1A in the supplemental material), results consistent with our previous findings (1). The expression patterns of ABC genes were also compared for DIM-resistant cell types. No differences in the expression of the ABC genes at the level of mRNA were observed between DIM-resistant cells and WT cells. For all cell types, the expression of the GAPDH gene was observed to be equivalent, and it served as the loading control.
FIG. 2.

Gene expression in WT parasites and parasites resistant to DIM (LdDR50), CPT (Ld20C), SAG (LdGE1), or miltefosine (Ld30M). mRNA expression levels in WT, LdDR50, Ld20C, LdGE1, and Ld30M parasites were determined by real-time RT-PCR, carried out for 35 cycles using RNA isolated from these parasite strains. CT values were expressed accordingly.
Reduced expression of topoisomerase I in DIM-resistant Leishmania parasites.
The basis for DIM resistance in Leishmania parasites was studied by examining topoisomerase I gene expression at the mRNA and protein levels. Real-time RT-PCR analysis of total RNA extracted from exponentially growing WT Leishmania parasites and parasites resistant to DIM (LdDR50), CPT (Ld20C), SAG (LdGE1), or miltefosine (Ld30M) revealed 4.6-fold and 3-fold decreased expression of the large subunit and 4.3-fold and 3.7-fold decreased expression of the small subunit of mRNA in DIM-resistant and CPT-resistant parasites, respectively, compared to WT cells (Fig. 2; see also Fig. S1A in the supplemental material). There were no such changes in the expression of either subunit at the level of mRNA in SAG- or miltefosine-resistant parasites. For all cell types, the expression of the GAPDH gene was observed to be equivalent, and it served as the loading control. Western blot analysis was performed to assess the levels of topoisomerase I protein expression in both WT and LdDR50 cells (see Fig. S1B in the supplemental material). The expression of the large and small subunits of LdTOP1LS in LdDR50 cells was 4.0-fold and 3.8-fold less than that in WT cells. The mRNA expression results are consistent with the protein expression results, suggesting that the downregulation of topoisomerase I in DIM-resistant parasites occurs at the transcriptional level.
Topoisomerase I gene sequencing.
Two point mutations in the large subunit of L. donovani topoisomerase I confer CPT resistance (13). To address whether point mutations confer resistance, we cloned the genes from WT and mutant parasites by RT-PCR and sequenced them. By sequence alignment of the WT and DIM-resistant genes for topoisomerase I, two amino acid substitutions resulting from two single-nucleotide mutations were observed in LdTOP1DRL and one amino acid substitution resulting from one single-nucleotide mutation was observed in LdTOP1DRS (see Table S1 and Fig. S2 in the supplemental material). Two amino acids found in WT parasites, phenylalanine 270 (F270) and lysine 430 (K430), were mutated by site-directed mutagenesis to leucine (L) and asparagine (N) by C-to-G and A-to-T transitions in the LdTOP1DRL of LdDR50 cells, respectively. Based on the crystal structure of Leishmania topoisomerase I (7), we observed that F270 in the large subunit is located near the active-site tyrosine 222 of the small subunit (Fig. 3) and that the K430N mutation occurs far from the active-site tyrosine. This observation suggests that amino acid F270 of the large subunit mediates the interaction between DIM and the enzyme. In addition, a mutation of asparagine 184 (N184) to serine (S) was observed for the small subunit in DIM-resistant parasites due to an A-to-G transition (see Table S1 in the supplemental material). None of the three mutations F270L, K430N, and N184S are conserved in human topoisomerase I.
FIG. 3.

Close-up view of the active-site Tyr222 of L. donovani topoisomerase I from WT parasites based on the crystal structure of L. donovani topoisomerase I. The effect of the F270L mutation in the large subunit of L. donovani topoisomerase I was modeled using the Swiss-Pdb Viewer, version 3.7. (A) Diagram produced using RasMol. The structure of proteins (WT enzyme) is shown in a side view. The large subunit (LdTOP1L) is shown in sky blue, the small subunit (LdTOP1S) in gold, DNA in green, residue Tyr222 in magenta, and F270 in red. (B) Schematic representation of the domain structure of L. donovani topoisomerase I with mutated residues. Nt, N terminus; Ct, C terminus.
Transfection of WT L. donovani with mutated LdTOP1DRLS.
To investigate the potential resistance mechanism and to evaluate the contribution of the three mutations observed to DIM resistance, we cloned the mutated LdTOP1DRL and LdTOP1DRS genes into a Leishmania expression vector. We also performed site-directed mutagenesis on the LdTOP1L and LdTOP1S genes to obtain amino acid substitutions resulting in the formation of mutated LdTOP1L and LdTOP1S. As a control, we also transfected Leishmania parasites with overexpressed WT LdTOP1L and LdTOP1S in order to eliminate the possible effect of the endogenous enzyme in conferring resistance on the parasites. When WT parasites were transfected with LdTOP1DRLS containing three amino acid substitutions (F270L and K430N in the large subunit and N184S in the small subunit), they showed a higher level of DIM resistance, similar to that shown by LdDR50 parasites (Fig. 4). Furthermore, we found that transfection of WT parasites with an F270L (large subunit with Phe270Leu reconstituted with the WT small subunit), F270L K430N (large subunit with Phe270Leu and Lys430Asp reconstituted with the WT small subunit), or F270L N184S (large subunit with Phe270Leu reconstituted with the small subunit with Asp184Ser) mutant individually resulted in the formation of cells that also conferred strong levels of resistance to DIM. The growth of the resulting cells is inhibited by 34%, 34%, and 31%, respectively, at 20 μM DIM.
FIG. 4.

Effects of DIM on the growth of LdTOP1L and LdTOP1S mutant parasites. The parasites were grown in the presence of increasing concentrations of DIM (1 to 20 μM) for 24 h. WT parasites were transfected with mutated genes encoding LdTOP1DRLF270L/K430N/LdTOP1DRSN184S, LdTOP1DRLF270L/K430N/LdTOP1S, LdTOP1DRLF270L/LdTOP1DRSN184S, LdTOP1DRLK430N/LdTOP1DRSN184S, LdTOP1DRLF270L/LdTOP1S, LdTOP1DRLK430N/LdTOP1S, or LdTOP1L/LdTOP1DRSN184S. The proliferation of WT parasites in the absence of any treatment was considered the maximal-growth control. The growth of the parasites was expressed as a percentage of the optical density of the control.
Transfection of WT cells individually with (i) a K430N mutation in the large subunit and a WT small subunit (LdTOP1S), (ii) a K430N mutation in the large subunit and an N184S mutation in the small subunit, or (iii) a WT large subunit (LdTOP1L) and an N184S mutation in the small subunit did not induce significant resistance to DIM. The growth of these mutants was inhibited 94%, 86%, and 91%, respectively, in the presence of 20 μM DIM. These results, taken together, suggest that the F270L mutation in the large subunit is responsible for the acquisition of resistance to DIM by the parasites.
Mutant L. donovani topoisomerase I is insensitive to DIM.
To investigate the effect of DIM on resistant L. donovani topoisomerase I (LdTOP1DRLS), we cloned, overexpressed, and purified DIM-resistant mutated topoisomerase I and performed plasmid relaxation assays (Fig. 5) as described in Materials and Methods. WT recombinant LdTOP1LS was purified as described previously (6). The relaxation experiments were performed under standard assay conditions where the plasmid DNA and the enzyme (LdTOP1DRLS or LdTOP1LS) were mixed at a molar ratio of 3:1. Under this condition (ratio of DNA to LdTOP1DRLS, 3:1), DIM was used at increasing concentrations from 1 μM to 50 μM. There was no catalytic inhibition of LdTOP1DRLS in the presence of DIM (Fig. 5, lanes 4 to 8), whereas the WT enzyme, LdTOP1LS, was completely inhibited at 10 μM DIM (Fig. 5, lane 12). It can be inferred from these results that the DIM-resistant mutant enzyme LdTOP1DRLS is insensitive to DIM.
FIG. 5.

Effect of DIM on relaxation activity with the WT (LdTOP1LS) and mutated (LdTOP1DRLS) reconstituted enzymes. The relaxation of supercoiled pBS SK(+) DNA with reconstituted LdTOP1LS and LdTOP1DRLS was assayed at a molar ratio of 3:1. Lane 1, 90 fmol of pBS SK(+) DNA; lane 2, same as lane 1, but the DNA was simultaneously incubated with 30 fmol of LdTOP1DRLS for 30 min at 37°C; lane 3, same as lane 2 but in the presence of 4% DMSO; lanes 4 to 8, same as lane 2 but in the presence of 1, 5, 10, 20, or 50 μM DIM, respectively; lane 9, same as lane 1; lane 10, same as lane 9, but the DNA was simultaneously incubated with 30 fmol of LdTOP1LS for 30 min at 37°C; lane 11, same as lane 10 but in the presence of 4% DMSO; lane 12, same as lane 10 but in the presence of 10 μM DIM. The positions of the supercoiled monomer (SM) and the relaxed and nicked monomers (RL/NM) are indicated. The reaction products were resolved in a 1% agarose gel and visualized after ethidium bromide staining.
To investigate the interacting residues of the enzyme that bind with DIM, we purified the subunits generated by site-directed mutagenesis, reconstituted the enzymes, and performed a standard relaxation assay (Fig. 6) in the presence of DIM. The enzymes showed no sensitivity to DIM when the mutated subunits were reconstituted as follows: the large subunit with F270L plus K430N and the small subunit with N184S (Fig. 6, lane 6), the large subunit with F270L and the WT small subunit (lane 8), the large subunit with F270L plus K430N and the WT small subunit (lane 12), and the large subunit with F270L and the small subunit with N184S (lane 14). Moreover, DIM sensitivity was restored when the large subunit with K430N was reconstituted with the WT small subunit (Fig. 6, lane 10) or with the small subunit with N184S (lane 16) and when the WT large subunit was reconstituted with the small subunit with N184S (lane 18). These results suggest that F270 in the large subunit is responsible for the sensitivity of the enzyme to DIM.
FIG. 6.

Effects of DIM on the relaxation activities of WT and site-specific mutated topoisomerase I enzymes. The sensitivities to DIM of WT and mutant topoisomerase I enzymes were determined by a plasmid DNA relaxation assay. Lane 1, 90 fmol of pBS SK(+) DNA; lane 2, same as lane 1, but DNA was simultaneously incubated with 30 fmol of LdTOP1LS for 30 min at 37°C; lane 3, same as lane 2 but in the presence of 4% DMSO; lane 4, same as lane 2 but in the presence of 10 μM DIM. Lanes 5, 7, 9, 11, 13, 15, and 17, same as lane 1, but DNA was simultaneously incubated with 30 fmol of LdTOP1DRLS, LdTOP1DRLF270L/LdTOP1S, LdTOP1DRLK430S/LdTOP1S, LdTOP1DRLF270L-K430N/LdTOP1S, LdTOP1DRLF270L/LdTOP1DRSN184S, LdTOP1DRLK430N/LdTOP1DRSN184S, and LdTOP1L/LdTOP1DRSN184S, respectively, for 30 min at 37°C as indicated. Lanes 6, 8, 10, 12, 14, 16, and 18, same as lanes 5, 7, 9, 11, 13, 15, and 17, respectively, but in the presence of 10 μM DIM. The positions of the supercoiled monomer (SM) and the relaxed and nicked monomers (RL/NM) are indicated. The reaction products were resolved in a 1% agarose gel and visualized after ethidium bromide staining.
F270 is responsible for the DIM-induced stabilization of the cleavable complex.
To investigate the mechanism of DIM resistance in the mutant enzymes, the stability of the topoisomerase I-DNA covalent complex was analyzed using the 25-mer full duplex oligonucleotide substrate in the presence of increasing concentrations of DIM (5 to 20 μM). After 30 min of incubation, the reactions were stopped with SDS, the samples were treated with trypsin, and the products were analyzed by polyacrylamide-urea gel electrophoresis (Fig. 7). In the absence of DIM, the cleavage-religation equilibrium was shifted toward religation for the WT enzyme; little trapped cleavable complex was found, and most of the cleaved product was religated and migrated as an uncleaved 25-mer (Fig. 7A, lane 2). When the WT enzyme was exposed to increasing concentrations of DIM, the cleavage-religation equilibrium was shifted toward cleavage, as reported previously (16). The band corresponding to the 12-mer cleaved product is clearly detectable, and the band intensity increases with increasing concentrations of DIM (Fig. 7A, lanes 3 to 6). For the F270L mutant, the equilibrium shifted toward religation in the absence of DIM, as for the WT enzyme (Fig. 7B, lane 2). But in the presence of increasing concentrations of DIM, the equilibrium was not shifted toward cleavage; there are no bands of 12-mer cleaved products in the gel (Fig. 7B, lanes 3 to 6). This observation revealed that the F270L mutant is insensitive to DIM and that DIM is unable to stabilize its cleavable complex. Moreover, in the presence of increasing concentrations of DIM, the cleavage-religation equilibrium shifted toward cleavage for the K430N mutant, as for the WT enzyme (Fig. 7C). However, for the N184S mutant, the cleavage equilibrium also shifted to religation at lower concentrations of DIM, and stabilization of cleavage occurred only at higher concentrations (Fig. 7D, lane 6). These results indicate that the K430N mutation in the large subunit is not responsible for the stabilization of the cleavable complex but might play some role in DIM binding. Moreover, the N284S mutant enzyme has some role in stabilizing the formation of the cleavable complex only at the highest concentration (20 μM) of DIM.
FIG. 7.

Cleavage/religation equilibrium of the duplex oligonucleotide substrate. The cleavage reactions and electrophoresis in a denaturing polyacrylamide gel were performed with WT topoisomerase I and three mutant enzymes as described in Materials and Methods. (A) Lane 1, 10 nM 25-mer duplex oligonucleotides 5′ end labeled with γ-32P as indicated at the top. Lane 2, same as lane 1, but the oligonucleotides were incubated with 0.2 μM LdTOP1LS in the absence of DIM. Lanes 3 to 6, same as lane 2, but the oligonucleotides and LdTOP1LS were incubated with 5, 10, 15, and 20 μM DIM, respectively, for 30 min at 23°C. (B through D) The same experiments were performed with the F270L (B), K430N (C), and N184S (D) mutant enzymes, as indicated.
DIM cannot interfere with the religation step in the F270L mutant topoisomerase I reaction.
To investigate whether DIM interferes with the religation reaction carried out by DIM-resistant mutant topoisomerase I, we performed time-dependent single-turnover religation experiments in the absence and in the presence of DIM (20 μM) with mutant enzymes (Fig. 8). In these experiments, religation was studied by assaying the ability of the covalent intermediate to attach a 5′-OH-terminated 11-mer to the cleaved oligonucleotide, forming a 23-mer product. Since we have observed that the F270 residue is solely responsible for the DIM-induced stabilization of the cleavable complex, the religation reactions were performed with WT LdTOP1LS and mutant LdTOP1LF270L/S as described in Materials and Methods. Reactions were stopped at various time points by the addition of an equal volume of 1% SDS. The samples were treated with trypsin to remove all but a short trypsin-resistant peptide from the topoisomerase I-DNA covalent complex before analysis in a sequencing gel. The WT and mutant enzymes were incubated for 60 min with a suicide substrate in order to generate the cleavable complexes. Once cleavage had occurred, 11-mer religation oligonucleotides were added for different time periods in the absence or presence of 20 μM DIM to start the religation process. As shown in Fig. 8, in the absence of DIM, the religation rate of the F270L mutant enzyme (Fig. 8B) was higher than that of the WT enzyme (Fig. 8A). Moreover, the presence of DIM did not affect the religation rate of the F270L mutant (Fig. 8B, lanes 5 to 7), but it inhibited the religation of the WT enzyme (LdTOP1LS) (Fig. 8A, lanes 5 to 7), as reported previously (16). We also performed the religation experiment with the N184S mutant (Fig. 8C) and found that the religation reaction was inhibited in the presence of DIM (Fig. 8C, lanes 5 to 7), as with the WT enzyme. These results indicate that DIM is able to bind the WT enzyme and inhibit its religation process but is unable to stabilize the enzyme-DNA covalent complex, and fails to inhibit the religation process, for the DIM-resistant LdTOP1F270L/S mutant enzyme.
FIG. 8.

Effect of DIM on single-turnover cleavage and religation by WT and mutant enzymes. Shown are results of time course religation experiments with the 32P-5′-end-labeled suicide DNA substrate (14-mer/25-mer) as indicated and the WT enzyme (LdTOP1LS) (A) or the F270L (B) or N184S (C) mutant enzyme (C). The DNA substrate was incubated with WT or mutant topoisomerase I in the absence or presence of DIM (20 μM) at 23°C as described in Materials and Methods. Lanes 1, suicide DNA substrate with the enzyme. Lanes 2 to 4, same as lanes 1, but the enzyme was incubated with 11-mer religation oligonucleotides for 30, 60, and 120 s, respectively, in the absence of DIM. Lanes 5 to 7, same as lanes 2 to 4, respectively, but in the presence of DIM. All the reactions were stopped by the addition of 2% (wt/vol) SDS. The samples were precipitated with ethanol, digested with trypsin, and analyzed by denaturing polyacrylamide sequencing gel electrophoresis. The uncleaved suicidal oligonucleotide, the covalent complex, and the religation products are indicated.
The binding affinity of DIM for the WT enzyme is higher than that for the DIM-resistant mutant enzyme.
From the crystal structure of the large subunit of L. donovani topoisomerase I, it is found that F270 is close to the active-site tyrosine residue of the small subunit and is a DNA binding residue, present in the DNA binding core domain. To investigate the binding affinity of DIM for the mutant enzyme, we performed equilibrium dialysis experiments as described in Materials and Methods. The number of DIM molecules bound to the proteins was plotted against increasing concentrations of DIM (Fig. 9). The dissociation constant (KD) was calculated from the equilibrium dialysis experiments. The KD of DIM-LdTOP1DRLS is 2.4 × 10−4 M. Previously, we found that the KD of the WT enzyme, LdTOP1LS, bound to DIM was 9.7 × 10−9 M (16). From these results it was calculated that the affinity of DIM for LdTOP1DRLS is 2.5 × 104-fold less than that for the WT enzyme. This finding reveals that DIM interacts very weakly with the DIM-resistant mutant enzyme, perhaps explaining the insensitivity of the enzyme to DIM in the relaxation assays.
FIG. 9.

Saturation plots of interactions between different mutated proteins of LdTOP1LS and DIM by equilibrium dialysis. Equilibrium dialysis experiments were performed as described in Materials and Methods. Shown are plots of the amount of ligand (DIM) bound to the mutated enzyme LdTOP1DRLS (A), LdTOP1DRLF270L-K430N (B), LdTOP1DRLF270L (C), or LdTOP1DRSN184S (D) versus the total ligand concentration (1, 5, 10, 15, and 20 μM).
To find out the differential affinity of DIM for the mutated subunits of LdTOP1LS, we also investigated the binding of both the mutated subunits to DIM by equilibrium dialysis experiments. The numbers of DIM molecules bound by LdTOP1LF270L-K430N/SN184S, LdTOP1LF270L-K430N, LdTOP1LF270L, and LdTOP1SN184S were plotted against increasing concentrations of DIM (Fig. 9). The binding constants of DIM-LdTOP1LF270L-K430N/SN184S (Fig. 9A), DIM-LdTOP1LF270L-K430N (Fig. 9B), DIM-LdTOP1LF270L (Fig. 9C), and DIM-LdTOP1SN184S (Fig. 9D) were 2.4 × 10−5 M, 7.6 × 10−4 M, 5.3 × 10−4 M, and 3.8 × 10−4 M, respectively. It was reported previously that the binding constants of DIM-LdTOP1L and DIM-LdTOP1S were 4.28 × 10−7 M and 5 × 10−8 M, respectively (16). The results obtained here suggest that the affinity of DIM for the large subunit is reduced 1.2 × 103-fold by the F270L mutation and 7.6 × 103-fold by the N184S mutation. Therefore, the results indicate that the low affinity of DIM for the mutated holoenzyme is due to the F270L mutation of the large subunit and the N184S mutation of the small subunit.
DISCUSSION
The structure-function analysis of the DIM-topoisomerase I interaction might be exploited in the development of rational approaches to chemotherapy for human leishmaniasis and also in the study of drug resistance. Resistance to traditional first-line drugs underlines the urgent need for new treatments. The unusual bisubunit topoisomerase I of Leishmania parasites is an attractive chemotherapeutic target. Various topoisomerase I inhibitors have been tested for their efficacies as antileishmanial agents. Recently, we have reported that DIM is a potent noncompetitive topoisomerase I inhibitor (16).
The possible potential mechanisms of drug resistance in a population of parasites include the following: (i) differential selection of resistant individuals from a mixed population, (ii) physiological adaptations, such as increased efflux or decreased influx, (iii) conversion of the drug to an inactive form by an enzyme, (iv) changes in gene expression, and (v) spontaneous mutations followed by selection. Hence, by developing DIM-resistant Leishmania parasites, a better understanding of the various mechanisms of development of chemoresistance can be obtained. Because the Leishmania plasma membrane represents the first cellular barrier that DIM encounters before reaching its specific target, we determined the efficiency of the drug at accumulating inside WT and resistant parasites in order to verify differences in the kinetics of DIM transport. The experiment on the transport of DIM in LdDR50 and WT parasites revealed no alteration in drug accumulation for the resistant parasites. Eukaryotic ABC transporters are involved in the translocation of various substances across the cell membrane. Drug resistance arising out of altered influx or efflux of drugs involves different classes of ABC transporters. Resistance to SAG is manifested by overexpression of the ABCC3 transporter, while overexpression of the ABCG4 transporter is involved in miltefosine resistance and overexpression of the ABCG6 transporter is involved in CPT resistance, in Leishmania parasites (1).
In the present study, we have characterized the mechanism of resistance of Leishmania parasites to DIM. LdDR50 parasites were about 42-fold more resistant to DIM than the WT strain. As with other drugs, resistance to DIM might involve alterations in cellular drug accumulation, the drug target, or the response to the drug-target interaction. The present study indicates that ABC transporters were not overexpressed in DIM-resistant Leishmania parasites. However, this study shows that without changes in the drug-trafficking pathway, these parasites develop a chemoresistance phenotype by harboring random mutations in the target genes.
To further study the mechanisms for the development of resistance in LdDR50, a series of quantitative and qualitative assays for topoisomerase I was performed. Real-time RT-PCR and Western blot analysis showed lower expression of the large and small subunits at both the mRNA and protein levels in LdDR50 parasites than in their WT counterparts. This result was consistent with that of a previous report where expression of topoisomerase I was reported to be decreased in CPT-resistant tumor cell lines (19). Moreover, the study with CPT-resistant Leishmania parasites identified two point mutations in the large-subunit gene of LdTOP1LS (13). To address this possibility, we sequenced both the large- and small-subunit genes (LdTOP1L and LdTOP1S) of DIM-resistant parasites. LdDR50 parasites had two amino acid substitutions (F270L and K430N) in the core domain of LdTOP1L and one amino acid substitution (N184S) in LdTOP1S. Based on mutations that we have identified in both the large and the small subunit, we hypothesized that these amino acid residues of the enzyme are crucial for the cytotoxic action of DIM against Leishmania parasites. Despite substantial resistance conferred by each amino acid substitution individually, site-directed mutagenesis studies indicated that all three substitutions were required to reach the highest level of the DIM-resistant phenotype. From a homology modeling study of L. donovani topoisomerase I and human topoisomerase I, we determined that F270 and K430 of LdTOP1L and N184 of LdTOP1S are equivalent to Y448, D614, and K642 of human topoisomerase I, respectively, residues that are predicted to interact with the essential indole moieties of DIM. The crystal structure of L. donovani topoisomerase I (7) revealed that K430 is quite far from the active-site tyrosine (Tyr222) of the small subunit, which is involved in cleavage activity, but that F270 is very close to the active-site Tyr222. The distance between the carbon of F270 and the closest atom of Tyr222, the phenolic oxygen atom, is 3.28 Å. F270 plays a role in stabilizing the phenolic anion during the attack on the scissile phosphate group. The distance between the carbon atom of F270 and the phosphate of bound DNA is 7.41 Å. Moreover, in a previous study of a CPT-11-resistant human lung cancer cell line, it was demonstrated that a mutation in residue 729, near the active site of the enzyme, is associated with decreased sensitivity (12). The equilibrium cleavage/religation experiment revealed that K430 has no role in the DIM-induced stabilization of the covalent complex, whereas this stabilization is inhibited for the F270L mutant enzyme compared to the WT enzyme. The results presented here provide a molecular explanation for the DIM resistance of the mutants of topoisomerase I.
The religation experiment indicates that DIM strongly inhibits the religation process for the native enzyme but it is inefficient at inhibiting religation for a mutant enzyme (F270L). Moreover, in the absence of DIM, the religation process of the mutant enzyme is faster than that of WT. Previously it was reported that the Ala653Pro mutation of human topoisomerase I exhibits enhanced DNA strand religation and inhibits the CPT-induced formation of the topoisomerase I-DNA cleavable complex (5). Based on these findings, we hypothesize that the increased religation rate of the F270L mutant enzyme results from conformational changes within the topoisomerase I catalytic pocket that enhance the binding of DNA to the covalent complex. In particular, mutation of the highly flexible phenylalanine might extend the α-helical structure of residues N-terminal to the active-site tyrosine, as seen in the structure shown in Fig. 3. The insensitivity of the F270L mutant to DIM might be due to the high affinity of DIM for the phenyl ring of the aromatic residue F270 in the WT enzyme. Our studies also suggest that mutation-induced alterations in the enzyme active-site architecture coincide with decreased DIM sensitivity.
In conclusion, we have shown for the first time the mechanism of DIM resistance in Leishmania parasites and the occurrence of a novel point mutation, F270L, in the large subunit. F270 is very close to the active-site tyrosine and is responsible for DIM-induced cleavage. The inability to induce topoisomerase I-DNA cleavable complexes with the DIM-resistant topoisomerase I enzyme demonstrates that DIM is a promising non-CPT topoisomerase I inhibitor for therapeutic development and a useful reagent for mapping topoisomerase I cleavage complexes in the Leishmania genome. Interestingly, this is the first report concerning the mechanism of resistance to DIM in any living organism. Moreover, we have identified three topoisomerase I mutation sites conferring resistance to DIM. These mutations provide new insight into the molecular interactions between DIM and the enzyme. This study advances our understanding of the mechanism of cell poisoning by the dietary phytochemical compound DIM and suggests specific modifications to the drug that may improve the efficacy of the molecule.
Supplementary Material
Acknowledgments
We thank S. Roy, director of the Indian Institute of Chemical Biology (IICB), Kolkata, India, for his interest in this work.
This work was supported by grants from Network Project NWP-38 of the Council of Scientific and Industrial Research (CSIR), Government of India, and the Department of Biotechnology; Government of India (BT/PR6399/BRB/10/434/2005), to H.K.M. A.R. was supported by a Senior Research Fellowship from the CSIR, Government of India.
Footnotes
Published ahead of print on 30 March 2009.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.BoseDasgupta, S., A. Ganguly, A. Roy, T. Mukherjee, and H. K. Majumder. 2008. A novel ATP-binding cassette transporter, ABCG6 is involved in chemoresistance of Leishmania. Mol. Biochem. Parasitol. 158:176-188. [DOI] [PubMed] [Google Scholar]
- 2.Champoux, J. J. 1976. Evidence for an intermediate with a single-strand break in the reaction catalyzed by the DNA untwisting enzyme. Proc. Natl. Acad. Sci. USA 73:3488-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Champoux, J. J. 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70:369-413. [DOI] [PubMed] [Google Scholar]
- 4.Chang, J. Y., J. F. Liu, S. H. Juang, T. W. Liu, and L. T. Chen. 2002. Novel mutation of topoisomerase I in rendering cells resistant to camptothecin. Cancer Res. 62:3716-3721. [PubMed] [Google Scholar]
- 5.Chrencik, J. E., B. L. Staker, A. B. Burgin, P. Pourquier, Y. Pommier, L. Stewart, and M. R. Redinbo. 2004. Mechanisms of camptothecin resistance by human topoisomerase I mutations. J. Mol. Biol. 339:773-784. [DOI] [PubMed] [Google Scholar]
- 6.Das, B. B., N. Sen, A. Ganguly, and H. K. Majumder. 2004. Reconstitution and functional characterization of the unusual bi-subunit type I DNA topoisomerase from Leishmania donovani. FEBS Lett. 565:81-88. [DOI] [PubMed] [Google Scholar]
- 7.Davies, D. R., A. Mushtaq, H. Interthal, J. J. Champoux, and W. G. Hol. 2006. The structure of the transition state of the heterodimeric topoisomerase I of Leishmania donovani as a vanadate complex with nicked DNA. J. Mol. Biol. 357:1202-1210. [DOI] [PubMed] [Google Scholar]
- 8.Eng, W. K., F. L. McCabe, K. B. Tan, M. R. Mattern, G. A. Hofmann, R. D. Woessner, R. P. Hertzberg, and R. K. Johnson. 1990. Development of a stable camptothecin-resistant subline of P388 leukemia with reduced topoisomerase I content. Mol. Pharmacol. 38:471-480. [PubMed] [Google Scholar]
- 9.Fiorani, P., A. Bruselles, M. Falconi, G. Chillemi, A. Desideri, and P. Benedetti. 2003. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 278:43268-43275. [DOI] [PubMed] [Google Scholar]
- 10.Fiorani, P., G. Chillemi, C. Losasso, S. Castelli, and A. Desideri. 2006. The different cleavage DNA sequence specificity explains the camptothecin resistance of the human topoisomerase I Glu418Lys mutant. Nucleic Acids Res. 34:5093-5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanzawa, F., Y. Sugimoto, K. Minato, K. Kasahara, M. Bungo, K. Nakagawa, Y. Fujiwara, L. F. Liu, and N. Saijo. 1990. Establishment of a camptothecin analogue (CPT-11)-resistant cell line of human non-small cell lung cancer: characterization and mechanism of resistance. Cancer Res. 50:5919-5924. [PubMed] [Google Scholar]
- 12.Kubota, N., F. Kanzawa, K. Nishio, Y. Takeda, T. Ohmori, Y. Fujiwara, Y. Terashima, and N. Saijo. 1992. Detection of topoisomerase I gene point mutation in CPT-11 resistant lung cancer cell line. Biochem. Biophys. Res. Commun. 188:571-577. [DOI] [PubMed] [Google Scholar]
- 13.Marquis, J. F., I. Hardy, and M. Olivier. 2005. Topoisomerase I amino acid substitutions, Gly185Arg and Asp325Glu, confer camptothecin resistance in Leishmania donovani. Antimicrob. Agents Chemother. 49:1441-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pommier, Y., P. Pourquier, Y. Urasaki, J. Wu, and G. S. Laco. 1999. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist. Updat. 2:307-318. [DOI] [PubMed] [Google Scholar]
- 15.Robinson, K. A., and S. M. Beverley. 2003. Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol. Biochem. Parasitol. 128:217-228. [DOI] [PubMed] [Google Scholar]
- 16.Roy, A., B. B. Das, A. Ganguly, S. BoseDasgupta, N. V. Khalkho, C. Pal, S. Dey, V. S. Giri, P. Jaisankar, S. Dey, and H. K. Majumder. 2008. An insight into the mechanism of inhibition of unusual bi-subunit topoisomerase I from Leishmania donovani by 3,3′-di-indolylmethane, a novel DNA topoisomerase I poison with a strong binding affinity to the enzyme. Biochem. J. 409:611-622. [DOI] [PubMed] [Google Scholar]
- 17.Roy, A., A. Ganguly, S. BoseDasgupta, B. B. Das, C. Pal, P. Jaisankar, and H. K. Majumder. 2008. Mitochondria dependent ROS-mediated programmed cell death (PCD) induced by 3,3′-diindolylmethane (DIM) through inhibition of FoF1-ATP synthase in unicellular protozoan parasite Leishmania donovani. Mol. Pharmacol. 74:1292-1307. [DOI] [PubMed] [Google Scholar]
- 18.Staker, B. L., K. Hjerrild, M. D. Feese, C. A. Behnke, A. B. Burgin, Jr., and L. Stewart. 2002. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 99:15387-15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugimoto, Y., S. Tsukahara, T. Oh-hara, T. Isoe, and T. Tsuruo. 1990. Decreased expression of DNA topoisomerase I in camptothecin-resistant tumor cell lines as determined by a monoclonal antibody. Cancer Res. 50:6925-6930. [PubMed] [Google Scholar]
- 20.Tan, K. B., M. R. Mattern, W. K. Eng, F. L. McCabe, and R. K. Johnson. 1989. Nonproductive rearrangement of DNA topoisomerase I and II genes: correlation with resistance to topoisomerase inhibitors. J. Natl. Cancer Inst. 81:1732-1735. [DOI] [PubMed] [Google Scholar]
- 21.Wang, J. C. 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 3:430-440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.