

Abstract
The activities of protease inhibitors in vivo may depend on plasma concentrations and viral susceptibility. This nonrandomized, open-label study evaluated the relationship of the inhibitory quotient (IQ [the ratio of drug exposure to viral phenotypic susceptibility]) to the human immunodeficiency virus type 1 (HIV-1) viral load (VL) change for ritonavir-enhanced protease inhibitors (PIs). Subjects on PI-based regimens replaced their PIs with ritonavir-enhanced indinavir (IDV/r) 800/200 mg, fosamprenavir (FPV/r) 700/100 mg, or lopinavir (LPV/r) 400/200 mg twice daily. Pharmacokinetics were assessed at day 14; follow-up lasted 24 weeks. Associations between IQ and VL changes were examined. Fifty-three subjects enrolled, 12 on IDV/r, 33 on FPV/r, and 8 on LPV/r. Median changes (n-fold) (FC) of 50% inhibitory concentrations (IC50s) to the study PI were high. Median 2-week VL changes were −0.7, −0.1, and −1.0 log10 for IDV/r, FPV/r, and LPV/r. With FPV/r, correlations between the IQ and the 2-week change in VL were significant (Spearman's r range, −0.39 to −0.50; P ≤ 0.029). The strongest correlation with response to FPV/r was the IC50 FC (r = 0.57; P = 0.001), which improved when only adherent subjects were included (r = 0.68; P = 0.001). In multivariable analyses of the FPV/r arm that included FC, one measure of the drug concentration, corresponding IQ, baseline VL, and CD4, the FC to FPV was the only significant predictor of VL decline (P < 0.001). In exploratory analyses of all arms, the area under the concentration-time curve IQ was correlated with the week 2 VL change (r = −0.72; P < 0.001). In conclusion, in PI-experienced subjects with highly resistant HIV-1, short-term VL responses to RTV-enhanced FPV/r correlated best with baseline susceptibility. The IQ improved correlation in analyses of all arms where a greater range of virologic responses was observed.
Ritonavir (RTV)-enhanced protease inhibitors (PIs) are a cornerstone of therapy for human immunodeficiency virus (HIV)-infected treatment-experienced patients with resistant virus (8, 11, 12, 17, 19). The antiretroviral response to RTV-enhanced PIs is related to the susceptibility of the individual's virus to the specific agent. Multiple studies of PIs have demonstrated a relationship between the drug concentration and the virologic response (reviewed in reference 5), although whether there is an upper threshold of virus resistance that cannot be overcome with increased PI concentration is not known.
Measured drug exposure may enhance the predictive value of virus susceptibility as assessed, for example, by the 50% inhibitory concentration (IC50). A ratio of drug exposure to virus susceptibility, termed the inhibitory quotient (IQ) ratio, may predict the short- and long-term virologic responses to PI-based therapy better than susceptibility alone. Conflicting results have been reported, with some studies demonstrating no added benefit to incorporating pharmacokinetics information (reviewed in references 13 and 16). In studies of lopinavir-RTV (LPV/r), an independent relationship between IQ and outcome has been seen in some studies (20), but not others (4, 14, 15). Published studies of amprenavir (APV) or fosamprenavir (FPV [APV's prodrug]) IQs have been small (10). One study showed an independent relationship between an APV genotype-based IQ and virologic response (21), two showed that the IQ did not add significantly to baseline genotypic resistance (7, 26), and a third showed a correlation with the 2-week viral load (VL) response that was lost when a single outlier was removed from the analysis (4).
Additional rigorous prospective evidence is needed to test whether the IQ adds additional predictive information to baseline resistance testing of antiretroviral response in patients with substantial PI resistance. Such data would support the use of PI concentrations for monitoring antiretroviral therapy. We hypothesized that the ratio of the PI concentration over the susceptibility of an individual's virus to that PI could predict antiretroviral response more robustly than HIV type 1 (HIV-1) drug susceptibility alone in a short-term, focused clinical trial. In this study, our objective was to evaluate the correlation between the IQ ratios (concentration at 12 h [C12]/IC50) for each PI and the short- and long-term antiretroviral activities of three RTV-enhanced PIs in individuals who had failed previous PI-based regimens.
(The results of this study were presented in part in abstract/poster L-123 at the 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO, 5 to 8 February 2006 [ClinicalTrials.gov identifier NCT00027339].)
MATERIALS AND METHODS
Study design, subject selection, and dosing.
This was a phase II, nonrandomized, open-label, parallel-arm study conducted at 16 sites in the United States. Antiretroviral-experienced, HIV-infected subjects were eligible if they were ≥18 years of age, had plasma HIV-1 RNA levels of >2,500 copies/ml, had received a PI-based regimen for ≥12 weeks immediately before the study, had ≥48 weeks of PI-based treatment experience, had safety laboratory values within prespecified ranges, and had phenotypic confirmation within the prior 60 days of decreased susceptibility (>2.5-fold) to at least two of three PIs to be used in the study. Exclusion criteria included recent receipt of HIV vaccines, investigational drugs, immunotherapies, hydroxyurea, or significant renal disease. The study was approved by the Institutional Review Board at each study site. All subjects gave written informed consent.
At study entry, the subjects discontinued their PIs, continued all other background antiretrovirals, and began a 14-day period on one of the following treatments: arm A, indinavir 800 mg plus RTV 200 mg (IDV/r) twice daily (BID); arm B, FPV 700 mg plus RTV 100 mg (FPV/r) BID; or arm C, LPV/r 400 mg/100 mg BID plus additional RTV 100 mg BID (LPV/r/r). Subjects who received IDV/r or LPV/r immediately before entry were ineligible for arm A or arm C, respectively, whereas subjects on APV/RTV or FPV/r prestudy were ineligible for arm B. On day 15, tenofovir disoproxil fumarate was added, and background therapy could be optimized. Enfuvirtide (Fuzeon) and/or a nonnucleoside reverse transcriptase inhibitor was allowed. Treatment continued for 24 weeks. IDV 800 mg was supplied as two 400-mg Crixivan capsules (Merck & Co., Inc., Whitehouse Station, NJ), LPV/r 400 mg/100 mg as Kaletra capsules (Abbott Laboratories, North Chicago, IL) containing LPV/r 133.3 mg/33.3 mg, FPV 700 mg as Lexiva 700-mg tablets (GlaxoSmithKline, Research Triangle Park, NC), and RTV as 100-mg Norvir capsules (Abbott Laboratories, North Chicago, IL). The planned sample size was 36 subjects per arm to detect a minimum correlation of 0.5 between the IQ and the VL response with ≥80% power at a 0.05 level of significance.
Pharmacokinetics analysis.
On day 14, blood samples were collected just before the morning dose (zero hour) and at 1, 2, 3, 6, 9, and 12 h postdose. The samples were analyzed using validated liquid chromatography-tandem mass spectrometry with lower limits of quantification for IDV, APV, LPV, and RTV of 10, 44, 40, and 25 ng/ml, respectively. The pharmacokinetics parameters, assessed for each PI by standard noncompartmental methods included the area under the concentration-time curve from 0 to 12 h postdose (AUC0-12), C12, the maximum plasma concentration (Cmax), the minimum plasma concentration (Cmin), time to Cmax (Tmax), and the trough concentration obtained immediately prior to the morning dose (Cτ). Cτ was also determined at study weeks 4, 8, and 24, prior to an observed dose. AUC0-12 was calculated by the linear trapezoidal method. Protein binding (PB) was assessed by equilibrium dialysis for APV and LPV on samples taken twice on day 14, at the estimated peak concentration (2 h postdose for APV and 6 h postdose for LPV), at 12 h, and at week 24 using validated assays with interday and intraday variation of ≤12%. The percentage of free drug was then calculated. The individual free-drug-adjusted C12 and AUC0-12 were determined based on the average of the two free-drug percentages multiplied by the total C12 and AUC0-12 values obtained at week 2.
Pharmacodynamic analysis.
Before entry, the subjects had an antiretroviral phenotype obtained using the PhenoSense assay (Monogram Biosciences). The resistance parameters included the measured IC50 and the IC50 change (n-fold) (FC). For each subject, six IQs were calculated based on their pharmacokinetics and resistance results. Four sets were based on the absolute IC50 either without a correction for PB (IQnIC) or with different correction factors, including multiplication of the IC50 by the attenuation by 50% human serum (IQcIC) (17a), multiplication of the pharmacokinetics parameter by population PB (IQpIC) (2, 15a, 18, 24), and, for LPV and APV, multiplication by the actual measured individual free-drug percentage for the individual subject (IQfIC). Two sets were based on FC, in which a wild-type IC50 (the mean IC50 of three standard variants measured in 50% human serum [22]) was multiplied by the FC from the PhenoSense assay. One FC IQ used the total measured drug (IQFC); the other, for LPV and APV, multiplied the pharmacokinetics parameter by the measured individual free-drug percentage (IQfFC). These IQs were calculated using one of several pharmacokinetics parameters, including C12, AUC0-12, and Cτ.
Efficacy analysis.
VLs were obtained at screening (day −60), preentry (day −14), entry (day 0), days 7 and 14 (two values), and weeks 4, 8, 16, and 24 (Roche UltraSensitive assay; lower limit of quantification, 50 copies/ml). The baseline VL was the geometric mean of the screening, preentry, and entry values. CD4 and CD8 cell counts were obtained at preentry, entry, and weeks 2, 8, and 24.
Safety analysis.
Adverse events, including signs, symptoms, and laboratory abnormalities, were graded using the standardized Division of AIDS, National Institute of Allergy and Infectious Disease, toxicity grading criteria. Safety analyses focused on grade 3 (severe) and higher adverse events.
Adherence analysis.
Adherence was measured using the electronic medication event monitoring system (MEMS Cap) attached to RTV bottles.
Statistical methods. (i) Pharmacokinetics data analysis.
Differences in PI Cτ levels within arms were compared using the Kruskal-Wallis test. A paired comparison of Cτ levels between weeks 2 and 24 for arm B (FPV) was also performed using the Wilcoxon signed-rank test. For FPV and LPV, differences in percentages of free APV and LPV between pairs of scheduled times were evaluated using the Wilcoxon signed-rank test. PI AUC0-12, C12, Cmax, and Cτ were compared for subjects in each treatment arm who experienced toxicities of grade <3 with values for subjects with toxicities of grade ≥3 using the Wilcoxon rank-sum test.
(ii) Pharmacodynamic-efficacy relationship data analysis.
For each PI, the resistance markers among the treatment arms were compared by the Kruskal-Wallis test. Correlation of short-term (day 14) and long-term (week 24) VL changes from baseline with IQ ratios, IC50, FC, AUC0-12, C12, Cmax, and Cτ were tested for significance using Spearman's rank correlation test and simple linear regression. Differences in CD4 and CD8 cell count changes between weeks 2 and 24 were evaluated by the Wilcoxon signed-rank test.
(iii) Efficacy-adherence relationship data analysis.
The significance of correlations between VL changes and adherence was evaluated using Spearman's rank correlation test and linear regression analysis. All statistical tests were two-tailed using a 0.05 level of significance.
RESULTS
Baseline characteristics, resistance, and treatment disposition.
Fifty-three subjects enrolled in the study: 12 on IDV/r, 33 on FPV/r, and 8 on LPV/r/r. The subjects were racially and ethnically diverse: 30% were white, 36% black, and 30% Hispanic. Eleven percent (6/53) were women. In the IDV/r, FPV/r, and LPV/r/r arms, the median baseline plasma HIV-1 RNAs were 5.0, 4.6, and 4.3 log10 copies/ml; the CD4 counts were 43, 142, and 287 cells/mm3; the times from initiation of first antiretroviral therapy to study entry were 6.6, 8.4, and 7.1 years; the numbers of previous PIs were 4, 5, and 3; and the median total years of prior PI treatment were 6.6, 7.2, and 5.3, respectively.
The subjects generally had highly PI-resistant virus. The baseline median FCs for LPV were 157 in the IDV/r arm, 79 in the FPV/r arm, and 20.5 in the LPV/r arm; for IDV, they were 33, 42, and 16 in these arms, respectively; and for FPV, they were 72.5, 12, and 37, respectively. The treatment arm was chosen by local investigators based on preentry resistance testing, and FCs differed significantly among the treatment arms (P < 0.02). In each arm, more than 50% of the subjects had FCs of >10 to the chosen agent.
Forty seven (89%) subjects completed the protocol on their assigned PI (94% [31/33] in the FPV/r arm). Three subjects (one in each arm) did not complete the 24-week protocol. Optimized regimens included a median of 2.5 nonnucleoside reverse transcriptase inhibitors. Only five subjects, all in the FPV/r arm, initiated enfuvirtide as a new agent.
Pharmacokinetics.
Forty-nine subjects had 12-hour pharmacokinetics assessments at day 14, with results from two subjects each missing in the IDV/r and FPV/r arms. The data for the FPV/r arm are in Table 1. Median APV Cτ values for the FPV/r arm were stable over time and similar to the day 14 median C12 value. Within-subject APV Cτ values were also stable over time (data not shown). The median C12 (ng/ml) for IDV (n = 10) was 806.0 (interquartile range [IQR], 469.00, 1,140.00), and for LPV (n = 8), the median was 6,880.0 (IQR, 5,914.00, 9,235.00).
TABLE 1.
Pharmacokinetics parameters at day 14 and trough concentrations over time for the FPV/r arma
| Parameter | n | Median | IQR |
|---|---|---|---|
| AUC(0-12) (ng · h/ml) | 31 | 35,370 | 26,622, 51,740 |
| C12 (ng/ml) | 2,060.00 | 1,300.00, 2,850.00 | |
| Cmax (ng/ml) | 4,970.00 | 3,960.00, 7710.00 | |
| Tmax (h) | 3.00 | 2.00, 6.00 | |
| Cτ (ng/ml) | |||
| Week 2 | 25 | 2,450 | 1,910, 3,100 |
| Week 4 | 21 | 2,330 | 1,780, 3,480 |
| Week 8 | 21 | 2,480 | 1,890, 3,740 |
| Week 24 | 20 | 2,020 | 1,475, 2,855 |
The longitudinal data for IDV and LPV are not shown due to the limited sample sizes for these arms. Pharmacokinetics data following FPV/r dosing refer to APV.
For the FPV/r and LPV/r arms, the percentages of drug free of PB are shown in Table 2, along with the protein-adjusted AUC0-12s for FPV and LPV. The percentages free for each drug were similar across time points. For FPV, the percentage of free drug at peak was slightly but significantly higher than at 12 h (P = 0.011; Wilcoxon signed-rank test). Values from individual subjects at Cmax and Cτ were correlated (r = 0.61; P = 0.001). The percentage free at the week 24 Cτ was not significantly different than at 12 h on day 14, and the values were highly correlated (r = 0.86; P < 0.001), with the estimated linear regression line virtually overlapping a 45° straight line representing one-to-one correlation (data not shown).
TABLE 2.
Percentages of drug free of PB and PB-adjusted AUCa
| Arm | Sampling time | % Free drug (not protein bound)b
|
||
|---|---|---|---|---|
| n | Median | Range (minimum, maximum) | ||
| FPV/r | Week 2, h 2 | 28 | 8.36 | 4.77, 13.01 |
| Week 2, h 12 | 31 | 7.53 | 5.10, 11.58 | |
| Week 24, trough | 23 | 6.60 | 3.93, 13.25 | |
| Overall | 82 | 7.87 | 3.93, 13.25 | |
| LPV/r | Week 2, h 6 | 8 | 1.03 | 0.64, 1.51 |
| Week 2, h 12 | 8 | 0.94 | 0.56, 1.15 | |
| Week 24, trough | 5 | 1.03 | 0.46, 1.18 | |
| Overall | 21 | 1.01 | 0.46, 1.51 | |
The PB-adjusted AUC0-12 values (ng · h/ml) for the FPV/r (n = 30) and LPV/r (n = 8) arms [median (range)], respectively, were as follows: 287.7 (153.4, 717.4) and 110.4 (67.2, 151.2).
The longitudinal data for IDV are not shown, as % free drug was not measured for IDV arms. Pharmacokinetics data following FPV/r dosing refer to APV.
IQ.
IQ values using day 14 C12 concentrations (Table 3) were highest when PB was ignored and varied between treatment arms when different methods were used to correct for PB. The values were most consistent between arms when estimates for free-drug percentages from population studies were used (C12_IQpIC), and these IQs were similar to values obtained when measured individual-subject free-drug concentrations were used (C12_IQfIC) for the FPV/r and LPV/r arms.
TABLE 3.
IQs
| Method | Arm | IQ
|
Correlation coefficient of IQ with VL change at wk 2 | P | ||
|---|---|---|---|---|---|---|
| n | Median | Range | ||||
| C12_IQnICa | IDV/r | 10 | 9.091 | (1.870, 61.495) | 0.02 | 0.96 |
| FPV/r | 31 | 19.556 | (1.802, 105.570) | −0.39 | 0.029 | |
| LPV/r | 8 | 156.735 | (16.969, 831.806) | −0.38 | 0.36 | |
| C12_IQFCb | IDV/r | 10 | 0.756 | (0.168, 7.378) | 0.09 | 0.80 |
| FPV/r | 31 | 0.541 | (0.039, 2.484) | −0.44 | 0.014 | |
| LPV/r | 8 | 5.320 | (0.740, 32.415) | −0.24 | 0.58 | |
| C12_IQcICc | IDV/r | 10 | 4.329 | (0.890, 29.284) | 0.02 | 0.96 |
| FPV/r | 31 | 5.146 | (0.474, 27.782) | −0.39 | 0.029 | |
| LPV/r | 8 | 40.189 | (4.351, 213.283) | −0.38 | 0.36 | |
| C12_IQpICd | IDV/r | 10 | 3.636 | (0.748, 24.598) | 0.02 | 0.96 |
| FPV/r | 31 | 1.956 | (0.180, 10.557) | −0.39 | 0.029 | |
| LPV/r | 8 | 2.351 | (0.255, 12.477) | −0.38 | 0.36 | |
| C12_IQfICe | FPV/r | 30 | 1.494 | (0.145, 7.949) | −0.47 | 0.01 |
| LPV/r | 8 | 1.179 | (0.095, 7.960) | −0.38 | 0.36 | |
| C12_IQfFCf | FPV/r | 30 | 0.039 | (0.003, 0.187) | −0.50 | 0.005 |
| LPV/r | 8 | 0.056 | (0.005, 0.310) | −0.26 | 0.54 | |
C12_IQnIC = C12/IC50.
C12_IQFC = C12/[mean wild-type IC50 (22) × IC50 FC].
C12_IQcIC = C12/[IC50 × attenuation by 50% human serum (18)].
C12_IQpIC = [population PB-corrected C12 (2, 14, 18, 24)]/IC50].
C12_IQfIC = [individual PB-corrected C12]/IC50].
C12_IQfFC = [individual PB-corrected C12]/[mean wild-type IC50 (22) × IC50 FC].
Treatment response.
Median changes in plasma HIV-1 RNA levels from baseline were modest. At 2 weeks, the FPV/r arm had a median change in plasma HIV RNA of −0.1 log10 copies/ml (range, −1.1 to +0.9 log10 copies/ml). For the IDV/r arm, the median change in plasma HIV RNA was −0.7 (range, −1.6 to +0.6), and for the LPV/r/r arm, it was −1.0 (range, −1.7 to −0.1). Of the 32 subjects treated with FPV/r, 21 had switched from LPV/r and 6 had previous APV exposure. Week 24 results varied widely among individuals. Median changes within arms were small. In the FPV/r arm, the median change for plasma HIV RNA was −0.3 (range, −3.2 to +1.0). In the IDV/r arm, it was −0.4 (range, −2.9 to +0.3), and in the LPV/r/r arm, it was −0.3 (range, −2.9 to +0.2).
Correlations for week 2 treatment response in the FPV/r arm.
Given that the sample size in the FPV/r arm approached the prespecified target, our analyses focused on this arm. The correlation between the FPV C12 IQ and 2-week VL response was modest and significant with or without correction for PB and when measured free-drug concentrations were used (Table 3). However, the strongest correlation with the 2-week treatment response to FPV/r substitution was the baseline IC50 FC (Fig. 1a) (Spearman r = 0.57; P = 0.001), which improved further when the analysis was limited to adherent subjects who took ≥80% of the RTV doses in the first 14 days (Spearman r = 0.68; P = 0.001) (Fig. 1b). Accounting for FPV exposure using IQ ratios did not improve on these correlations (Table 3). All correlations between the IQ (using C12, Cτ, or AUC0-12 and various measures of susceptibility) and the 2-week response, though significant, had less significant R values, with the exception of the IQ using AUC and FC susceptibility (AUC_IQFC), which had the same correlation (r = −0.57; P = 0.001) (Fig. 2a) and was more robust than the correlation observed with C12_IQFC (r = −0.44; P = 0.014) (Fig. 2b). Incorporating individual measured percentages free for APV improved the IQ correlation using FC for the C12_IQfFC (r = −0.5; P = 0.005), though not for the AUC_IQfFC (r = −0.51; P = 0.004). Accounting for adherence modestly improved the observed correlation for most comparisons. No single pharmacokinetics parameter (Cmax, AUC0-12, C12, or average Cτ) showed an inverse relationship between an increasing concentration and a greater decrease in change in VL at 2 weeks. All correlation coefficients were positive and nonsignificant (Cmax, r = 0.27; AUC0-12, r = 0.26; C12, r = 0.20; and Cτ, r = 0.18).
FIG. 1.
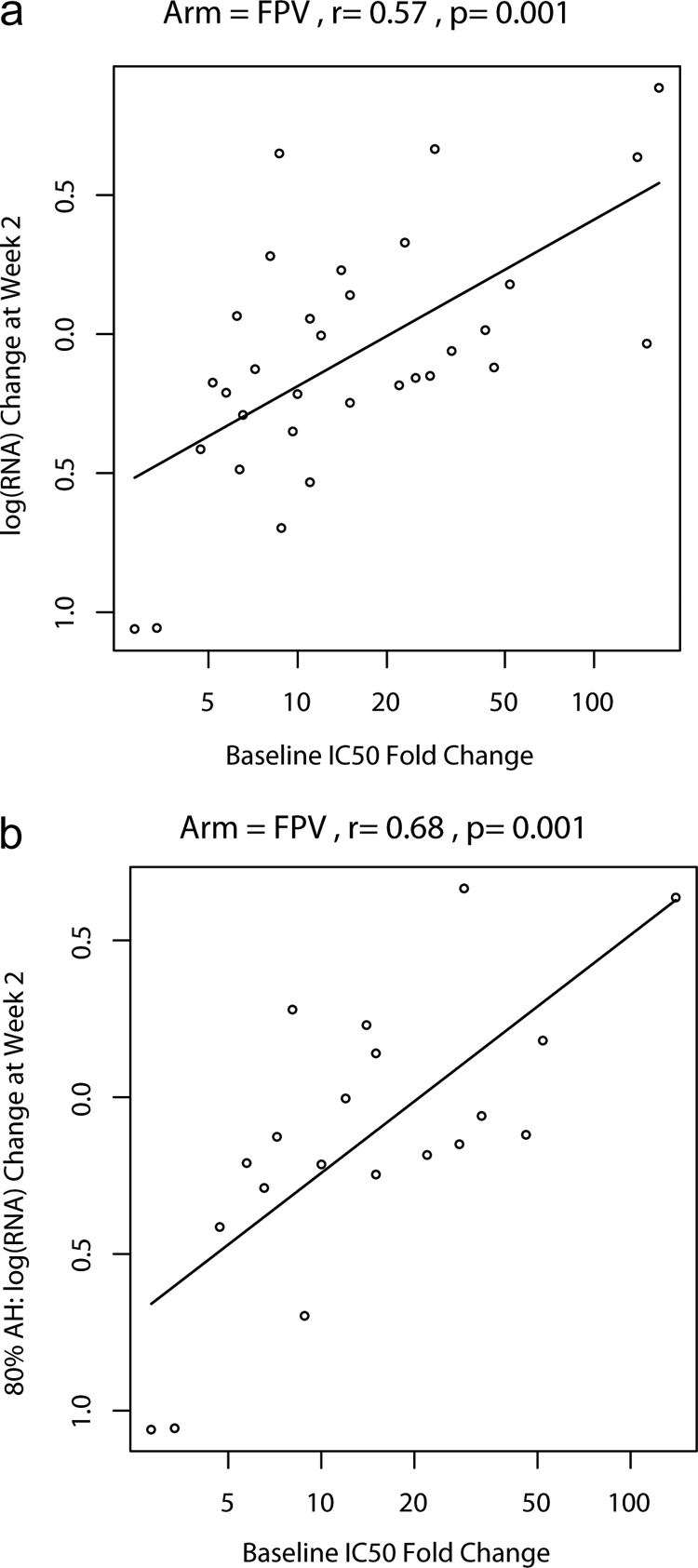
(a) Plot of log RNA change versus IC50 FC (on a loge scale) and estimated log RNA change as a function of IC50 FC (linear regression model). (b) Plot of log RNA change versus IC50 FC (on a loge scale) and estimated log RNA change as a function of IC50 FC for subjects with at least 80% adherence (AH) (linear regression model).
FIG. 2.
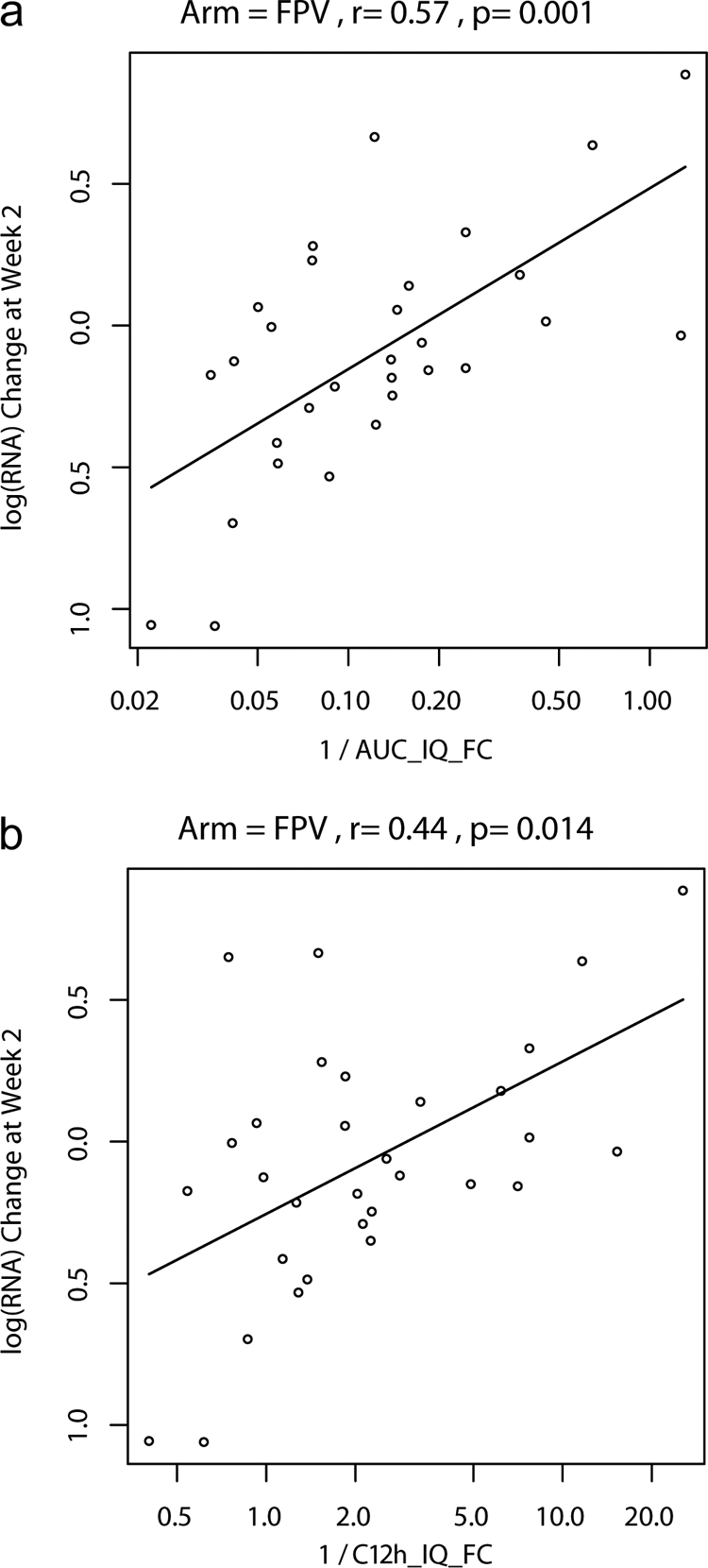
(a) Plot of log RNA change versus the reciprocal of AUC_IQFC (on a loge scale) and estimated log RNA change as a function of the reciprocal of AUC_IQFC (linear regression model). AUC_IQFC = AUC/[mean wild-type IC50 of reference strains × IC50 FC]. (b) Plot of log RNA change versus the reciprocal of C12_IQFC (on a loge scale) and estimated log RNA change as a function of the reciprocal of C12 _IQFC (linear regression model).
The relationships of baseline susceptibility or IQ with change in VL from baseline at 24 weeks in the FPV/r arm had correlations similar to those of the short-term results; most were not significant. Examining the VL change at day 7 in the FPV/r arm did not change the relationships. The correlation coefficients and P values for the relationships between week 2 VL responses and IQ ratios are listed in Table 3 for LPV/r and IDV/r. None were significant, nor were correlations between FC and response (data not shown). Inverse relationships between measures of concentration and treatment response were also not observed.
Using FPV/r data, multivariate regression analyses by stepwise selection investigated the relationships of VL changes at week 2 with potential predictors of response. The variables included baseline VL and CD4 and CD8 cell counts; one measure of susceptibility (IC50, FC, or mean wild-type IC50 × FC); one measure of the week 2 drug concentration; and the corresponding IQ ratio. Baseline susceptibility to FPV was the only significant predictor of VL decline in all models that included the FC variable (P < 0.001). In one model that included the IQ based on individual measured free-APV AUC and the actual IC50, the AUC_IQfIC was the only significant predictor (P < 0.001).
When data from all three arms were combined in an exploratory analysis, the IQ based on C12 and FC susceptibility (C12_IQFC) was correlated with the week 2 change in VL (r = −0.53; P < 0.001), and when the LPV/r and FPV/r arms were combined, the IQ based on the measured free C12 value and FC (C12_IQfFC) was also significantly correlated with the week 2 and week 24 VL responses (r = −0.47 for both; P = 0.003 and 0.004). The combined-arm analysis using the IQ based on the AUC and FC (AUC_IQFC) gave the strongest correlation (r = −0.72; P < 0.001) of any comparison. Unlike the analysis for the FPV/r arm, the combined analysis showed only modest correlation between either the IC50 or the FC and the week 2 VL response (r = 0.41, P = 0.003 and r = 0.25, P = 0.078, respectively).
Tolerability/toxicity results.
Few subjects had grade 3 or greater signs, symptoms, or laboratory abnormalities through 24 weeks (4/12 on IDV/r, 9/33 on FPV/r, and none on LPV/r). Approximately one-half were considered possibly or definitely study drug related. None were unanticipated. No significant relationships between drug concentrations and toxicity were found.
DISCUSSION
This study was designed to rigorously assess the relationships of pharmacokinetics parameters (including measured free-drug concentrations for the more highly protein-bound PIs), PI susceptibility, and IQ with short- and long-term treatment responses. Only the PI component of the regimen was changed over the first 14 days to limit confounding, and adherence over this period was measured by MEMS Caps. The FPV/r arm accrued enough subjects to reliably evaluate treatment response. Most subjects had highly PI-resistant virus, resulting in limited treatment responses. In the FPV/r arm, most subjects had FC IC50s of >10, designated to be above the upper treatment response cutoff in the PhenoSense assay. Somewhat greater 2-week treatment responses were observed in the IDV/r and LPV/r arms, despite higher median FCs at baseline; however, the nonrandomized nature of the study precludes comparisons among arms. Despite the limitations, several important observations can be made.
The pharmacokinetics results for IDV/r, FPV/r, and LPV/r are consistent with previous reports (1, 9). APV AUC values were similar to those noted in a recent large pharmacokinetics study, although we observed higher APV C12 and Cτ values (23). APV concentrations 12 h postdose were similar to predose Cτ and median Cτ in FPV/r-treated subjects. Within-subject APV Cτ values were consistent over time. The percentages of free APV and LPV were consistent with previous reports (3, 6), and these values remained stable over time. In the FPV/r arm, we demonstrated that at Cmax, the proportion of free APV was modestly but significantly higher than at 12 h postdose and that the percentages of free APV within individuals on the FPV/r arm were highly correlated and stable over time. APV percentages free were less variable than previously reported in a smaller study of APV/r (3).
We demonstrated that IQs vary widely for a given PI and across different PIs depending upon whether and how one controls for PB. For APV and LPV, the PB correction method using average free-drug percentages obtained from previous studies (2, 15a, 18, 24) resulted in IQ ratios that were numerically closest to the ratio created using measured free-drug percentages from the individual subjects.
In the evaluation of predictors of treatment response in the FPV/r arm, short-term (2-week) antiretroviral treatment responses to FPV/r correlated most strongly with baseline FC susceptibility to FPV (Fig. 1a). Accounting for adherence improved the relationship further (Fig. 1b). The correlation with the treatment response was not improved by incorporating drug exposure information. Multiple different IQ formulas were evaluated, and only the IQ that incorporated AUC0-12 and FC susceptibility had a correlation coefficient that was the same as the FC susceptibility (Fig. 2a). Similarly, in multivariate regression models that included baseline susceptibility (measured as FC), drug exposure, IQ, baseline HIV RNA, and baseline CD4, only baseline FC significantly predicted the outcome. Although PI concentration ranges for patients on boosted PI are broad, higher plasma APV concentrations did not appear to contribute to inhibition of highly PI-resistant virus, which was present in most of our FPV/r-treated subjects, suggesting that increasing PI plasma concentrations cannot overcome very high-level PI resistance for a given PI, even when the amount of free drug present is accounted for. We used the approved dose of FPV/r (700/100 mg BID), and perhaps a higher dose could improve the inhibition of highly resistant virus, although the drug levels others have achieved at 1,400/100 mg BID were not dose-proportionally increased (25). In exploratory analyses using all three arms, where the range of week 2 VL responses was greater, accounting for concentration and susceptibility (IQ) produced stronger correlations than with susceptibility alone. The IQ using AUC and FC resulted in the strongest correlation we observed. These results suggest that when there is a broader range of variant susceptibility and treatment responses, incorporating the drug concentration and susceptibility improves the correlation with virologic response over susceptibility alone.
Our findings, when evaluating the FPV/r arm, are consistent with some but vary from other previous observations from studies that were not prospectively designed to evaluate IQ. In an early study of LPV/r when efavirenz was also administered as a new agent, the LPV IQ was a significant predictor of antiretroviral treatment response in multivariate regression models. These models did not include baseline susceptibility as a separate variable (15). In a large study evaluating a genotype IQ for LPV (GIQ), both the genotype and GIQ were predictive of the 6-month outcome in multivariable models, and in contrast to our results with FPV/r and phenotype-based IQ, in subjects with high-level LPV resistance, the GIQ was the only significant predictor of the outcome (20). In two studies of phenotype-based and genotype-based APV IQs, a higher IQ was associated with an improved outcome, but statistical analyses were limited (10, 21). In one study of APV/r in subjects with high levels of resistance at baseline, the genotype and not the GIQ was predictive of the response (26), consistent with our results, although in another, larger study of FPV/r in treatment-experienced subjects with a broad range of genotypic resistance to FPV, a significant independent relationship between GIQ and 12-week treatment outcome was demonstrated (23). Consistent with our exploratory analysis, the relationship between IQ and treatment response is most apparent when there is a broad range of susceptibility in the population studied, which would be more likely for PIs with activity across a large FC range.
Our study was limited, as only the FPV/r arm accrued sufficient subjects to make robust comparisons within an arm, and the levels of baseline APV resistance blunted the range of treatment responses. Conversely, our study was unique in that it was prospectively designed to address the correlation of IQ with outcome, included a full pharmacokinetics analysis with measurement of free-drug percentages, and measured adherence. The 2-week treatment response reflected the single substitution and was not confounded by changes to other regimen components. Overall, we could not demonstrate an improvement in the correlation of treatment response with IQ compared to baseline virus susceptibility to FPV, though an exploratory analysis across the three arms was suggestive. Whether the IQ may provide a better predictor of outcome in individuals with more sensitive virus requires further study.
Acknowledgments
We are extremely grateful to the individuals who volunteered for this study. We also acknowledge the contributions of other A5126 protocol team members: Courtney N. Ashton (Frontier Science Technology Research Foundation), laboratory data coordinator; Robin DiFrancesco (Laboratory for Antiviral Research, Buffalo, NY), laboratory technologist; Karin L. Klingman (Division of AIDS, National Institute of Allergy and Infectious Disease), medical officer; Bernadette Jarocki (Frontier Science Technology Research Foundation), data manager; Beverly Putnam (University of Colorado Health Science Center), field representative; Rose D. Todd-Stanford (AIDS Clinical Trials Group Community Constituency Group); Paul T. Tran (Division of AIDS, National Institute of Allergy and Infectious Disease), pharmacist; Dale Kempf, Scott C. Brun, and William Chris Woodward (Abbott Laboratories); Odin J. Naderer and Keith A. Pappa (GlaxoSmithKline); Marianne Poblenz and James F. Rooney (Gilead); Jon H. Condra and Michael N. Robertson (Merck Research Laboratories); and Michael P. Bates, Eoin Coakley, and Rick L. Pesano (Monogram Biosciences).
We acknowledge John G. Gerber and the University of Colorado Health Sciences pharmacology laboratory staff and Patricia S. Lizak and the University of California at San Francisco pharmacology laboratory staff for their contributions to this work. Abbott donated study medication, MEMS Caps, and MEMS data analysis; Gilead provided study medication.
We thank investigators and staff at these participating clinical research sites: Frances Canchola and Laura Felix at the University of Southern California (A1201), CTU Grant 5U01AI069428-02; Charles Hicks and Margaret McDaniel at Duke University Medical Center (A1601), CTU Grant 5U01, AI069484-02; Mark Packard and JoAnn Kuruc at the University of North Carolina (A3201), CFAR Grant AI50410, CTU Grant AI69423-01, GCRC Grant RR00046; Donna Mildvan and Manuel Revuelta at Beth Israel Medical Center (A2851), CTU Grant AI46370; Elizabeth Race and Todd Morgan at UT Southwestern Medical Center at Dallas (A3751), CTU Grant 3, UO1 AI046376; Deborah O'Connor and Mitchell Goldman at Indiana University (A2601), CTU Grant AI025859; Margie Vasquez and Charles Gonzalez at New York University/NYC HHC at Bellevue Hospital Center (A0401), CTU Grants AI-27665 and AI069532, GCRC Grant M01-RR00096; Janet Nicotera and Vicki Bailey at Vanderbilt University Therapeutics Clinical Research Site (A3652), CTU Grant AI069439, GCRC Grant RR024977; Beverly Putnam and John Koeppe at the University of Colorado Hospital (A6101), GCRC Grant RR00051, CTU Grant AI69450; Sandra Valle and Jane Norris at Stanford University (A0501), CTU Grant AI069556-02; Jorge Santana and Olga Méndez at the University of Puerto Rico (A5401), CTU Grant 5 U01 A1069415-02; Linda Meixner and Kathleen Nuffer at the University of California, San Diego (A0701), CTU Grant AI 69432, CFAR Grant AI36214; Margaret A. Fischl and Hector Bolivar at the University of Miami AIDS Clinical Research Unit (A0901), CTU Grant AI069477; Richard Pollard and Abby Olusanya at the University of California, Davis Medical Center (A3852), CTU Grant 1U01AI069483-03; William O'Brien and Gerianne Casey at the University of Texas, Galveston (A6301), CTU Grant UO1Al32782.
This work was supported in part by the AIDS Clinical Trials Group funded by the National Institute of Allergy and Infectious Diseases under the following grants: AI38858, AI68636, AI68634, and AI64086 (R.H.). This work was also supported in part by the General Clinical Research Center Units funded by the National Center for Research Resources. GlaxoSmithKline provided study medication and supportive funding for pharmacologic assays; Merck supplied study medications and supportive funding for phenotypic assays; and Monogram Biosciences provided supportive funding for phenotypic and genotypic assays.
Conflicts of interest: J. J. Eron is the principal investigator on research grants to the University of North Carolina from Boehringer Ingelheim, GlaxoSmithKline, Merck, and Panacos. He served as a consultant to Avexa, Bristol-Myers Squibb, GlaxoSmithKline, Merck, Monogram, Panacos, Pfizer, Tibotec, and Tobira. He was on the speakers' bureaus for or received honoraria from Bristol-Myers Squibb, Merck, Roche, and Tibotec. R. Haubrich reported research funding to the University of California at San Diego from Abbott, Gilead, and GlaxoSmithKline and he received honoraria from Abbott, Gilead, and Merck for consultation. F. Aweeka, H. Wu, J.-G. Park, B. Bastow, and S. Yu report no conflicts. D. D. Richman has served as a consultant to Anadys Pharmaceuticals, Inc.; Biota; Bristol-Myers Squibb, Inc.; Gilead; Idenix; Koronis Pharmaceuticals; Merck & Co, Inc.; Monogram Biosciences, Inc.; Pfizer, Inc.; Roche Pharmaceuticals; and Tobira Therapeutics, Inc. G. Pakes is employed by and owns stock in GlaxoSmithKline.
Footnotes
Published ahead of print on 23 March 2009.
REFERENCES
- 1.Acosta, E. P., H. Wu, S. M. Hammer, S. Yu, D. R. Kuritzkes, A. Walawander, J. J. Eron, C. J. Fichtenbaum, C. Pettinelli, D. Neath, E. Ferguson, A. J. Saah, and J. G. Gerber. 2004. Comparison of two indinavir/ritonavir regimens in the treatment of HIV-infected individuals. J. Acquir. Immune Defic. Syndr. 37:1358-1366. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, P. L., R. C. Brundage, L. Bushman, T. N. Kakuda, R. P. Remmel, and C. V. Fletcher. 2000. Indinavir plasma protein binding in HIV-1-infected adults. AIDS 14:2293-2297. [DOI] [PubMed] [Google Scholar]
- 3.Barrail, A., C. Le Tiec, S. Paci-Bonaventure, V. Furlan, I. Vincent, and A. M. Taburet. 2006. Determination of amprenavir total and unbound concentrations in plasma by high-performance liquid chromatography and ultrafiltration. Ther. Drug Monit. 28:89-94. [DOI] [PubMed] [Google Scholar]
- 4.Barrail-Tran A., L. Morand-Joubert, G. Poizat, G. Raguin, C. Le Tiec, F. Clavel, E. Dam, G. Chêne, P. M. Girard, A. M. Taburet, and the Puzzle-1 (ANRS 104) Study Group. 2008. Predictive values of the human immunodeficiency virus phenotype and genotype and of amprenavir and lopinavir inhibitory quotients in heavily pretreated patients on a ritonavir-boosted dual-protease-inhibitor regimen. Antimicrob. Agents Chemother. 52:1642-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boffito, M., E. Acosta, D. Burger, C. V. Fletcher, C. Flexner, R. Garaffo, G. Gatti, M. Kurowski, C. F. Perno, G. Peytavin, M. Regazzi, and D. Back. 2005. Current status and future prospects of therapeutic drug monitoring and applied clinical pharmacology in antiretroviral therapy. Antivir. Ther. 10:375-392. [PubMed] [Google Scholar]
- 6.Boffito, M., P. G. Hoggard, W. E. Lindup, S. Bonora, A. Sinicco, S. H. Khoo, G. Di Perri, and D. J. Back. 2004. Lopinavir protein binding in vivo through the 12-hour dosing interval. Ther. Drug Monit. 26:35-39. [DOI] [PubMed] [Google Scholar]
- 7.Clevenbergh, P., R. Boulme, M. Kirstetter, and P. Dellamonica. 2004. Efficacy, safety and predictive factors of virological success of a boosted amprenavir-based salvage regimen in heavily antiretroviral-experienced HIV-1-infected patients. HIV Med. 5:284-288. [DOI] [PubMed] [Google Scholar]
- 8.Clotet, B., N. Bellos, J. M. Molina, D. Cooper, J. C. Goffard, A. Lazzarin, A. Wohrmann, C. Katlama, T. Wilkin, R. Haubrich, C. Cohen, C. Farthing, D. Jayaweera, M. Markowitz, P. Ruane, S. Spinosa-Guzman, and E. Lefebvre. 2007. Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet 369:1169-1178. [DOI] [PubMed] [Google Scholar]
- 9.Corbett, A. H., M. L. Lim, and A. D. Kashuba. 2002. Kaletra (lopinavir/ritonavir). Ann. Pharmacother. 36:1193-1203. [DOI] [PubMed] [Google Scholar]
- 10.Duval, X., C. Lamotte, E. Race, D. Descamps, F. Damond, F. Clavel, C. Leport, G. Peytavin, and J. L. Vilde. 2002. Amprenavir inhibitory quotient and virological response in human immunodeficiency virus-infected patients on an amprenavir-containing salvage regimen without or with ritonavir. Antimicrob. Agents Chemother. 46:570-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grinsztejn, B., B. Y. Nguyen, C. Katlama, J. M. Gatell, A. Lazzarin, D. Vittecoq, C. J. Gonzalez, J. Chen, C. M. Harvey, and R. D. Isaacs. 2007. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 369:1261-1269. [DOI] [PubMed] [Google Scholar]
- 12.Hicks, C. B., P. Cahn, D. A. Cooper, S. L. Walmsley, C. Katlama, B. Clotet, A. Lazzarin, M. A. Johnson, D. Neubacher, D. Mayers, and H. Valdez. 2006. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatment-experienced HIV-1-infected patients at 48 weeks in the randomized evaluation of strategic intervention in multi-drug resistant patients with tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 368:466-475. [DOI] [PubMed] [Google Scholar]
- 13.Hoefnagel, J. G., P. P. Koopmans, D. M. Burger, R. Schuurman, and J. M. Galama. 2005. Role of the inhibitory quotient in HIV therapy. Antivir. Ther. 10:879-892. [PubMed] [Google Scholar]
- 14.Hoefnagel, J. G., M. J. van der Lee, P. P. Koopmans, R. Schuurman, S. Jurriaans, A. I. van Sighem, L. Gras, F. de Wolf, J. M. Galama, and D. M. Burger. 2006. The genotypic inhibitory quotient and the (cumulative) number of mutations predict the response to lopinavir therapy. AIDS 20:1069-1071. [DOI] [PubMed] [Google Scholar]
- 15.Hsu, A., J. Isaacson, S. Brun, B. Bernstein, W. Lam, R. Bertz, C. Foit, K. Rynkiewicz, B. Richards, M. King, R. Rode, D. J. Kempf, G. R. Granneman, and E. Sun. 2003. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 47:350-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15a.Hsu, A., R. Bertz, D. Hickman, M. Emery, G. Kumar, J. Denissen, S. Vasavanonda, A. Molla, D. Kempf, G. R. Granneman, and E. Sun. 2001. 8th Conf. Retrovir. Opportunistic Infect., abstr. 753.
- 16.Khoo, S. H., A. Winston, and D. Back. 2007. Combining resistance and pharmacology for optimum patient care. Curr. Opin. HIV AIDS 2:157-168. [DOI] [PubMed] [Google Scholar]
- 17.Lazzarin, A., T. Campbell, B. Clotet, M. Johnson, C. Katlama, A. Moll, W. Towner, B. Trottier, M. Peeters, J. Vingerhoets, G. de Smedt, B. Baeten, G. Beets, R. Sinha, and B. Woodfall. 2007. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 370:39-48. [DOI] [PubMed] [Google Scholar]
- 17a.Limoli, K., L. Trinh, G. Heilek-Snyder, N. Hellmann, and C. Petropoulos. 2002. 14th Int. AIDS Conf., abstr. TuPeB4574.
- 18.Livington, D. J., S. Pazhanisamy, D. J. Porter, J. A. Partaledis, R. D. Tung, and G. R. Painter. 1995. Weak binding of VX-478 to human plasma proteins and implications for anti-human immunodeficiency virus therapy. J. Infect. Dis. 172:1238-1245. [DOI] [PubMed] [Google Scholar]
- 19.Madruga, J. V., P. Cahn, B. Grinsztejn, R. Haubrich, J. Lalezari, A. Mills, G. Pialoux, T. Wilkin, M. Peeters, J. Vingerhoets, G. de Smedt, L. Leopold, R. Trefiglio, and B. Woodfall. 2007. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-1: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 370:29-38. [DOI] [PubMed] [Google Scholar]
- 20.Marcelin, A. G., I. Cohen-Codar, M. S. King, P. Colson, E. Guillevic, D. Descamps, C. Lamotte, V. Schneider, J. Ritter, M. Segondy, H. Peigue-Lafeuille, L. Morand-Joubert, A. Schmuck, A. Ruffault, P. Palmer, M. L. Chaix, V. Mackiewicz, V. Brodard, J. Izopet, J. Cottalorda, E. Kohli, J. P. Chauvin, D. J. Kempf, G. Peytavin, and V. Calvez. 2005. Virological and pharmacological parameters predicting the response to lopinavir-ritonavir in heavily protease inhibitor-experienced patients. Antimicrob. Agents Chemother. 49:1720-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcelin, A. G., C. Lamotte, C. Delaugerre, N. Ktorza, H. Ait Mohand, R. Cacace, M. Bonmarchand, M. Wirden, A. Simon, P. Bossi, F. Bricaire, D. Costagliola, C. Katlama, G. Peytavin, and V. Calvez. 2003. Genotypic inhibitory quotient as predictor of virological response to ritonavir-amprenavir in human immunodeficiency virus type 1 protease inhibitor-experienced patients. Antimicrob. Agents Chemother. 47:594-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molla, A., S. Vasavanonda, G. Kumar, H. L. Sham, M. Johnson, B. Grabowski, J. F. Denissen, W. Kohlbrenner, J. J. Plattner, J. M. Leonard, D. W. Norbeck, and D. J. Kempf. 1998. Human serum attenuates the activity of protease inhibitors toward wild-type and mutant human immunodeficiency virus. Virology 250:255-262. [DOI] [PubMed] [Google Scholar]
- 23.Pellegrin, I., D. Breilh, G. Coureau, S. Boucher, D. Neau, P. Merel, D. Lacoste, H. Fleury, M. C. Saux, J. L. Pellegrin, E. Lazaro, F. Dabis, and R. Thiebaut. 2007. Interpretation of genotype and pharmacokinetics for resistance to fosamprenavir-ritonavir-based regimens in antiretroviral-experienced patients. Antimicrob. Agents Chemother. 51:1473-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadler, B. M., C. Gillotin, Y. Lou, and D. S. Stein. 2001. In vivo effect of α1-acid glycoprotein on pharmacokinetics of amprenavir, a human immunodeficiency virus protease inhibitor. Antimicrob. Agents Chemother. 45:852-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shelton, M. J., M. B. Wire, Y. Lou, B. Adamkiewicz, and S. S. Min. 2006. Pharmacokinetic and safety evaluation of high-dose combinations of fosamprenavir and ritonavir. Antimicrob. Agents Chemother. 50:928-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valer, L., D. Gonzalez de Requena, C. de Mendoza, L. Martin-Carbonero, J. Gonzalez-Lahoz, and V. Soriano. 2004. Impact of drug levels and baseline genotype and phenotype on the virologic response to amprenavir/ritonavir-based salvage regimens. AIDS Patient Care STD 18:1-6. [DOI] [PubMed] [Google Scholar]