

Abstract
Clostridium perfringens is a gram-positive anaerobe and a pathogen of medical importance. The detection of acid phosphatase activity is a powerful diagnostic indicator of the presence of C. perfringens among anaerobic isolates; however, characterization of the enzyme has not previously been reported. Provided here are details of the characterization of a soluble recombinant form of this cell-associated enzyme. The denatured enzyme was ∼31 kDa and a homodimer in solution. It catalyzed the hydrolysis of several substrates, including para-nitrophenyl phosphate, 4-methylumbelliferyl phosphate, and 3′ and 5′ nucleoside monophosphates at pH 6. Calculated Kms ranged from 0.2 to 0.6 mM with maximum velocity ranging from 0.8 to 1.6 μmol of Pi/s/mg. Activity was enhanced in the presence of some divalent cations but diminished in the presence of others. Wild-type enzyme was detected in all clinical C. perfringens isolates tested and found to be cell associated. The described enzyme belongs to nonspecific acid phosphatase class C but is devoid of lipid modification commonly attributed to this class.
Clostridium perfringens is a ubiquitous gram-positive, endospore-forming anaerobic bacterium. While found in soil, the organism is often a constituent of the intestinal flora and is capable of causing severe gastrointestinal and histotoxic infections in humans and animals. As a species, C. perfringens is a very heterogeneous group of organisms with respect to metabolic by-products produced in vitro and pathogenic potential (25). Although the literature is replete with descriptions of the immunological, bio- and physicochemical attributes of C. perfringens-encoded toxins, as well as molecular and morphological details of sporulation, detailed analysis of other constituent enzymes, including those involved in acquisition and regulation of inorganic phosphate, has received relatively little attention. This lack of information is notable. A growing cadre of investigators (1, 7) has recognized the clinical utility of acid phosphatase as a biochemical marker for the identification of C. perfringens isolated from water and other sources and its use in the differentiation of C. perfringens from ∼95% of all currently recognized clostridial species in the genus (42).
Acid phosphatases (EC 3.1.3.2) are ubiquitous and catalyze, at an acidic pH, the transfer of phosphoryl groups from phosphomonoesters to water (48). These enzymes play essential roles in the generation, acquisition, and mobilization of inorganic phosphate and critical roles in phosphoryl relay systems intimately involved in signal transduction pathways in both prokaryotes and eukaryotes. A subgroup of phosphatases has been designated nonspecific acid phosphatases (NSAPs) by Thaller and coworkers (45) and includes those bacterial polyspecific phosphatases that are secreted across the cytoplasmic membrane and exhibit optimum catalytic activity at an acidic pH (41). NSAPs are recognized as a distinct group of phosphatases and categorized into three classes (46). The class C enzymes are characterized by the presence of four invariant aspartic acid residues (in bold type) contained within a bipartite signature motif consisting of (I/V)-(V/A/L)-D-(I/L)-D-E-T-(V/M)-L-X-(N/T)-X-X-Y near the N terminus and (I/V)-(L/M)-X-X-G-D-(N/T)-L-X-D-F near the C terminus, separated by a variable-length polypeptide linker region (46). Class C enzymes purified and/or characterized thus far include those from Bacillus anthracis (9), Haemophilus influenzae (e [P4]) (37, 38), Streptococcus equisimilis (LppC) (24), Staphylococcus aureus (SapS) (6), Helicobacter pylori (HppA) (35), and Elizabethkingia meningosepticum (previously termed Chryseobacterium meningosepticum) (OlpA) (29). Conserved attributes of these enzymes include a metal-containing polypeptide component of 25 to 30 kDa and broad substrate specificity. Members of class C, with one exception (6), are cell associated, a property achieved through addition of an N-terminal lipid modification which anchors the protein in the membrane (24, 37). Most recently, the recombinant Haemophilus influenzae e (P4) acid phosphatase was crystallized (28), and its three-dimensional structure was determined (8). The crystal structure confirmed the presence of a divalent cation in the active site and showed that e (P4), and presumably other class C enzymes, belong to the haloacid dehalogenase superfamily (3, 8, 30).
Provided here are the details of the purification and biochemical characterization of a soluble recombinant form of the class C acid phosphatase from C. perfringens hereafter designated rCppA (for recombinant CppA). Furthermore, the presence of an active form of the enzyme is demonstrated in clinical isolates and localized cytochemically and immunologically to the membrane of the C. perfringens type strain. To our knowledge, this is the first report of the existence of a membrane-associated class C acid phosphatase devoid of an N-terminal consensus lipoprotein motif.
(Preliminary results of this work were presented to The Anaerobe Society of the Americas in Annapolis, MD.)
MATERIALS AND METHODS
Bacterial strains and materials.
The overexpression vector, E. coli strains, and recombinant DNA constructs described in this study are listed in Table 1. Clostridium perfringens strain ATCC 10543 and ATCC 13124 were obtained from Mark McIntosh (Department of Molecular Microbiology and Immunology, University of Missouri, Columbia) (17). Clinical Clostridium perfringens isolates were randomly selected samples from those submitted to the Veterinary Medical Diagnostic Laboratory at the University of Missouri. Plasmid DNA was isolated from 5-ml Escherichia coli overnight cultures using a Wizard miniprep kit (Promega, Madison, WI). C. perfringens chromosomal DNA was isolated using QIAamp DNA blood minikit (Qiagen, Valencia, CA). Standard manipulations of DNA were performed using restriction enzymes and T4 DNA ligase (New England Biolabs, Beverly, MA) and Pfu DNA polymerase (Stratagene, La Jolla, CA). Bacteriological media were from Difco (Detroit, MI) and purchased from Fisher Scientific (Chicago, IL). Oligonucleotide primers used throughout this study were purchased from Integrated DNA Technologies (Coralville, IA). All other chemicals including secondary antibody, were obtained from Sigma Chemical (St. Louis, MO) and were of the highest purity available. Chromatography experiments were conducted using ion-exchange, HiTRAP metal-chelating, and gel filtration chromatography resins (GE Biosciences, Piscataway, NJ). Protein electrophoresis was performed on NuPAGE Bis-Tris gels (Invitrogen, Carlsbad, CA). The molecular mass standards for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were Perfect Protein markers (15 to 150 kDa) (Novagen, Madison, WI).
TABLE 1.
Oligonucleotide primers, strains, and plasmids used in this study
| Primer, strain, or plasmid | Purpose | Source or reference |
|---|---|---|
| Oligonucleotide primers | ||
| CP201/NcoI (AAAACCATGGTTTTACGAAAACAACCAAGAAGCA)a | Amplification of CPE0201 | Integrated DNA Technologies |
| CP201/XhoI (AAACCGCTCGAGTTTAAATGACTTTATACTATTTAA)a | ||
| Forward (GCTAAACCATATGCAGTTACCTTAGCTATTGATGAAACAATAATAGATAATTCACC)b | Mutagenesis of Asp 100 of the DDDD motif | Integrated DNA Technologies |
| Reverse (GGTGAATTATCTATTATTGTTTCATCAATAGCTAAGGTAACTGCATATGGTTTAGC)b | ||
| Bacterial strains | ||
| Clostridium perfringens ATCC 13124 | Genomic DNA source | 17 |
| C. perfringens random clinical isolates | Clinical specimens | |
| Escherichia coli DH5α and E. coli Top10 | Host strain for cloning | Invitrogen |
| E. coli BL21 AI | Host strain for protein expression | Invitrogen |
| Plasmids | ||
| pET20b | Expression vector | Novagen |
| pRCppA and pRCppAD100A | CppA expression | This study |
The boldface letters indicate incorporated restriction sites of CPE201 amplification primers.
The individual boldface letters represent mutated nucleotides.
Culture methods.
Plasmid constructs were maintained in E. coli DH5α grown in LB broth or agar containing ampicillin at 50 μg/ml. C. perfringens clinical strains and type strains were grown for approximately 2 days at 37°C under anaerobic conditions generated by inclusion of AnaeroPack (Mitsubishi Gas Chemical Co. Inc.), on prereduced Remel blood agar plates. For determination of specific C. perfringens acid phosphatase activity, bacterial colonies were scraped from blood agar plates, suspended in 50 mM sodium acetate (pH 6.0) containing 150 mM NaCl, and used in the enzyme assay.
Enzyme assays.
Routine phosphomonoesterase assays were performed along with those to determine the pH optimum of the enzyme for p-nitrophenylphosphate (pNPP) as previously described (35). One unit of activity was defined as the amount of enzyme necessary to catalyze conversion of 1 nmol substrate to product per hour at 37°C. Enzyme activity for substrates other than pNPP was determined by discontinuous colorimetric assay as reported by Lanzetta et al. (22) using previously described kinetic assays (35).
Protein determination.
Protein concentrations were determined using bicinchoninic acid (BCA) protein assay reagent (Pierce, Rockford, IL) as previously described (43). Bovine serum albumin (1 mg/ml) was used as the protein assay standard.
Estimation of molecular mass and oligomeric state.
Estimation of the recombinant enzyme's molecular mass in the denatured state was performed on a NuPAGE 4 to 12% Bis-Tris gel (Invitrogen) utilized per the manufacturer's recommendation. A modified version of the zymogram technique of Rossolini et al. (40) following SDS-PAGE and renaturation was used to qualitatively assess the in situ phosphomonoesterase activity of the purification samples. 4-Methylumbelliferyl phosphate (4MUP) at a final concentration of 1 mM was used as the substrate in the zymogram assay buffer. The reaction was stopped with 10 volumes of 2 M NaOH. Fluorescent bands were observed using a UV transilluminator at 302 nm. Recombinant acid phosphatase was subjected to matrix-assisted laser desorption ionization-time of flight mass spectrometry at the University of Missouri Proteomics Center. Estimation of the oligomeric state of rCppA was performed by gel filtration chromatography and analytical ultracentrifugation as previously described (34).
Immunological methods.
Rabbit anti-rCppA antiserum was obtained using standard immunological methods approved by the University of Missouri Animal Care and Use Committee.
Construction and expression of plasmids pRCppA and pRCppA D100A.
Query of the complete genome sequence (26) of Clostridium perfringens strain ATCC 13124 with class C acid phosphatase genes identified an open reading frame, designated CPE0201, which was annotated as a probable acid phosphatase. The corresponding open reading frame from ATCC strain 10543 was amplified by PCR primers CP201/NcoI and CP201/XhoI (Table 1) under standard cycling parameters and cloned as a NcoI-XhoI fragment into pET20b. The primer CP201/NcoI was designed to anneal to a position within cppA to generate a open reading frame devoid of its endogenous signal sequence for secretion. The missing secretion signal sequence was ultimately replaced with the pelB leader sequence of the pET20b plasmid. To generate a specific mutation in the putative DDDD active site motif, oligonucleotide primers (Table 1) to position D100 were used with universal T7 reverse and forward oligonucleotide primers (Novagen) to generate N-terminal and C-terminal amplicons, respectively. The DNA products were gel purified and used as templates in overlapping extension PCR (15, 16) with the T7 universal primers. The PCR product, ∼1.0-kb fragment, was then cloned as a NcoI-XhoI fragment into pET20b. This final product was designated pRCppA D100A. The presence of the desired mutation and lack of any additional substitutions was verified by double-stranded DNA sequencing.
Protein purification.
Generation of recombinant protein was performed in E. coli BL21 AI through the arabinose autoinduction system as described by Studier (44). Two 0.5-liter cultures of autoinduced E. coli BL21 AI harboring pRCppA were centrifuged at 5,000 × g for 10 min, suspended in 20 to 25 ml of 20 mM NaH2PO4 (pH 7.0) containing 500 mM NaCl (buffer A), and stirred for approximately 30 min at room temperature in the presence of ∼1 mg each of lysozyme and DNase. All subsequent purification procedures were conducted at 4°C unless otherwise noted. The bacteria were treated by disruption with a French press followed by centrifugation at 31,000 × g for 10 min to remove unbroken cells and debris. The supernatant produced by low-speed centrifugation was subjected to ultracentrifugation at 192,000 × g for 1.0 h to remove bacterial membranes and associated proteins. The ultracentrifugation supernatant was applied to a Ni-charged HiTRAP metal chelate chromatography resin and washed with buffer A containing 20 mM imidazole. Adsorbed proteins were eluted from the Ni-charged resin using a linear 20 mM to 1 M imidazole gradient. Active fractions were pooled, dialyzed, and applied to Q-Sepharose anion-exchange resin followed by elution in a linear 0 to 1 M NaCl gradient. Active fractions were pooled and subjected to further biochemical analysis. A similar scheme was employed for the purification of rCppA D100A.
N-terminal sequence of rCppA.
Approximately 20 μg of purified rCppA was subjected to gel electrophoresis on a 10% (vol/vol) Tris-Tricine polyacrylamide gel, electroblotted onto a polyvinylidene difluoride membrane, and submitted to the Nucleic Acid Chemistry Laboratory of Washington University School of Medicine, St. Louis, MO, for N-terminal sequencing.
Localization of C. perfringens phosphomonoesterase.
The C. perfringens type strain ATCC 13124 was plated on prereduced blood agar plates and incubated under anaerobic conditions at 37°C. At 48 h, cells were scraped off the plates and suspended in 20 mM glycine buffer (pH 6.0). Lysozyme was added at a final concentration of 1 mg/ml. The C. perfringens preparation was subjected to disruption with a French press and low-speed and ultracentrifugation steps as described above. Samples were taken at each step of the fractionation and assessed for specific antigen by Western blot analysis.
Cytochemical localization of CppA activity by TEM.
Whole cells of C. perfringens type strain 10543 were harvested from blood agar plates after 24 h of growth, suspended in 0.1 M sodium acetate buffer (pH 6.0), and then encased in 10% gelatin matrix (175 bloom strength; Electron Microscopy Sciences). Solidified samples were evaluated for localization of phosphatase activity employing a Gomori stain strategy whereby liberated phosphate is captured as lead precipitates in situ (35). Gelatin-encased samples were exposed for 10 min to 1 mM AMP in buffer and then to freshly filtered 3 mM lead nitrate in water. Following several buffer rinses, specimens were prepared for transmission electron microscopy (TEM) by either standard fixation with osmium tetroxide as the secondary fixative and processed into Epon Araldite resin or microwave-assisted immunofixation with ruthenium red in the primary fixative and no secondary fixation, processing into LR White resin (4). Thin-section resin-embedded samples, unstained or stained briefly with uranyl acetate and lead citrate, were examined on a JEOL 1200EX transmission electron microscope at 80 kV.
RESULTS
Identification of cppA, cloning, and expression of rCppA.
Analysis of C. perfringens cppA and deduced amino acid product from C. perfringens strain ATCC 10543 revealed an open reading frame encoding a protein with similarity to class C acid phosphatase orthologs (46). In particular, the cppA-encoded protein contained the characteristic bipartite sequence motif of L-D-I-D-E-T at residues 99 to 104 and G-D-N-L-G-D-F at positions 215 to 221 (the invariant aspartic acid residues indicated in bold type) (Fig. 1). While the predicted protein contained the essential residues to categorize CppA catalytically into NSAP class C, its sequence, like those from other gram-positive organisms (Fig. 1) Staphylococcus aureus (NP 370831) and Clostridium novyi (YP 879060) (NCBI Entrez database), is devoid of the requisite cysteine residue between positions 15 and 35 of the protein, a residue that is essential for lipid modification (18). The absence of this residue precluded the designation of these enzymes as lipoproteins. Results from hydropathy analysis (data not shown) of the CppA sequence suggested the presence of a stretch of amino acids at the N terminus with sufficient hydrophobic character to anchor the protein to the cytoplasmic membrane (window of 9) (21), a supposition supported by fractionation experiments and confirmed with the anti-rCppA antibody (see below).
FIG. 1.

Amino acid sequence alignment of bacterial NSAP class C proteins. The top amino acid sequence of the alignment is that of the H. influenzae class C acid phosphatase lipoprotein e (P4) designated Hi. Directly above the H. influenzae sequence are black numbers (from 1 to 270 in multiples of 10) designating the residue number of this prototype class C enzyme. Above the residue numbers are symbols designating stretches of α-helices and β-strands as determined by structural analysis (8). Other sequences, including that from C. perfringens CppA (Cp), are Pasteurella multocida (Pm), Helicobacter pylori (Hp), Bacillus anthracis (Ba), Staphylococcus aureus (Sa), Streptococcus equisimilis (Se), Elizabethkingia (Chryseobacter) meningosepticum (Em), Clostridium novyi (Cn), and Francisella tularensis (Ft). Residues identically conserved in all sequences are denoted by white letters on a red background. Other regions of high sequence conservation are indicated by boxed red letters on a white background. Black bars under the alignment indicate residues of the DDDD motif. This figure was created with ESPript (12). Gaps introduced to maximize alignment are indicated by periods.
The gene denoted cppA was PCR amplified and modified to remove the N-terminal 25 amino acids for expression from pET20b in T7 E. coli expression strains. Results from DNA sequencing of the pRCppA expression vector verified the insertion of PCR-amplified/modified cppA juxtaposed 5′ to the T7 promoter and 3′ and in frame with the hexahistidine C-terminal region and the transcription terminator. The recombinant protein expressed from this construct is hereafter referred to as rCppA.
Recombinant phosphomonoesterase purification.
Large-scale preparation of rCppA was carried out as described in Materials and Methods. High levels of recombinant phosphatase activity, ∼198,000 units/mg, were found in the soluble fraction of E. coli BL21 AI following French press disruption of cells and ultracentrifugation. Consistent with the purification of other recombinant class C enzymes (8, 35, 38), no significant difference in total activity was noted between cells before and after treatment with a French press. The recombinant enzyme remained soluble through all centrifugation steps, consistent with the absence of the N-terminal hydrophobic domain. The soluble protein was subjected to Ni-chelate and anion-exchange chromatography using standard techniques resulting in a final increase in purity of approximately 14.2-fold and a recovery of 42% of the total activity (Table 2). When samples from the purification steps were subjected to SDS-PAGE and stained, a prominent protein band of approximately 31 kDa was enriched throughout the purification scheme (Fig. 2A, lanes 2 to 6). This protein constituted greater than 95% of stained protein in the final preparation (Fig. 2A, lane 6). A protein of similar size was also observed on Western blots stained with either mouse antihexahistidine (Fig. 2B, lane 6, anti-His tag) or rabbit anti-recombinant CppA immunoglobulin G antibody (Fig. 2B, lane 6, anti-rCppA). When samples of the purification procedure were subjected to SDS-PAGE, renatured, and assessed for enzymatic activity using a modification of the zymogram technique, a single fluorescent band indicative of enzyme-catalyzed dephosphorylation of 4MUP was positioned coincident with the purified, immunoreactive polypeptide (Fig. 2C). N-terminal sequencing of the protein identified the first 10 residues of the recombinant enzyme as MVYENNQEAK. These residues were identical to the probable acid phosphatase encoded by cppA starting at valine 26. The N-terminal methionine was intentionally integrated into the recombinant construct during PCR-mediated amplification. The above results are consistent with successful expression and purification of recombinant C. perfringens CppA. With the exception of lack of detectable activity, similar results were obtained for the active site mutant of CppA (Fig. 2A, B, and C, lanes 7).
TABLE 2.
Purification table for recombinant C. perfringens acid phosphatase
| Purification step | Total activity (units, 107)a | Total protein (mg) | Sp act (units, 104/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| After French press | 19.2 | 970.3 | 19.8 | 1.0 | |
| Supernatant after low-speed centrifugation | 23.0 | 809.7 | 28.4 | 1.4 | 119 |
| Supernatant after ultracentrifugation | 15.1 | 313.3 | 48.1 | 2.4 | 78 |
| After Ni-chelate chromatography | 8.6 | 29.9 | 288.9 | 14.6 | 44 |
| After Q-Sepharose chromatography | 8.1 | 28.8 | 281.2 | 14.2 | 42 |
Units expressed as the amount of enzyme required to convert 1 nmole of the pNPP substrate to product per hour.
FIG. 2.
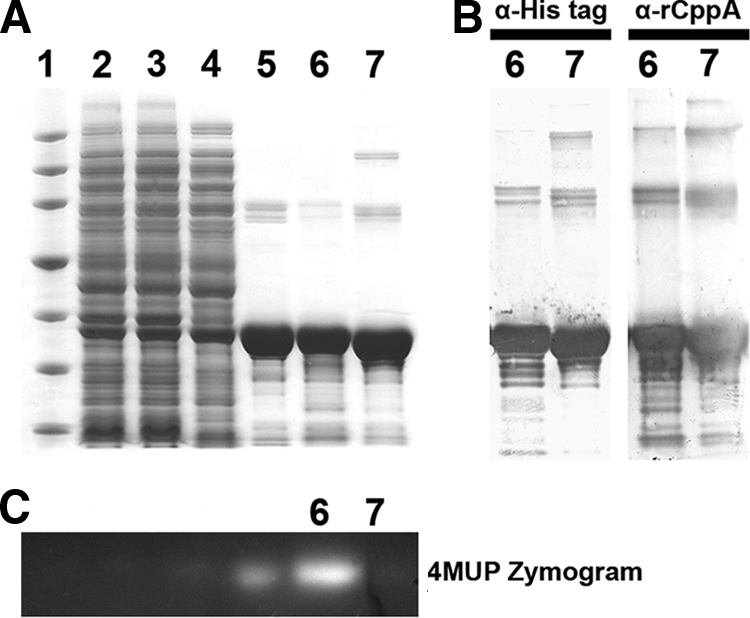
SDS-PAGE separation and detection of rCppA and active site mutant purification samples. All sample lanes contain ∼25 μg protein. (A) Lane 1, Novagen Perfect Protein standards (150, 100, 75, 50, 35, 25, and 15 kDa); lane 2, E. coli BL21 AI transformed with pRCppA, after autoinduction; lane 3, preparation after treatment with a French press; lane 4, supernatant after ultracentrifugation; lane 5, pooled fractions after Ni-chelate chromatography; lane 6, pooled fractions after Q-Sepharose chromatography; lane 7, purified rCppA D100A. (B) Western blot detection of active recombinant rCppA (lane 6) and mutant rCppA D100A (lane 7) with anti-His tag (α-His tag) and detection of active rCppA (lane 6) and mutant rCppA D100A (lane 7) with anti-rCppA (α-rCppA) (1:25,000 dilution). (C) 4MUP-mediated zymogram detection of acid phosphatase activity of rCppA purification samples, following SDS-PAGE and renaturation. Lanes are identical to those in panel A.
Molecular mass and oligomeric state determination.
The monomeric molecular mass of recombinant CppA estimated by matrix-assisted laser desorption ionization-time of flight mass spectrometry was 31,196.36 Da, a value within 1.0% of the theoretical value calculated from the deduced amino acid sequence of the recombinant enzyme with ExPASy (10). A value of approximately 31 kDa was obtained by SDS-PAGE. Analytical centrifugation and gel filtration chromatography were used to determine the oligomeric state of the recombinant protein in solution. Results from Superdex 200 gel filtration chromatography of rCppA yielded a molecular mass of approximately 61 kDa (data not shown). Equilibrium analytical ultracentrifugation using standard methods fit a single-species model with an apparent molecular weight of 62,000 (data not shown). As the monomer molecular mass was empirically demonstrated to be 31 kDa by mass spectrometry and SDS-PAGE, results from analytical centrifugation and gel filtration chromatography indicate that the prevalent form of rCppA is that of a homodimer, a state consistent with the oligomeric state of other well-characterized class C enzymes (8, 35, 37).
Enzymatic properties of recombinant C. perfringens CppA.
In addition to its physical properties, a number of enzymatic properties of rCppA were examined. These properties included pH optimum, substrate specificity, effects of potential phosphomonoesterase inhibitors, and kinetic parameters for various substrates. A mutant form of the enzyme was also generated in which an alanine residue was substituted for the first aspartic acid residue of the DDDD motif at amino acid position 100.
Purified rCppA behaved like an acid phosphatase (acid pH optimum) in all buffers tested. Maximum phosphomonoesterase activity was found in sodium acetate containing 1 mM MgCl2 at approximately pH 5.6 for pNPP and slightly higher for 5′ UMP (Fig. 3). While enzymatic activity was diminished at 1 pH unit to either side of the optimum, the effect was more pronounced on the acidic side of the profile (Fig. 3). Results from pH kinetic experiments with pNPP as the substrate suggest that the observed decrease in phosphatase activity with increasing [H3O+] correlated with an increase in Km for the substrate pNPP. In addition, as higher concentrations of acetic acid were necessary for the preparation of more acidic conditions, it was necessary to examine the effect of increasing concentrations of the acetate anion on the activity of the enzyme. At pH 5 and 6, the increasing acetate anion concentration (50 mM to 0.6 M) had negligible effect on the phosphatase activity of the enzyme for catalysis of pNPP hydrolysis (Fig. 3, inset A). As the pH optimum for the physiologic substrate 5′ UMP was approximately 6.0, all subsequent enzymatic analyses were conducted at this pH.
FIG. 3.

Effect of pH on the phosphomonoesterase activity of rCppA. Effect of pH on the phosphomonoesterase activity of rCppA for pNPP (•) and 5′ UMP (□) both at a final concentration of 1 mM. Max, maximum. (Inset A) Effect of acetate concentration on rCppA activity for pNPP at pH 5.0 and 6.0. (Inset B) Effect of pH on the Km of pNPP for rCppA at six pHs as assessed by standard kinetic assays as described in Materials and Methods.
Like other class C acid phosphatases, the C. perfringens enzyme was found to catalyze the hydrolysis of a number of different phosphomonoesters, including nucleoside 2′, 3′, and 5′ monophosphates, amino acid phosphates, and at least two phosphoanhydride-containing substrates. The highest levels of activity were observed for those phosphomonoesters conjoined with aliphatic ring structures. Significant activity was observed for physiologically relevant substrates (reported as percent hydrolysis of pNPP): 5′ UMP (75%), 5′ GMP (56%), 5′ AMP (50%), and 3′ TMP (45%). However, the nonphysiological substrates pNPP, 4MUP (99%), phenophthalein bisphosphate (88%), and phenophthalein monophosphate (81%) were the best substrates tested at final concentrations of 1 mM at pH 6.0 in the presence of 1 mM MgCl2. Results from assessment of initial rate data (Table 3) suggest that while rCppA has greatest activity for pNPP, the affinity of the enzyme for the nucleoside 5′ monophosphates is greater by 2.5- to 3-fold compared to the Km of the enzyme for the colorimetric substrate. All substrates examined for initial velocity data had Km values below 1 mM, the concentration used to assess activity in the substrate specificity assay. The Vmax values ranged from approximately 0.7 μmol Pi produced/second/mg of protein for 5′ AMP to just over 1.6 μmol Pi produced/s/mg of protein for the pNPP substrate. Substrates hydrolyzed by rCppA at less than 20% of pNPP in the initial assessment included ATP, 3′ AMP, flavin mononucleotide, ribose 5-phosphate, pyridoxal phosphate, ADP, 2′ AMP, and nicotinamide phosphates, as well as serine, threonine, and tyrosine phosphates, and were not assessed further (data not shown).
TABLE 3.
Kinetics of rCppA for selected substrates
| Substrate | Km (mM) (mean ± SD) | Vmax (μmol Pi/s/mg) (mean ± SD) |
|---|---|---|
| pNPP | 0.658 ± 0.05 | 1.639 ± 0.03 |
| 5′ UMP | 0.221 ± 0.03 | 1.19 ± 0.031 |
| 5′ GMP | 0.283 ± 0.05 | 0.868 ± 0.03 |
| 5′ AMP | 0.269 ± 0.08 | 0.776 ± 0.07 |
| 5′ TMP | 0.222 ± 0.04 | 0.791 ± 0.07 |
Various compounds, including known acid phosphatase inhibitors and mono- and divalent metals, were tested for their effect on enzymatic activity. Each compound tested at a final concentration of 1 mM was assayed in the presence of 0.2 M sodium acetate (pH 6.0) containing 1 mM MgCl2 and checked for its effect on the pH of the assay mixture. The effects of these compounds fell into the following four groups: (i) compounds that enhanced phosphomonoesterase activity for pNPP, including Cr, Co, and Cu ions; (ii) compounds that had little effect (retention of 75 to 100% activity) on enzyme-mediated catalysis, including Mg2+, Mn2+, Ba2+, Zn2+, Ca2+ and K1+; (iii) compounds that diminished enzymatic activity for pNPP from 25 to 75% of control, including ribose, tartrate, guanosine, molybdate, and vanadate; and (iv) compounds that diminished activity to below 25%, including Cd, Pb, Hg, Sm, and Ce ions. With respect to the first group, the observed enhancement of activity by Cr, Co, and Cu ions was enzyme dependent, as no increase in the product (p-nitrophenol) was noted in the presence of both metal- and heat-inactivated enzyme. Whether exogenously added metals enhance enzymatic activity through displacement of the originally bound metal or through some other mechanism is currently unknown. Of those inhibitors in the fourth group, all were found to inhibit the enzyme activity for pNPP by 50% when present at micromolar concentrations (data not shown). One such inhibitor, Hg, inhibited activity by 50% at 1.2 μM; the least effective of these inhibitors, NaF, inhibited activity by 50% when present at approximately 0.5 mM. Interestingly, of the compounds that most effectively inhibit enzyme activity, these were also metals (Sm and Ce). In addition, the cation lead was an effective inhibitor when provided in the assay as lead nitrate, lead chloride, or lead acetate; lead inhibited the enzyme when present at a final concentration of 1 mM by approximately 80% in all three forms. Barium nitrate and barium chloride at similar concentrations had no significant inhibitory effect on the enzyme, suggesting that the lead-mediated inhibition of the enzyme was cation dependent. While assumed to be a metalloenzyme related to H. influenzae phosphomonoesterase e (P4) and other class C NSAPs, the activity of rCppA was not significantly altered when dialyzed against 50 mM sodium acetate or glycine buffers containing 10 mM EDTA and assessed for activity in the presence or absence of 1 mM MgCl2.
Initial assessment of the necessity of the recombinant C. perfringens CppA DDDD motif was examined by mutagenesis of the first aspartic acid residue of the motif to alanine. Confirmation of the alteration of this residue at position D100, the equivalent position of the purported nucleophile of recombinant e (P4), was confirmed by DNA sequencing of the mutant open reading frame within the overexpression vector. Induction and purification of the mutant protein were achieved in a manner similar to that of the active recombinant protein (data not shown). The protein preparation contained a strongly staining band of approximately 30 kDa (Fig. 2A, lane 7) which was recognized by both the monoclonal antihexahistidine antibody and the rabbit polyclonal antibody raised against the active recombinant protein (Fig. 2B, lane 7, anti-His-tag and anti-rCppA, respectively). In addition, while the mutant protein was present on the SDS-polyacrylamide gel at quantities approximately equivalent to those of the active recombinant enzyme (Fig. 2A, lane 7 versus lane 6), zymogram detection of activity was significantly reduced compared to that of the control (Fig. 2C, lane 7 versus lane 6). In addition, the specific activity of purified rCppA D100A, as assessed by both the standard colorimetric assay using pNPP as the substrate and the more sensitive fluorescence assay (39) using 4MUP as the substrate, was less than 1% of that for the wild-type enzyme. These observations suggest, as for other class C enzymes tested (35, 36), that the first aspartic acid in the DDDD motif of C. perfringens CppA is required for full enzymatic activity.
Identification and localization of C. perfringens CppA.
Previous investigators have noted copious levels of C. perfringens-associated acid phosphatase activity when 4MUP or 2-naphthyl-phosphate were used as substrates in semisolid selective media and have suggested the enzyme's use as a specific marker for the presence of C. perfringens in food (7) or water (42). However, quantitation of enzymatic activity relative to total bacterial protein and purification and characterization of the enzyme have not been reported.
To address this issue, 10 veterinary isolates and 2 C. perfringens type strains were tested and found to be positive for acid phosphatase activity in our standard biochemical assay. The enzymatic activity was heat-labile and linear with increasing time of incubation or protein concentration. In clinical isolates, the specific acid phosphatase activity ranged from less than 1,200 to greater than 6,000 units/mg protein when pNPP was employed as the substrate. The average specific activity in these isolates was ∼3,300 units/mg with a standard deviation among the 10 isolates of 1,400 units/mg protein. The levels of observed activity were unaltered in the presence of sodium phosphate. Although the total number of samples is small, no correlation between specific activity and clinical source (food animal versus companion animal) was readily apparent. We noted substantially higher specific activity in the C. perfringens type strains; the specific activities in strains ATCC 10543 and ATCC 13124 were 19,000 units/mg and 15,000 units/mg protein, respectively. No change in specific activity for the type strains was noted with repeated passage (n = 10) on prereduced blood agar media incubated under anaerobic conditions for ∼48 h on each passage (data not shown).
When whole C. perfringens clinical isolates were separated by SDS-PAGE and subjected to Western blot analysis using anti-rCppA antibody, a heavily stained band at approximately 34 kDa was observed in each of the lanes containing clinical strains (Fig. 4A, lanes 2 to 9). The ExPASy predicted molecular mass of the unmodified gene product encoded by the cppA open reading frame was 34.2 kDa, a value consistent with the calculated molecular mass of the cross-reactive antigen detected by Western blotting.
FIG. 4.

Immunological detection and localization of C. perfringens CppA. (A) SDS-PAGE and Western blot detection (rabbit anti-rCppA [1:25,000]) of recombinant CppA (lane 1) and wild-type C. perfringens (lanes 2 to 9) in eight clinical isolates. (B) Western blot detection (rabbit anti-rCppA [α-rCppA] [1:25,000]) of CppA in the ultracentrifugation pellet (CP ultra pellet) (lane 1) and rCppA (lane 2) after SDS-PAGE separation.
Preliminary biochemical data were obtained for the phosphomonoesterase activity of C. perfringens type strain ATCC 10543. No increase in total activity was noted after cell disruption with a French press suggesting that the substrate had access to the enzyme. In fractionation experiments, inorganic phosphate-resistant acid phosphatase activity (data not shown) and an antigenically reactive band of approximately 34 kDa were recovered in the pellet after ultracentrifugation following French press disruption of cells (Fig. 4B). Attributes, such as phosphatase activity below pH 7, intimate cell association, and resistance to phosphate-mediated inhibition, were consistent with a phosphomonoesterase belonging to the group of NSAPs designated class C and consistent with expression of the cppA gene product to whose surrogate the rabbit antibody was produced.
Cytochemical localization of C. perfringens acid phosphatase activity employed TEM with a modified Gomori stain approach. Gelatin-encased whole cells treated with substrate and then phosphate capture agent prior to standard fixation or immunofixation and embedding demonstrated concentrated zones of electron-dense precipitates indicative of extracellular enzymatically liberated phosphate (Fig. 5A and B, respectively); no such precipitates were present in the absence of either lead or substrate (insets in Fig. 5A and B, respectively). As illustrated in these representative thin-section micrographs, significant phosphatase activity was observed nonuniformly distributed in the bacterial membrane and cell wall for some, but not all, cells within a population. These data are consistent with a membrane-bound, cell-associated acid phosphatase.
FIG. 5.

Transmission electron micrographs depicting detection and localization of acid phosphatase activity of C. perfringens type strain ATCC 10543. Thin sections of gelatin-encased whole-cell suspensions treated with AMP as a substrate and then with lead nitrate as a phosphate capture agent prior to processing for TEM. The images are from a standard fixation and processing method with no additional staining after thin sectioning (inset, without lead) (A) and a microwave-assisted immunofixation protocol with staining after thin sectioning (inset, minus substrate) (B). Bars, 0.2 μm.
Taken together, analyses of C. perfringens extracts and whole cells suggest the presence of an enzyme with not only biochemical and immunological properties similar to those of the recombinant enzyme generated for the study but also a protein that partitions with the membrane fraction. The presence of a similar-sized antigenic band in the clinical strains suggests the ubiquitous nature of the enzyme in C. perfringens isolates, a property noted by other investigators interested in the clinical utility of this enzyme in C. perfringens identification (7, 42).
DISCUSSION
Reported here are the details of the characterization of the recombinant C. perfringens acid phosphatase along with confirmation of its presence in clinical isolates and localization of the wild-type enzyme to the bacterial membrane. While not the first gram-positive class C acid phosphatase to be characterized, CppA represents the first to be examined from an anaerobic source. CppA is of particular importance, as it serves as a clinically reliable and sensitive enzyme marker to differentiate C. perfringens from other clostridial species (42). The rapid conversion of pNPP to p-nitrophenol by cell-associated acid phosphatase from a colony of obligately anaerobic gram-positive rods is indicative in 95 of 100 isolates of the presence of C. perfringens (1, 7, 42).
To date, a number of enzymes in this class have been identified including enzymes from Pasteurella multocida (6), Bacillus anthracis (9), Streptococcus equisimilis (24), and Francisella tularensis (unpublished data) or purified and characterized enzymes from H. pylori (35), E. meningosepticum (29), S. aureus (6), Mycoplasma bovis (33), and H. influenzae (8, 37, 38). Results from this study suggest that the class C acid phosphatase of C. perfringens possesses many attributes common to those previously examined; however, CppA is devoid of a lipid modification signal sequence. Aside from C. perfringens, the only other clostridial species that encodes a homolog is C. novyi strain NT (Fig. 1). Sequences encoding a class C acid phosphatase were not detected in the complete or draft genome sequences of 17 different Clostridium species, including C. botulinum and C. tetani. The predicted acid phosphatase from C. novyi is 45% identical to CppA at the protein level (within a 234-amino-acid aligned region) and interestingly, contains a LysM domain within the N-terminal region, resulting in a protein that is 52 amino acids longer than CppA. As LysM domains are reported to be involved in peptidoglycan binding (2), this region of the putative C. novyi acid phosphatase may represent a previously unrecognized form of surface anchoring for this enzyme class.
Previous work from this laboratory and others has demonstrated that class C enzymes are metalloenzymes, possess subunit molecular masses of ∼25 to 30 kDa, exist as multimers in solution, are most active between pH 5 and 6, and catalyze the hydrolysis of 5′ and 3′ nucleoside monophosphates in vivo (8, 33, 35, 37); known exceptions are noted in Table 4. Polypeptide sequence alignment of CppA to other class C enzymes and the presence of critical and invariant aspartic acid residues of the DDDD motif suggest that CppA is a bona fide member of the NSAP class C group of enzymes.
TABLE 4.
Properties of class C acid phosphatases
| Enzyme [reference(s)] | dMMa (kDa) | nMMb (kDa) | pH(s) | EDTAc | Cationd | 5′ nucleotidase activitye | 3′ nucleotidase activitye |
|---|---|---|---|---|---|---|---|
| e (P4) (36, 37) | 28 | 55 | 5.1 | − | Cu2+ | − | − |
| HppA (34) | 24.3 | 72 | 4.6, 5.2 | − | Cu2+ | + | − |
| LppC (24) | 32 | UD | 5.0 | + | UD | + | + |
| OlpA (28) | 30 | 30 | 6.0 | − | Mg2+ | + | + |
| SapS (6) | 30 | UD | 5.0 | − | Mg2+ | UD | UD |
| CppA | 31.1 | 62 | 5.6, 6.0 | + | Cr2+ | + | + |
Denatured molecular mass (dMM) by SDS-PAGE.
Native molecular mass (nMM) without N-terminal lipid. UD, undetermined.
−, absent; +, present.
Exogenous cations providing maximum activity.
−, absent; +, present; UD, undetermined.
The CppA subunit molecular mass is 31.1 kDa, slightly higher than most enzymes of the class but less than that of S. equisimilis LppC. Recombinant CppA exists as a dimer in solution as indicated by results from two independent experimental methods. Dimerization is a common and perhaps essential feature of class C enzymes (8, 33, 35, 37, 38). From the e (P4) structure (8), residues implicated to be critical to dimerization include L22, Q39, Y75, W78, R93, W94, and W251. Residues at corresponding positions in CppA are L57, E67, H111, Y114, E129, W130, and F276. Of these, H111 and E129, not accounting for conservative substitutions, differ substantially with regard to hydrophobic character (21), suggesting that at least these two residues may be of less importance with regard to dimer formation in class C enzymes.
While class C enzymes have been shown to be active over the pH range 5 to 7, showing the “typical” bell-shaped pH rate profile as found in a myriad of acid phosphatases, the highest activities for class C enzymes have generally been observed between pH 5.5 and 6.0. While the highest activity of CppA for pNPP was observed at pH 5.6, Van Etten (47) has suggested that values such as this are “misleading as the effects of pH on both Km and Vmax are mixed even at high substrate concentration.” In addition, pNPP is not a physiological substrate and is unusually acidic because of the electron-withdrawing effect of the p-nitro group leading to an ionization behavior that differs drastically from those of physiological substrates. To address such concerns, the pH optimum of rCppA was determined for 5′ UMP, a physiological substrate devoid of the electron-withdrawing group. The pH optimum for this substrate was approximately 6.0, a value consistent with other class C phosphomonoesterases. In addition, results from our kinetic inspection of the [H+] effect on Km are consistent with the findings of Van Etten (47) in that the bell-shaped pH rate profile, as often described for acid phosphatases, is the result of increased Km values associated with substrate ionization.
An intriguing property of rCppA was its retention of activity in the presence of EDTA (Table 4). Such results have also been noted for recombinant S. equisimilis LppC using crude preparations of enzyme and the zymogram technique of activity assessment (24). These results are suggestive that a spectrum of EDTA sensitivity may exist within the class C enzymes. In addition, it has been noted for CppA, as well as other class C enzymes, that activity is enhanced by the addition of divalent cations, most often Mg2+ and Cu2+. The triad of metal enhancers of CppA activity were Cu2+, Co2+, and Cr2+. Whether these metal ions enhance activity by replacement of the active site Mg2+ as observed in the three-dimensional structure of e (P4) (8) or by association with the enzyme at other sites is currently unknown.
The polyspecific nature of class C phosphomonoesterases and their predilection for the catalysis of arylphosphate hydrolysis are properties also exhibited by recombinant CppA. The subject of this study efficiently catalyzed the hydrolysis of both 5′ and 3′ nucleoside monophosphates and displayed detectable activity for phosphoanhydride bonds, properties not shared by all class C enzymes, most notably e (P4) (Table 4). Of the few kinetic parameters known for class C enzymes, the Km constants are approximately 0.5 to 1 mM for 5′ nucleoside monophosphates substrates for the H. pylori enzyme (35) and approximately 0.01 to 0.015 mM for similar substrates of the E. meningosepticum enzyme (29). The observed kinetic parameters of recombinant CppA for physiological substrates (∼0.2 mM [Table 3]) are between those of the former and latter.
Whether C. perfringens CppA plays a critical role in the survival of the bacterium in vivo or in vitro has yet to be determined. Like most other class C phosphomonoesterases, CppA appears to be membrane associated, but via a mechanism independent of the typical fatty acid modification of the N terminus. The absence of an essential cysteine between residues 15 and 35 precludes this polypeptide's inclusion in the standard set of lipoproteins as discussed by Juncker et al. (18). At least one report found in the Soviet literature (19) suggests the presence of a C. perfringens cell wall-associated acid phosphatase; however, we note that CppA lacks the gram-positive cell wall-anchoring motif (LPXTG) as promulgated by Okumura et al. (27). Multiple mechanisms are available to anchor a given polypeptide to a bacterial membrane. For example, Hoopman et al. have demonstrated the existence of an acid phosphatase domain present as the N-terminal portion of the Moraxella catarrahlis MapA membrane-associated autotransporter protein (14). Irrespective of its anchoring mechanism, the preponderance of our data suggests CppA membrane association. While the asymmetric distribution of the enzyme was noted, no gram-positive signal peptides for envelope localization, as discussed by DeDent et al. (5), are present in CppA. As signaling complexes, composed in part by phosphatases, are known to cluster in diverse species of bacteria (11), the polar deposition of this phosphatase may be suggestive of a role in signaling. The presence of the enzyme on the membrane makes CppA a potentially important immunological target, analogous to the e (P4) antigen used in the nontypeable H. influenzae vaccine (13).
The biological role of C. perfringens acid phosphatase CppA remains elusive. A logical choice of function is CppA-catalyzed hydrolysis of 3′ and/or 5′ nucleoside monophosphates, concomitant with or prior to acquisition of either or both products of the hydrolytic reaction, serving perhaps as an important constituent of a nucleotide biosynthetic salvage pathway much like the role ascribed to LppC of S. equisimilis (23). Perhaps like the acid phosphatase of H. influenzae e (P4), to which the transport of nicotinamide mononucleotide (20) or heme (32) into H. influenzae or into specific E. coli auxotrophs (36) has been ascribed, an alternative substrate may be disclosed upon further examination of the clostridial enzyme. Recent results by Philippe et al. (31) suggest that inorganic phosphate concomitantly induces spore morphogenesis and enterotoxin production in C. perfringens. It will be of interest to ascertain whether such induction occurs in the presence of known CppA substrates in C. perfringens strains with active and inactive (D100A) cppA gene products. The nonuniform distribution of phosphatase activity within the cell walls of gelatin-encased suspended clostridial cells observed by TEM in this study suggests that further investigation of this membrane-associated enzymatic activity with respect to cell orientation within a colony structure, at a site of attachment, and relative to a nutrient source may provide needed clues to the biological role of this very active enzyme in C. perfringens.
While much remains to be understood about the CppA acid phosphatase, this report describes the purification and biochemistry of the recombinant enzyme and the native enzyme's presence and activity in type strains and clinical isolates. These findings support the use of this enzyme as a diagnostic tool for differentiation of the clostridial species and suggest its potential for use in C. perfringens vaccine development.
Acknowledgments
We thank the following individuals or entities: Mark Crankshaw and the Protein Sequencing Facility at Washington University (St. Louis, MO); Sherrie Neff and the University of Missouri-Columbia (UM-C) Office of Animal Research; and UM-C Proteomics Center Research Core Facility for services provided; J. G. Songer, M. A. McIntosh, and M. B. Heidari for the C. perfringens strains used in this study; Edward Voss for his enthusiastic guidance and immunochemistry advice; Mark Rudelson for translation of articles published in Russian; Venkataseshu Ganjam for his advice, counsel, and reading of this article; and Howard Wilson for his diligent efforts in preparation of the figures in this article.
Electron microscopy was performed by the authors at the UM-C Electron Microscopy Core Facility hosted by the Department of Veterinary Pathobiology.
Footnotes
Published ahead of print on 10 April 2009.
REFERENCES
- 1.Adcock, P. W., and C. P. Saint. 2001. Rapid confirmation of Clostridium perfringens by using chromogenic and fluorogenic substrates. Appl. Environ. Microbiol. 67:4382-4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman, A., and M. Bycroft. 2000. The structure of a LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD). J. Mol. Biol. 299:1113-1119. [DOI] [PubMed] [Google Scholar]
- 3.Calderone, V., C. Forleo, M. Benvenuti, M. C. Thaller, G. M. Rossolini, and S. Mangani. 2006. The first structure of a bacterial class B acid phosphatase reveals further structural heterogeneity among phosphatases of the haloacid dehalogenase fold. J. Mol. Biol. 365:708-721. [DOI] [PubMed] [Google Scholar]
- 4.Chance, D. L., C. A. Jensen, T. J. Reilly, and T. P. Mawhinney. 2005. Analysis of bacterial colony biology using microwave-assisted processing and LR white resin. Microsc. Microanal. 11(Suppl. 2):968-969. [Google Scholar]
- 5.DeDent, A., T. Bae, D. Missiakas, and O. Schneewind. 2008. Signal peptides direct surface proteins to two distinct envelope locations of Staphylococcus aureus. EMBO J. 27:2656-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.du Plessis, E. M., J. Theron, L. Joubert, T. Lotter, and T. G. Watson. 2002. Characterization of a phosphatase secreted by Staphylococcus aureus strain 154, a new member of the bacterial class C family of nonspecific acid phosphatases. Syst. Appl. Microbiol. 25:21-30. [DOI] [PubMed] [Google Scholar]
- 7.Eisgruber, H., P. Geppert, B. Sperner, and A. Stolle. 2003. Evaluation of different methods for the detection of Clostridium perfringens phosphatases. Int. J. Food Microbiol. 82:81-86. [DOI] [PubMed] [Google Scholar]
- 8.Felts, R. L., Z. Ou, T. J. Reilly, and J. J. Tanner. 2007. Crystallization and preliminary X-ray analysis of a recombinant class C acid phosphatase from Haemophilus influenzae. Biochemistry 46:11110-11116. [DOI] [PubMed] [Google Scholar]
- 9.Felts, R. L., T. J. Reilly, M. J. Calcutt, and J. J. Tanner. 2006. Cloning, purification, and crystallization of Bacillus anthracis class C acid phosphatase. Acta Crystallogr. Sect. F 62:705-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gasteiger, E., C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel, and A. Bairoch. 2005. Protein identification and analysis tools on the ExPASy Server, p. 571-608. In J. M. Walker (ed.), The proteomics protocols handbook. Humana Press, Totowa, NJ.
- 11.Gestwicki, J. E., A. C. Lamanna, R. M. Harshey, L. L. McCarter, L. L. Kiessling, and J. Adler. 2000. Evolutionary conservation of methyl-accepting chemotaxis protein location in Bacteria and Archaea. J. Bacteriol. 182:6499-6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gouet, P., E. Courcelle, D. I. Stuart, and F. Metoz. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305-308. [DOI] [PubMed] [Google Scholar]
- 13.Green, B. A., E. Barany, T. J. Reilly, A. L. Smith, and G. W. Zlotnick. 2005. Certain site-directed nonenzymatically active mutants of the Haemophilus influenzae P4 lipoprotein are able to elicit bactericidal antibodies. Infect. Immun. 73:4454-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoopman, T. C., W. Wang, C. A. Brautigam, J. L. Sedillo, T. J. Reilly, and E. J. Hansen. 2008. Moraxella catarrhalis synthesizes an autotransporter that is an acid phosphatase. J. Bacteriol. 190:1459-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horton, R. M., Z. Cai, S. N. Ho, and L. R. Pease. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528-535. [PubMed] [Google Scholar]
- 16.Horton, R. M., H. D. Hunt, M. M. Ho, J. K. Pullen, and L. R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61-68. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh, H. Y., M. J. Calcutt, L. F. Chapman, M. Mitra, and D. S. Smith. 2003. Purification and characterization of a recombinant α-N-acetylgalactosiminidase from Clostridium perfringens. Protein Expr. Purif. 32:309-316. [DOI] [PubMed] [Google Scholar]
- 18.Juncker, A. S., H. Willenbrock, G. Von Heijne, S. Brunak, H. Nielsen, and A. Krogh. 2003. Prediction of lipoprotein signal peptides in gram-negative bacteria. Protein Sci. 12:1652-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kachelkina, A. I., V. M. Kushanrec, and V. A. Blagoveshchenski. 1967. Localization of phosphatases in Clostridium perfringens cells. Dok. Akad. Nauk 174:700-703. (In Russian.) [PubMed] [Google Scholar]
- 20.Kemmer, G., T. J. Reilly, J. Schmidt-Brauns, G. W. Zlotnick, B. A. Green, M. J. Fiske, M. Herbert, A. Kraiβ, S. Schlör, A. Smith, and J. Reidl. 2001. NadN and e (P4) are essential for utilization of NAD and nicotinamide mononucleotide but not nicotinamide riboside in Haemophilus influenzae. J. Bacteriol. 183:3974-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 22.Lanzetta, P. A., I. J. Alvarez, P. S. Reinach, and O. A. Candia. 1979. An improved assay for detection of nanomole amounts of inorganic phosphate. Anal. Biochem. 100:95-97. [DOI] [PubMed] [Google Scholar]
- 23.Malke, H., and K. Steiner. 2000. Functional properties of the cytoplasmic membrane lipoprotein acid phosphatase of Streptococcus dysgalactiae subspecies equisimilis, p. 879-883. In D. R. Martin and J. R. Tagg (ed.), Streptococci and streptococcal diseases entering the new millennium: proceedings of the XIV Lancefield International Symposium on Streptococci and Streptococcal Diseases, October 11-15 1999, Auckland, New Zealand. Lancefield International Symposium on Streptococci and Streptococcal Diseases, Porirua, New Zealand.
- 24.Malke, H. 1998. Cytoplasmic membrane lipoprotein LppC of Streptococcus equisimilis functions as an acid phosphatase. Appl. Environ. Microbiol. 64:2439-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McClane, B. A., D. M. Lylerly, J. S. Moncrief, and T. D. Wilkins. 2000. Enterotoxic clostridia: Clostridium perfringens type A and Clostridium difficile, p. 551-562. In V. A. Fischetti, R. P. Novick, J. J. Ferretti, D. A. Portnoy, and J. I. Rood (ed.), Gram-positive pathogens. American Society for Microbiology, Washington, DC.
- 26.Myers, G. S. A., D. A. Rasko, J. K. Cheung, J. Ravel, R. Seshadri, R. T. Deboy, Q. Ren, J. Varga, M. M. Awad, L. M. Brinkac, S. C. Daugherty, D. H. Haft, R. J. Dodson, R. Madupu, W. C. Nelson, M. J. Rosovitz, S. A. Sullivan, H. Khouri, G. I. Dimitrov, K. L. Watkins, S. Mulligan, J. Benton, D. Radune, D. J. Fisher, H. S. Atkins, T. Hiscox, B. H. Jost, S. J. Billington, J. G. Songer, B. A. McClane, R. W. Titball, J. I. Rood, S. B. Melville, and I. T. Paulsen. 2006. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 16:1031-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okumura, K., H. I. Kawsar, T. Shimizu, T. Ohta, H. Hayashi, and T. Shimizu. 2005. Identification and characterization of a cell-wall anchored DNase gene in Clostridium perfringens. FEMS Microbiol. Lett. 242:281-285. [DOI] [PubMed] [Google Scholar]
- 28.Ou, Z., R. L. Felts, T. J. Reilly, J. C. Dixon, and J. J. Tanner. 2006. Crystallization and preliminary X-ray analysis of a recombinant class C acid phosphatase from Haemophilus influenzae. Acta Crystallogr. Sect. F 62:464-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Passariello, C., S. Schippa, P. Iori, F. Berlutti, M. C. Thaller, and G. M. Rossolini. 2003. The molecular class C acid phosphatase Chryseobacterium meningosepticum (OlpA) is a broad-spectrum nucleotidase with preferential activity on 5′-nucleotides. Biochim. Biophys. Acta 1648:203-209. [DOI] [PubMed] [Google Scholar]
- 30.Peisach, E., J. D. Selengut, D. Dunaway-Mariano, and K. N. Allen. 2004. X-ray crystal structure of the hypothetical phosphotyrosine phosphatase MDP-1 of the haloacid dehalogenase superfamily. Biochemistry 43:12770-12779. [DOI] [PubMed] [Google Scholar]
- 31.Philippe, V. A., M. B. Mendez, I. Huang, L. M. Orsaria, M. R. Sarker, and R. R. Grau. 2006. Inorganic phosphate induces spore morphogenesis and enterotoxin production in the intestinal pathogen Clostridium perfringens. Infect. Immun. 74:3651-3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reidl, J., and J. Mekalanos. 1996. Lipoprotein e(P4) is essential for hemin uptake by Haemophilus influenzae. J. Exp. Med. 183:621-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reilly, T. J., R. Madupu, B. Methe, and M. J. Calcutt. 2007. Biochemical characterization of a recombinant form of the acid phosphatase lipoprotein Mycoplasma bovis, abstr. G-011, p. 309. Abstr. 107th Gen. Meet. Am. Soc. Microbiol. American Society for Microbiology, Washington, DC.
- 34.Reilly, T. J., R. L. Felts, M. T. Henzl, M. J. Calcutt, and J. J. Tanner. 2006. Characterization of recombinant Francisella tularensis acid phosphatase A. Prot. Expr. Purif. 45:132-141. [DOI] [PubMed] [Google Scholar]
- 35.Reilly, T. J., and M. J. Calcutt. 2004. The class C acid phosphatase of Helicobacter pylori is a 5′ nucleotidase. Protein Expr. Purif. 33:48-56. [DOI] [PubMed] [Google Scholar]
- 36.Reilly, T. J., B. A. Green, G. W. Zlotnick, and A. L. Smith. 2001. Contribution of the DDDD signature motif of H. influenzae lipoprotein e (P4) to phosphomonoesterase activity and heme transport. FEBS Lett. 494:19-23. [DOI] [PubMed] [Google Scholar]
- 37.Reilly, T. J., D. L. Chance, and A. L. Smith. 1999. The outer membrane lipoprotein e (P4) of Haemophilus influenzae is a novel phosphomonoesterase. J. Bacteriol. 181:6769-6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reilly, T. J., and A. L. Smith. 1999. Overexpression, purification, and characterization of recombinant soluble Haemophilus influenzae e (P4) phosphomonoesterase. Protein Expr. Purif. 17:401-409. [DOI] [PubMed] [Google Scholar]
- 39.Reilly, T. J., G. S. Baron, F. E. Nano, and M. S. Kuhlenschmidt. 1996. Characterization and sequencing of a burst inhibiting acid phosphatase from Francisella tularensis. J. Biol. Chem. 271:10973-10983. [DOI] [PubMed] [Google Scholar]
- 40.Rossolini, G. M., M. C. Thaller, P. Pezzi, and G. Satta. 1994. Identification of an E. coli periplasmic acid phosphatase containing a 27 kDa-polypeptide component. FEMS Microbiol. Lett. 118:167-174. [DOI] [PubMed] [Google Scholar]
- 41.Rossolini, G. M., S. Schippa, F. Berlutti, L. E. Macaskie, and M. C. Thaller. 1998. Bacterial nonspecific acid phosphohydrolase: physiology, evolution and use as tools in microbial biotechnology. Cell. Mol. Life Sci. 54:833-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sartory, D. P., R. Waldock, C. E. Davies, and A. M. Field. 2006. Evaluation of acid phosphatase as a confirmation test for Clostridium perfringens isolated from water. Lett. Appl. Microbiol. 42:418-424. [DOI] [PubMed] [Google Scholar]
- 43.Smith, P. K., R. I. Krohn, G. T. Hemanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson, and D. C. Klenk. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76-85. [DOI] [PubMed] [Google Scholar]
- 44.Studier, W. F. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41:207-234. [DOI] [PubMed] [Google Scholar]
- 45.Thaller, M. C., F. Berlutti, S. Schippa, L. Selan, and G. M. Rossolini. 1998. Bacterial acid phosphatase gene fusions useful as targets for cloning-dependent insertional inactivation. Biotechnol. Prog. 14:241-247. [DOI] [PubMed] [Google Scholar]
- 46.Thaller, M. C., S. Schippa, and G. M. Rossolini. 1998. Conserved sequence motifs among bacterial, eukaryotic, and archaeal phosphatases that define a new phosphohydrolase superfamily. Protein Sci. 7:1647-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Etten, R. L. 1982. Human prostatic acid phosphatase: a histidine phosphatase. Ann. N. Y. Acad. Sci. 390:25-51. [DOI] [PubMed] [Google Scholar]
- 48.Vincent, J. B., M. W. Crowder, and B. A. Averill. 1992. Hydrolysis of phosphate monoesters: a biological problem with multiple chemical solutions. Trends Biochem. Sci. 17:S105-S110. [DOI] [PubMed] [Google Scholar]