

Abstract
The Hedgehog (Hh) family of secreted proteins regulates mammalian development and cancer formation through Gli transcription factors, which exist in both activator and repressor forms. In vertebrates, the primary cilia play an essential role in Hh signal transduction and are required for both the activator and repressor activities of Gli proteins. In the current study, we demonstrate that mouse Suppressor of Fused (Sufu) interacts with Gli proteins and inhibits Gli activator activity in the absence of cilia. Removal of Sufu in both Smoothened (Smo) and Ift88 mutants, respectively, leads to full activation of Hh signaling, suggesting that Smo-mediated repression of Sufu, but not the inhibitory function of Sufu, requires cilia. Finally, we show that Sufu is important for proper activator/repressor ratio of Gli3 protein in mice, both in the presence and absence of cilia.
Keywords: cilia, Gli, Hh signaling, IFT88, Smoothened, Sufu
INTRODUCTION
Hedgehog (Hh) proteins are critical for development of diverse animal species (Hooper and Scott, 2005). In Drosophila, Cubitus interruptus (Ci) mediates all transcriptional responses to Hh signaling (Methot and Basler, 2001). Hh inhibits the proteolytic processing of Ci that turns it into a transcriptional repressor. In addition, high levels of Hh convert Ci into a transcriptional activator. Intracellular transduction of Hh signals involves many proteins including transmembrane proteins Patched (Ptc), Smoothened (Smo), and cytoplasmic regulators such as Fused kinase, kinesin-like protein Costal-2 and Suppressor of Fused (Sufu). In vertebrates, there are three homologs of Ci, Gli1, 2 and 3. Gli3 is efficiently processed into a transcriptional repressor in vivo (Wang et al., 2000). Gli2 acts as the primary activator in vivo, in part due to inefficient processing (Pan et al., 2006). Gli1 expression is dependent on Hh signaling, hence it acts as a secondary activator to enhance pathway activation (Bai et al., 2004).
Sufu is largely dispensable for Hh signaling in Drosophila and its role in Hh signaling can only be fully revealed when a positive regulator of Hh signaling (e.g. Fu) is also mutated (Preat, 1992). By contrast, loss of Sufu function leads to full activation of Hh signaling in mammals (Cooper et al., 2005; Svard et al., 2006). In vertebrates, Sufu inhibits Gli-mediated transcriptional activation in the nucleus by recruiting a histone deacetylation complex to Gli target genes (Cheng and Bishop, 2002; Paces-Fessy et al., 2004). Sufu also sequesters Gli in the cytoplasm in the absence of Hh (Ding et al., 1999; Dunaeva et al., 2003; Kogerman et al., 1999; Merchant et al., 2004; Pearse et al., 1999; Stone et al., 1999), but the molecular mechanism underlying this sequestration remains elusive.
Through the study of mouse mutants for intraflagellar transport genes (IFT genes, including Ift88) that fail to form cilia, we have recently discovered an essential role for cilia in Hh signal transduction (Huangfu et al., 2003). Paradoxically, IFT mutants exhibit phenotypes characteristic of loss of transcriptional activator function of Gli proteins (GliA) despite the presence of full-length Gli3 proteins (Haycraft et al., 2005; Huangfu and Anderson, 2005; Liu et al., 2005).
Ciliary localization of key Hh signaling regulators may underlie cilia-dependent Hh signal transduction in mammals. Localization of Ptch1 and Smo in the cilia is regulated by Hh, and appears to be important for their function (Corbit et al., 2005; Rohatgi et al., 2007). Sufu and Gli are also localized to the cilia, but the significance of such localization remains to be addressed(Haycraft et al., 2005).
In the current study, we show that Sufu physically interacts with all three Gli proteins in both wild type and Ift88 mutant cells. We also found that Sufu is sufficient and required for inhibiting Gli-mediated transcriptional activation in Ift88 mutant cells, suggesting that Sufu remains functional in the absence of cilia. Furthermore, double mutant analyses indicate that Sufu is responsible for the loss of GliA function in both Ift88 and Smo mutants. Finally, we found that the ratio between the activator and repressor forms of Gli3 protein, as well as the total level of Gli3 protein in the embryos, is drastically affected by the loss of Sufu function, in the presence and absence of cilia. In conclusion, our data indicate that cilia are dispensable for the function of Sufu in regulating the activity, processing and level of Gli proteins.
MATERIAL AND METHODS
Mutant mice and analysis
Ift88 (Murcia et al., 2000), Sufu (Svard et al., 2006) and Smo (Zhang et al., 2001) mutant mice and embryos were genotyped as described. Immunohistochemistry is performed as described (Hoover et al., 2008).
Primary cell culture and immunocytochemistry
Mouse embryonic fibroblasts were generated from individual E9.5 embryos as described (Hoover et al., 2008; Svard et al., 2006). Specifically, cells from individual E9.5 embryos were dissociated by passage through Gauge 20 needles, and plated in DMEM/F12 supplemented with 15% fetal bovine serum (FBS), non-essential amino acid, sodium pyruvate, Glutamax and antibiotics, at 37°C and 5% CO2. In order to confirm the genotypes of the wild type and Ift88 mutant cells, cells are allowed to grow confluent in a 60mm dish and lysed, followed by polymerase chain reaction (PCR) and immunoblot assays.
To visualize primary cilia, cells growing on gelatin-coated glass coverslips for 48 hours in medium containing 0.5% FBS are labeled with antibodies against acetylated tubulin (Sigma, T7451) or γ-tubulin (Sigma, T5326), and visualized under a Nikon E600 microscope.
Cell transfection and reporter assays
Transient cell transfection was performed using Lipofectamine 2000 (Invitrogen). An 8×GliBS-firefly luciferase reporter was used for quantitation of GliA activity (Sasaki et al., 1997), Human Gli1, Gli3 and mouse Gli2, Sufu were cloned into mammalian expression vectors with Myc (pCS2+), FLAG (pFLAG-CMV2, Sigma) or GFP (pEGFPC1, Invitrogen) tags. Plasmids expressing shRNAs against Sufu were obtained from Dr. Taipale and were used as described (Varjosalo et al., 2006). pTK-RL (Renilla luciferase) was used as transfection control. Luciferase activity was detected using a Dual-luciferase reporter assay system (Promega), in a Modulus luminometer (Turner Biosys). Data analyses including the student t-tests were performed using Microsoft Excel. All assays were done in triplicates and repeated at least three times.
Coimmunoprecipitation and Western blotting
Co-immunoprecipitation between over-expressed proteins were performed using a FLAG Tagged protein Immunoprecipitation kit (Sigma). Endogenous Sufu was precipitated with a goat polyclonal antibody (M-15, Santa Cruz Biotech) immobilized on protein A-Agarose beads (Santa Cruz Biotech). Proteins are separated on SDS-PAGE for immunoblots with standard procedures. Quantitation of the immunoblots was performed using NIH ImageJ.
A rabbit polyclonal antibody against mouse IFT88 was generated against a synthesized peptide at the N-terminus of the protein as described (Taulman et al., 2001), and its specificity was confirmed by immunofluorescence and immunoblot assays with whole cell lysates of wild type and Ift88 mutant embryos. Gli3 was detected with two rabbit polyclonal antibodies (Chen et al., 2004; Wang et al., 2000). A rabbit polyclonal antibody (H-300, Santa Cruz Biotech) was used to detect endogenous Sufu in immunoblots to avoid cross-reaction with goat IgG used for immunoprecipitation. Actin (anti-Actin, Santa Cruz Biotech) and tubulin (anti-α/β-Tubulin, Cell Signaling) serve as loading controls.
RESULTS AND DISCUSSION
Cilia are absent on Ift88 mutant cells
In order to unambiguously address whether Sufu requires cilia to inhibit Gli activity and Hh signaling, it is critical to establish a tissue culture system in which cilia formation is completely disrupted. An apparent null allele for mouse Ift88, Ift88Δ2-3βGal (originally named Tg737Δ2-3βGal), generated through the replacement of the first two coding exons with a lacZ-expression cassette, produces severely truncated Ift88 mRNA and no IFT88 protein (Murcia et al., 2000; Taulman et al., 2001). As a result, cilia formation is completely disrupted in Ift88Δ2-3βGal homozygous mutant embryos (Murcia et al., 2000; and our unpublished data). We established primary embryonic fibroblast (MEF) culture from these mutant embryos as well as their wild type littermates as control. To test whether we have established pure culture of Ift88Δ2-3βGal homozygous mutants, we performed genomic PCR that distinguishes the Ift88Δ2-3βGal allele from the wild type allele. No wild type PCR product was detected in the mutant cell lysate after 55 cycles of amplification, indicating no contamination from wild type or heterozygous cells (Fig. 1A). We further tested these cells in an immunoblot assay with a mouse IFT88 specific antibody and confirmed the absence of IFT88 protein (Fig. 1B). Finally, we examined whether cilia formation is completely disrupted in these cells via immunocytochemistry. Using an antibody against acetylated α-tubulin, we labeled the axonemes and the basal bodies of the primary cilia (Fig. 1C, 308/356 = 86% ciliated). In Ift88Δ2-3βGal homozygous mutant cells, no axonemal staining was detected (Fig. 1D, n=388). Instead, we found punctuate doublet staining in many mutant cells characteristic of the basal bodies or centrosomes (Fig. 1D). We confirmed the presence of the basal bodies or centrosomes in the Ift88Δ2-3βGal mutant cells with the basal body/centrosome marker γ-tubulin (Fig. 1E, F). In summary, we had established an in vitro tissue culture system to specifically address the requirement for primary cilia. For the rest of this report, we will refer to homozygous Ift88Δ2-3βGal mutant (cells and embryos) as Ift88 mutant.
Figure 1.

Cilia do not form in Ift88 mutant MEFs. (A) Genomic PCRs using primers specific to wild type or Ift88 mutant allele are performed on MEF cell lysates. No wild type product is detected in the Ift88 mutant cells. DNA prepared from the tail of a heterozygous Ift88 mutant mouse is used as control. (B) Western blot is performed on MEF cell lysates using an IFT88-specific antibody. No IFT88 protein is detected in the Ift88 mutant MEFs. Tubulin is used as a loading control. (C) Acetylated α-tubulin is present in the basal body (arrowhead) and axoneme (arrow) of the cilia in wild type cells. (D) Acetylated α-tubulin is present in punctuate doublets (arrowheads) in Ift88 mutant cells. (E, F) γ-tubulin is present in punctuate doublets (arrowheads) in both wild type (E) and Ift88 mutant (F) cells. DNA is stained blue in C–F.
Sufu physically interacts with all Gli proteins in the absence of cilia
We first examined whether Sufu interacts with Gli proteins in the absence of cilia through coimmunoprecipitation analyses between Sufu and all three Gli proteins. As previously reported (Ding et al., 1999; Dunaeva et al., 2003; Kogerman et al., 1999; Pearse et al., 1999; Stone et al., 1999), over-expressed Gli1, Gli2 and Gli3 co-immunoprecipitate with Sufu in wild type mouse embryonic fibroblasts (MEFs; Fig 2A–C). Interestingly, Sufu co-immunoprecipitates with all three Gli proteins in Ift88 mutant MEFs, indicating the physical interactions between Sufu and Gli proteins are not dependent on cilia (Fig 2A–C).
Figure 2.

Sufu physically interacts with all Gli proteins in the absence of cilia. (A–C) Sufu co-immunoprecipitates with Gli1 (A), Gli2 (B) and Gli3 (C) when over-expressed in wild type and Ift88 mutant cells. (D) Endogenous Gli3 co-immunoprecipitates with Sufu, but not by unrelated antibodies (anti-Myc) in wild type and Ift88 mutant cells. Panels shown in D are assembled from one single experiment in which same experimental procedure was applied to both experimental and control samples. For each panel, the input lanes are loaded with 10% of protein lysate used for immunoprecipitation.
We next examined whether endogenous Sufu interacts with Gli proteins in the absence of cilia, using antibodies capable of detecting endogenous Sufu and Gli3 in immunoblots. We found that endogenous Sufu and Gli3 coimmunoprecipitate in both wild type and Ift88 mutant cells, indicating that their interaction does not depend on cilia (Fig 2D).
Sufu inhibits Gli-activated reporter expression in the absence of cilia
We next addressed whether Sufu inhibits GliA activity in the absence of cilia using a Gli-responsive reporter (Sasaki et al., 1999). Consistent with a previous report, over-expression of Gli1 significantly activates reporter expression in both wild type and Ift88 mutant cells (Fig 3A, p< 0.01)(Haycraft et al., 2005). Interestingly, when we co-transfected the cells with Gli1 and Sufu, the expression of the reporter is decreased in both wild type and Ift88 mutant cells compared to the cells transfected with Gli1 alone, suggesting that Sufu inhibits GliA function in the absence of cilia (Fig 3A, p<0.01).
Figure 3.

Sufu inhibits Gli activities in the absence of cilia. Wild type and Ift88 mutant MEFs are co-transfected with a Gli-responsive luciferase reporter and various effectors. Activities of the firefly luciferase are plotted to reflect the relative expression level of the reporter. (A) Sufu inhibits Gli1-mediated activation of the reporter in wild type and Ift88 mutant cells. (B) Sufu inhibits Gli2-mediated activation of the reporter in wild type and Ift88 mutant cells. (C) Reducing the level of Sufu protein by RNA interference activates a Gli-responsive reporter expression in both wild type and Ift88 mutant cells. Immunoblots below each chart shows the relevant proteins present in the same volume of lysate used for luciferase activity measurement. In C, the relative amount of Sufu protein is calculated and shown below the immunoblots. The amount in cells treated with scrambled shRNA is arbitrarily set to 1.
We next examined whether Gli2, the primary activator of the Hh signaling, is subject to inhibitory control by Sufu in the absence of cilia. We found that Gli2 activates reporter expression, and that this activation is inhibited by Sufu, in both wild type and Ift88 mutant cells (Fig 3B, p< 0.01).
Our result is different from a previous report that over-expression of Gli1, but not Gli2, activates Ptch1 expression in the absence of cilia (Haycraft et al., 2005). The Difference in the levels of Gli2 expression, which was not shown in the previous report, is one possible explanation. Alternatively, our use of a reporter gene solely responsive to GliA activation could underlie the differential outcome since the regulation of endogenous Ptch1 expression may be more complex. Finally, we normalized our reporter expression against a cotransfected Renilla-luciferase control in order to eliminate the impact of differing transfection efficiency. In contrast, in the previous report, Ptch1 expression was normalized against actin, which is expressed in both transfected and non-transfected cells.
Reducing Sufu protein level activates Hh signaling in the absence of cilia
If the inhibitory function of Sufu on Gli activity is dependent on cilia, reducing Sufu in Ift88 mutant cells should not affect Hh signaling. We hence reduced the level of Sufu in wild type and Ift88 mutant cells by RNA interference (RNAi). As reported previously (Varjosalo et al., 2006), a moderate reduction of Sufu protein (by 35%–60%) significantly activates the Gli-responsive reporter gene expression in wild type cells (Fig 3C, p<0.01). We found similar activation of the reporter gene expression in Ift88 mutant cells, indicating that Sufu retains its inhibitory function on GliA in the absence of cilia (Fig 3C, p<0.01).
The relationship between Sufu and cilia was examined in a recent study with MEFs derived from mouse embryos carrying a point mutation in Ift172, encoding another IFT complex component, or an in-frame deletion in Dnchc2, encoding a subunit of the retrograde IFT motor (Ocbina and Anderson, 2008). In contrast to our results, it was reported that mutations in these two genes completely abolished the ability of the cells to activate Hh signaling in response to the reduction of Sufu. This is surprising especially because ciliogenesis is not completely blocked and Hh signaling is less severely disrupted in Dnchc2 mutant embryos compared to the Ift88 mutant cells used in our study (Huangfu and Anderson, 2005). It is not yet clear what underlies the difference between the reported result and ours regarding the relationship between Sufu and cilia. However, as detailed below, our conclusion that Sufu remains functional in the absence of cilia is well supported by in vivo epistatic analysis. It is critical to determine whether these two genes (Ift172 and Dnchc2) are fully epistatic to Sufu, as suggested by their in vitro results, in a more stringent in vivo double mutant analysis.
Sufu inhibits Hh signaling in developing embryos in the absence of cilia
The ability of Sufu to inhibit GliA activity in cultured cells despite the complete loss of cilia suggests that the inhibitory function of Sufu may be responsible for the lack of GliA activity in Ift88 mutant embryos. We tested this hypothesis by examining Hh signaling in Sufu/Ift88 double homozygous mutants.
At E9.5, the homozygous Sufu and Ift88 mutant embryos are morphologically distinct (Cooper et al., 2005; Murcia et al., 2000; Svard et al., 2006). In Ift88 mutants, the neural tube closure defects (NTD) are restricted to the midbrain and posterior forebrain (Fig. 4A, B). Moreover, in the brain region with NTD, the neural ridges are elevated, suggesting the initiation of neurulation is not disrupted. In contrast, Sufu mutants exhibit extensive NTD including the entire brain and part of the spinal cord with no neural ridge elevation (Fig 4C). Sufu/Ift88 double mutant embryos are morphologically indistinguishable from Sufu single mutants, suggesting Sufu is epistatic to Ift88 (Fig 4D).
Figure 4.

Sufu is epistatic to Ift88. (A–D) lateral views of E9.5 mouse embryos. Arrowheads in B point to elevated neural ridges in the brain of an Ift88 mutant embryo. Note that there is no neural ridge elevation in the brains of the Sufu (C) and Sufu/Ift88 double (D) mutant embryos. (E–H) Ptch1 expression in E9.5 whole embryos detected by wholemount RNA in situ hybridization. Ptch1 expression is reduced in an Ift88 mutant embryo (F), but upregulated in the Sufu (G) and Sufu/Ift88 double (H) mutant embryos. (I–J) Gli1 expression in E9.5 whole embryos detected by wholemount RNA in situ hybridization. Gli1 expression is reduced in an Ift88 mutant embryo (J), but upregulated in the Sufu (K) and Sufu/Ift88 double (L) mutant embryos
To address whether the morphological similarity between Sufu and Sufu/Ift88 double mutants is associated with similar Hh signaling activity, we examined the expression of Hh target genes Gli1 and Ptch1 in these embryos. Both genes are expressed in tissues adjacent to the source of Hh ligands, such as the ventral neural tube and somites (Fig. 4E, I). In Ift88 mutants, expression of both genes is reduced, suggesting reduced Hh signaling and GliA function (Fig. 4F, J). In Sufu mutants, Gli1 and Ptch1 are highly expressed in broad regions of the embryo, indicating ectopic activation of the Hh signaling pathway (Fig. 4G, K). Sufu/Ift88 double mutants exhibit the same broad expression of Gli1 and Ptch1 as Sufu mutants, indicating that Hh signaling remains under the negative regulation of Sufu in the absence of cilia (Fig 4H, L).
Sufu regulates ventral spinal cord patterning in the absence of cilia
Hh signaling plays essential roles in establishing the dorsal-ventral patterning in the spinal cord (Dessaud et al., 2008). Therefore, by comparing the spinal cord patterning in Sufu/Ift88 double mutant embryos with those in Sufu and Ift88 single mutants, we can determine the genetic interaction between the two genes in Hh pathway regulation. At E9.5, the floorplate marker Foxa2 and a marker for V3 interneuron precursors Nkx2.2 are co-expressed in the ventral-most region of the spinal cord (Fig 5A,B). Pax6 is strongly expressed in the progenitor cells for the dorsal and lateral interneurons, as well as motor neurons (Fig 5C). In Ift88 mutant spinal cord, Foxa2 and Nkx2.2-expressing cells are absent, whereas Pax6 is expressed throughout the DV axis (Fig 5D–F). By contrast, the Sufu mutant spinal cords are severely ventralized, with Foxa2 and Nkx2.2-expression expanded into the dorsal spinal cord (Fig 5G,H). Pax6, on the other hand, is not expressed (Fig 5I). Sufu/Ift88 double mutants exhibit spinal cord ventralization indistinguishable from Sufu mutants, suggesting activation of Hh signaling in all spinal cord cells (Fig 5J–L).
Figure 5.
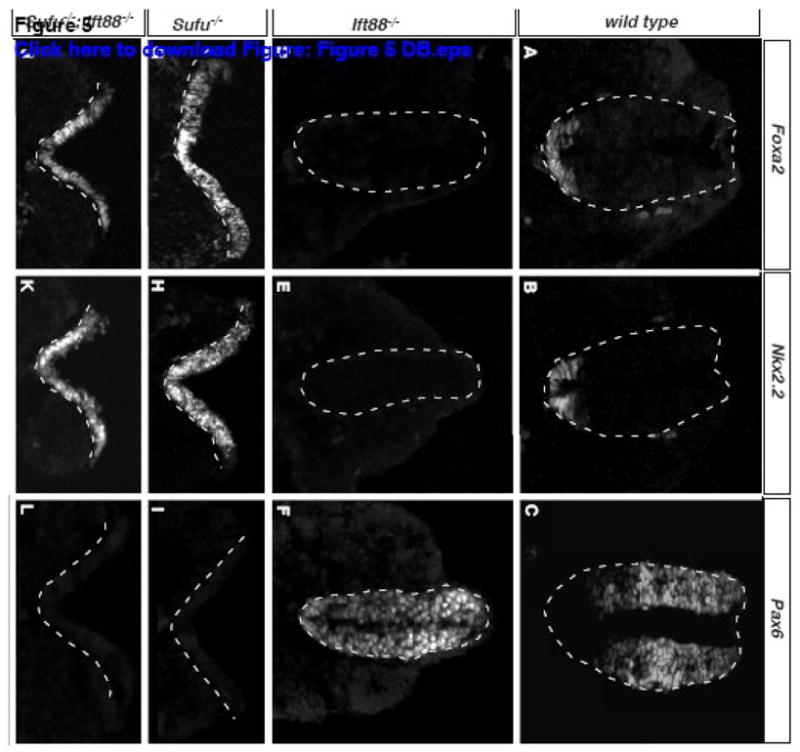
Sufu regulates spinal cord patterning in the absence of cilia. (A) Foxa2 labels the floor plate in wild type spinal cord. (B) Nkx2.2 labels the precursors for V3 interneurons. (C) Pax6 is present in the neural progenitor cells of the dorsal two-thirds of the spinal cord. (D) Foxa2 and (E) Nkx2.2 are absent in Ift88 mutants. (F) Pax6 is present throughout the spinal cord in Ift88 mutants. (G) Foxa2 and (H) Nkx2.2 are present throughout the spinal cord of the Sufu mutants. (I) Pax6 is absent in the Sufu mutant spinal cord. (J) Foxa2 and (K) Nkx2.2 are present throughout the spinal cord of Sufu/Ift88 double mutants. (L) Pax6 is absent in the Sufu/Ift88 double mutant spinal cord. Shown are immunofluorescent images of transverse sections of E9.5 embryos. Dashed lines outline the spinal cords.
The full activation of Hh signaling in Sufu/Ift88 double mutants strongly indicates that Sufu retains its inhibitory function in the absence of cilia and is likely responsible for the lack of GliA activity in Ift88 mutants. Therefore, we concluded that the inhibitory function of Sufu on GliA activity and Hh signaling does not rely on cilia, despite their co-localization to the tip of primary cilia in wild type cells (Haycraft et al., 2005).
Smo activates Hh signaling by antagonizing the inhibitory function of Sufu
Smo is translocated to the primary cilia in the presence of Hh ligand, and its ciliary localization is essential for Hh pathway activation (Corbit et al., 2005). We hypothesize that the ciliary translocation of Smo allows it to antagonize the function of Sufu, hence activates GliA function. This hypothesis predicts that the function of Smo can only be revealed in the presence of both cilia and Sufu. It has been shown that the removal of Smo has no effect on Hh signaling in the absence of cilia (Huangfu and Anderson, 2005). We sought to address the relationship between Smo and Sufu by characterizing the spinal cord patterning in Sufu/Smo double mutants.
In order to analyze the DV patterning of the spinal cord, we examined three genes that are expressed in neural progenitor cells at different positions along the DV axis of E9.5 spinal cord. Nkx2.2 is expressed in the precursors of V3 interneurons, which is the ventral-most neuronal cell type (Fig. 6A). Pax6 is expressed in dorsal two-thirds of the spinal cord (Fig. 6B). Pax7 labels dorsal-most region of the spinal cord (fig. 6C). In the Smo mutant spinal cord, lack of Hh signaling activity leads to the loss of Nkx2.2 expression (Fig. 6D), as well as the ventral expansion of Pax6 and Pax7 expression (Fig. 6E, F). In Sufu mutants, Nkx2.2 is expressed throughout the DV axis of the spinal cord (Fig. 6G), whereas the expression of Pax6 and Pax7 is absent (Fig. 6H, I). The Sufu/Smo double mutant spinal cords are indistinguishable from those in Sufu mutants, with widespread Nkx2.2 expression (Fig. 6J) at the expense of Pax6 and Pax7 expression (Fig. 6K, L). The identical phenotype between Sufu mutants and Sufu/Smo double mutants indicates that loss of Smo has no impact on Hh signaling and embryonic patterning in the absence of Sufu, suggesting that Smo activates GliA activity by antagonizing the inhibitory function of Sufu.
Figure 6.
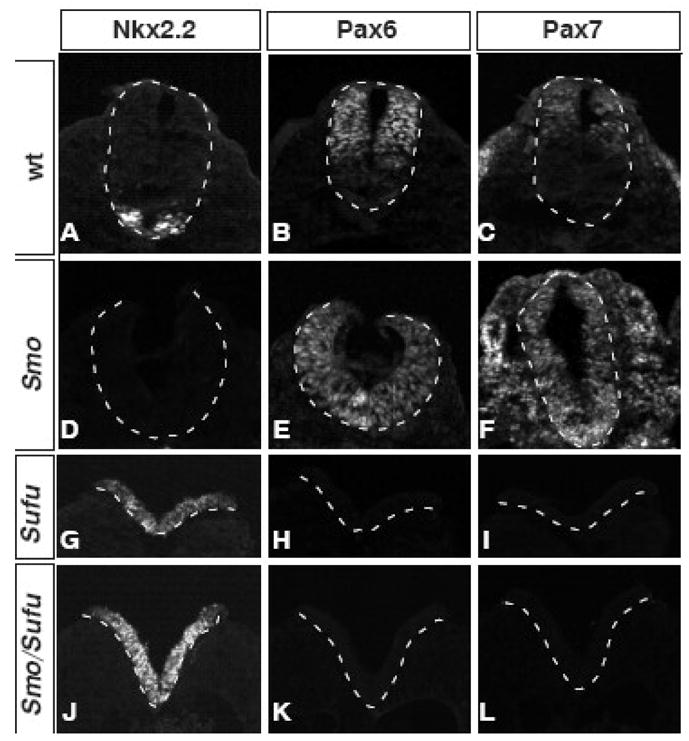
Sufu regulates spinal cord patterning downstream of Smo. (A) Nkx2.2 labels the precursors for V3 interneurons. (B) Pax6 is present in the neural progenitor cells of the dorsal two-thirds of the spinal cord. (C) Pax7 is present in the dorsal progenitor cells of the wild type spinal cord. (D) Nkx2.2 is absent in Smo mutants. (E) Pax6 is present throughout the spinal cord in Smo mutants. (F) Pax7 is present throughout the spinal cord in Smo mutants. (G) Nkx2.2 is present throughout the spinal cord of Sufu mutant. (H) Pax6 is absent in the Sufu mutant spinal cord. (I) Pax7 is absent in the Sufu mutant spinal cord. (J) Nkx2.2 is present throughout the spinal cord of Sufu/Smo double mutant. (K) Pax6 is absent in the Sufu/Smo double mutant spinal cord. (L) Pax7 is absent in the Sufu/Smo double mutant spinal cords. Shown are immunofluorescent images of transverse sections of E9.5 embryos. Dashed lines outline the spinal cord.
It is unclear how Smo antagonizes Sufu function molecularly. It could directly interact with Sufu. Alternatively, it might modify Gli proteins such that they are no longer subject to Sufu inhibition. In either scenario, genetic data suggest that this function of Smo requires cilia (Huangfu and Anderson, 2005). In the absence of cilia, Gli is under constitutive inhibition of Sufu, leading to the loss of GliA activity. At least in the case of Gli3, this occurs despite an increase in the level of full-length Gli3 protein as the result of reduced proteolytic processing.
Loss of Sufu alters the ratio between the activator and repressor forms of Gli3 protein
Gli3 is proteolytically processed from a 190 kD precursor (Gli3-190) into an 83-kD transcriptional repressor (Gli3-83) in vivo and this processing is regulated by Hh signaling (Wang et al., 2000; Fig 7). We sought to address whether the Gli3-190/Gli3-83 ratio is regulated by Sufu and whether this regulation is affected by the loss of cilia. For this purpose, we directly examined Gli3 protein by immunoblots in Sufu and Sufu/Ift88 double mutants.
Figure 7.

Sufu regulates the ratio between the activator and repressor forms of Gli3 protein independent of cilia. (A) Immunoblot showing the level of Gli3-190 and Gli3-83 in E9.5 wild type and different combinations of Sufu and Ift88 mutants. Each lane was loaded with protein extracted from a single embryo. (B) Immunoblot showing the levels of full length Gli3 (Gli3-190) and processed product (Gli3-83) in wild type E12.5 limb bud, and MEFs derived from E9.5 wild type, Sufu and Ptch1 mutant embryos. Tubulin was used as loading control in both A and B. (C) Graphical representation of the Gli3-190 versus Gli3-83 ratio in E9.5 wild type and different combinations of Sufu and Ift88 mutants. (D) Graphical representation of the relative level of total Gli3 protein (Gli3-190 and Gli3-83). The relative level of Gli3 protein in wild type was set to one. Note that the ratios in C and D may be under-estimated due to saturation of band density in A and B. Graphics in C and D summarize data from at least 3 embryos of each genotype.
We first examined Gli3 protein in E9.5 whole embryo extracts. In wild type embryos, there is more Gli3-83 compared to Gli3-190, due to efficient proteolytic processing (Fig. 7A, C). We found that in both Sufu and Ift88 mutants, there are more Gli3-190 than Gli3-83 (Fig. 7A and C). The Gli3-190/Gli3-83 ratio appears to be much higher in Sufu mutants compared to that in Ift88 mutants (Fig. 7A and C). The ratio between the two forms of Gli3 in Sufu−/−;Ift88−/− double mutants is indistinguishable from that in Sufu mutants, suggesting that Sufu regulates the ratio between these two forms of Gli3 protein in the absence of cilia (Fig. 7A, C). In conclusion, our results indicate that Sufu plays essential roles in regulating the ratio between Gli3-190 and Gli3-83, likely through regulating the proteolytic processing of Gli3. An alternative explanation is that in the absence of Sufu, Gli3-83 is highly unstable, leading to the change in the Gli3-190/Gli3-83 ratio.
We next examined Gli3 protein in MEFs derived from E9.5 mouse embryos. Similar to what we found in the whole embryo lysates, a significant amount of Gli3 protein is processed into Gli3-83 in wild type MEFs (Fig 7B). In contrast, Gli3-83 is barely detectable in Sufu−/− MEFs, drastically increasing the Gli3-190/Gli3-83 ratio (Fig. 7B and data not shown).
Interestingly, there is a drastic decrease in the total level of Gli3 protein in both Sufu and Sufu/Ift88 double homozygous mutant embryos, as well as in cultured Sufu mutant cells (Fig. 7A, B and D). The reduction of Gli3 protein in Sufu mutant embryos may partly result from the decrease in Gli3 transcription due to broad activation of Hh signaling (Svard et al., 2006). Additionally, Sufu may play important roles in the translation or degradation of Gli3 protein. Consistent with this additional role for Sufu, loss of Sufu in Drosophila leads to a drastic reduction in the level of Ci protein without affecting its transcription (Ohlmeyer and Kalderon, 1998). We also found that the level of Gli3 protein is largely normal in Ptch1 mutants, which exhibit similar activation of the Hh pathway to Sufu mutants, suggesting that the drastic decrease in Gli3 protein level in Sufu mutants is not solely the result of Hh pathway activation (Goodrich et al., 1997). Revealing the roles of Sufu in Gli3 post-transcriptional regulation will need the uncoupling of Gli3 transcription and Hh pathway activation. It is also interesting to investigate the roles for Sufu in the regulation of endogenous Gli1 and Gli2 when the antibodies are available.
In conclusion, we have provided evidence supporting cilia-independent roles for Sufu in regulating the activator activities of Gli proteins, as well as post-transcriptional regulation of Gli3. Molecularly, Sufu may interact with Gli prior to cilia entrance. Alternatively, as Sufu antagonizes Gli activity in the nucleus (Cheng and Bishop, 2002; Paces-Fessy et al., 2004), our data may be an indication that cilia localization of Sufu is not a prerequisite for its nuclear function. In either case, our data strongly suggest that the inhibitory function of Sufu on GliA and Hh signaling does not rely on cilia. Nevertheless, our current work, combined with previous studies (Corbit et al., 2005; Huangfu and Anderson, 2005), indicates that inactivation of Sufu by a Smo-mediated mechanism is key to the activation of GliA, and is likely dependent on cilia. Our current work provides a parsimonious explanation for the roles of Smo, Sufu and cilia in Gli activation, as well as a framework for further investigation of the molecular mechanisms underlying the cilia-dependent Hh signaling in vertebrates.
Acknowledgments
We thank Drs Z. Lai and D. Cavener for critically reading the manuscript and Dr. M. Kasper for technical advice. We thank Drs N. Murcia and A. McMahon for providing mouse mutants for Ift88 and Smo, and Dr. S. Mackem and Dr. B. Wang for Gli3 antibodies. We thank Dr. J. Taipale for shRNAs against mouse Sufu. The monoclonal antibodies against Foxa2, Nkx2.2 and Pax6 were obtained from the Developmental Studies Hybridoma Bank. This work is supported by grants from the Swedish Cancer Society, the Swedish Research Council, NIH Program Project Grant AR47898 and NIH/NCI MMHCC Grant U01 CA105491 to RT, a Swedish Research Council grant to ST, and a Penn State University new lab startup fund to AL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–15. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Chen Y, Knezevic V, Ervin V, Hutson R, Ward Y, Mackem S. Direct interaction with Hoxd proteins reverses Gli3-repressor function to promote digit formation downstream of Shh. Development. 2004;131:2339–47. doi: 10.1242/dev.01115. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Bishop JM. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc Natl Acad Sci U S A. 2002;99:5442–7. doi: 10.1073/pnas.082096999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, Bale AE. Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development. 2005;132:4407–17. doi: 10.1242/dev.02021. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–21. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173–86. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- Ding Q, Fukami S, Meng X, Nishizaki Y, Zhang X, Sasaki H, Dlugosz A, Nakafuku M, Hui C. Mouse suppressor of fused is a negative regulator of sonic hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol. 1999;9:1119–22. doi: 10.1016/s0960-9822(99)80482-5. [DOI] [PubMed] [Google Scholar]
- Dunaeva M, Michelson P, Kogerman P, Toftgard R. Characterization of the physical interaction of Gli proteins with SUFU proteins. J Biol Chem. 2003;278:5116–22. doi: 10.1074/jbc.M209492200. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–17. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- Hoover AN, Wynkoop A, Zeng H, Jia J, Niswander LA, Liu A. C2cd3 is required for cilia formation and Hedgehog signaling in mouse. Development. 2008 doi: 10.1242/dev.029835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–7. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, Toftgard R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–9. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–11. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Merchant M, Vajdos FF, Ultsch M, Maun HR, Wendt U, Cannon J, Desmarais W, Lazarus RA, de Vos AM, de Sauvage FJ. Suppressor of fused regulates Gli activity through a dual binding mechanism. Mol Cell Biol. 2004;24:8627–41. doi: 10.1128/MCB.24.19.8627-8641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, Basler K. An absolute requirement for Cubitus interruptus in Hedgehog signaling. Development. 2001;128:733–42. doi: 10.1242/dev.128.5.733. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127:2347–55. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- Ocbina PJ, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn. 2008;237:2030–8. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmeyer JT, Kalderon D. Hedgehog stimulates maturation of Cubitus interruptus into a labile transcriptional activator. Nature. 1998;396:749–53. doi: 10.1038/25533. [DOI] [PubMed] [Google Scholar]
- Paces-Fessy M, Boucher D, Petit E, Paute-Briand S, Blanchet-Tournier MF. The negative regulator of Gli, Suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem J. 2004;378:353–62. doi: 10.1042/BJ20030786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–77. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse RV, 2nd, Collier LS, Scott MP, Tabin CJ. Vertebrate homologs of Drosophila suppressor of fused interact with the gli family of transcriptional regulators. Dev Biol. 1999;212:323–36. doi: 10.1006/dbio.1999.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preat T. Characterization of Suppressor of fused, a complete suppressor of the fused segment polarity gene of Drosophila melanogaster. Genetics. 1992;132:725–36. doi: 10.1093/genetics/132.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–6. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124:1313–22. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–24. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- Stone DM, Murone M, Luoh S, Ye W, Armanini MP, Gurney A, Phillips H, Brush J, Goddard A, de Sauvage FJ, Rosenthal A. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J Cell Sci. 1999;112 (Pt 23):4437–48. doi: 10.1242/jcs.112.23.4437. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–97. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12:589–99. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M, Li SP, Taipale J. Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev Cell. 2006;10:177–86. doi: 10.1016/j.devcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–34. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell. 2001;106:781–92. [PubMed] [Google Scholar]