

Abstract
Shifts in microbial communities are implicated in the pathogenesis of a number of gastrointestinal diseases, but we have limited understanding of the mechanisms that lead to altered community structures. One difficulty with studying these mechanisms in human subjects is the inherent baseline variability of the microbiota in different individuals. In an effort to overcome this baseline variability, we employed a mouse model to control the host genotype, diet, and other possible influences on the microbiota. This allowed us to determine whether the indigenous microbiota in such mice had a stable baseline community structure and whether this community exhibited a consistent response following antibiotic administration. We employed a tag-sequencing strategy targeting the V6 hypervariable region of the bacterial small-subunit (16S) rRNA combined with massively parallel sequencing to determine the community structure of the gut microbiota. Inbred mice in a controlled environment harbored a reproducible baseline community that was significantly impacted by antibiotic administration. The ability of the gut microbial community to recover to baseline following the cessation of antibiotic administration differed according to the antibiotic regimen administered. Severe antibiotic pressure resulted in reproducible, long-lasting alterations in the gut microbial community, including a decrease in overall diversity. The finding of stereotypic responses of the indigenous microbiota to ecologic stress suggests that a better understanding of the factors that govern community structure could lead to strategies for the intentional manipulation of this ecosystem so as to preserve or restore a healthy microbiota.
A highly diverse, complex community of microorganisms inhabits the gastrointestinal tracts of mammals. This community, referred to as the indigenous microbiota, exists in a delicate symbiosis with the host (3, 15). A significant body of research has demonstrated that disturbances in this balance can disrupt intestinal homeostasis. Multiple disease states may arise, at least in part, in response to altered indigenous microbial communities in the gut (10, 47, 53, 55). Conversely, research on probiotics indicates that the normal balance between the indigenous microbiota and the host can be protected or restored through the administration of beneficial microbes (6, 45, 59).
The relationship between the indigenous microbiota and a host involves multiple interactions. The indigenous microbiota plays a central role in the digestion and nutrition of the host (30, 55). These microbes also affect the regulation and homeostasis of the host immune system (27, 46). As part of the innate defenses of the gastrointestinal tract, the community of indigenous microbes forms an ecological barrier that prevents the ingress of pathogenic microorganisms. For example, the development of Clostridium difficile-associated colitis following antibiotic administration arises from a loss of intrinsic “colonization resistance” against pathogenic organisms (60). Antibiotic disturbance of the normal community structure of the microbiota may allow the germination of environmentally acquired spores, with subsequent overgrowth of the pathogen and toxin production. Alternatively, C. difficile colitis may develop subsequent to the expansion of low-abundance C. difficile populations that normally produce insignificant levels of toxin. In either case, the disruption of the indigenous microbiota by antibiotic administration is a key component of pathogenesis (7).
Murine models have provided important insights into the interactions between the microbiota and the host. One consistent feature of microbiota studies with human subjects is that there is significant interindividual variation in the indigenous microbiota (12, 13). This variation likely arises from the accumulated effects of genetic and environmental influences on the gut microbial community (11). The significant baseline variation makes it difficult to conduct studies that follow the dynamics of the gut microbiota in humans, especially if the goal is to discern stereotypic responses to a given manipulation. Therefore, as with other areas of biomedical research, murine models offer unique advantages for microbiota experimentation.
Several recent studies have described murine models of disease in which altered indigenous gut microbial communities are generated through the administration of antibiotics. These altered communities can be either permissive or required for the development of the model disease state, although in other cases they appear to be protective (5, 9, 24, 26, 52). Although these studies have provided insight into many of the host responses to the indigenous microbiota, we have remarkably little information as to the exact nature of the effect of antibiotic administration on the microbial communities themselves. For example, these studies assume that genetically identical mice would harbor a consistent baseline microbiota. Furthermore, it is also assumed that the microbiota responds in a reproducible manner to the antibiotic administration, resulting in consistent changes in the structure and function of the microbiota that are responsible for the observed changes in the host response. These crucial assumptions have not been rigorously tested to date.
Early studies of the gut microbiota relied on culture-based techniques that characterize only a small fraction of the microbial diversity present (19). The introduction of molecular techniques, e.g., DNA sequencing of PCR amplicons from rRNA genes, allowed the detection and enumeration of microorganisms that are refractory to cultivation (41, 61). Each sequence serves as a proxy for the occurrence of a microbial genome in a microbial community. Most of the amplicon sequences from the human gut microbiota correspond to Firmicutes or Bacteroidetes (13), and their total complexity exceeds 15,000 different operational taxonomic units (OTUs) (42).
For most complex microbial communities, including the gut microbiota, a small number of phylotypes dominate population structures and mask the appearance of many distinct but low-abundance taxa in most molecular surveys (54). A meaningful comparison of microbial population structures for different complex communities requires analysis of many thousands of PCR amplicon sequences in order to estimate the relative abundance of different phylotypes and to detect the presence of rare taxa. Recent advances in DNA sequencing technology have permitted the development of methods for deep culture-independent surveys of microbial diversity at relatively low cost. In this study we conducted controlled experiments to characterize the changes in the community structure of the murine gastrointestinal microbiota during antibiotic administration and to monitor the response of this community after withdrawal of the drug. Using a high-throughput tag sequencing approach targeting the V6 hypervariable region of the 16S rRNA gene (21, 54), we gained an unprecedented view of the diversity present in the gut microbiota and were able to detail the dynamics of the gut microbial community during periods of ecologic stress brought on by antibiotic administration. We find that antibiotic administration results in reproducible, significant, and in some cases long-lasting changes in the community structure of the gut microbiota. These changes most likely disturb the balanced interactions between the indigenous microbiota and the host and account for observed changes in gut homeostasis that have been shown to result from antibiotic administration in both clinical and experimental settings.
MATERIALS AND METHODS
Mouse models and housing conditions.
C57BL/6 interleukin-10-deficient (IL-10−/−) mice were from a breeding colony maintained under specific-pathogen-free conditions at Michigan State University (MSU), derived from mice originally purchased from Jackson Laboratories (Bar Harbor, ME). Wild-type C57BL/6J mice were purchased directly from Jackson Laboratories and housed with autoclaved food, bedding, and water. For the antibiotic therapy experiments, selected 4- to 6-week-old mice were treated with antibiotics added either into their food (amoxicillin [3.0 mg], metronidazole [0.69 mg], and bismuth [0.185 mg] formulated per 5-g tablet/day/average [20-g] mouse; BioServ, Frenchtown, NJ) or into their drinking water (cefoperazone [0.5 mg/ml]; Sigma- Aldrich). Experiments with the combination of amoxicillin, metronidazole, and bismuth (AMB) were carried out at the University Research Containment Facility at MSU, and experiments with cefoperazone were carried out in the Unit for Laboratory Animal Medicine at the University of Michigan. All experimental protocols were approved by the animal use and care committees at the respective institutions.
Sample collection and DNA extraction.
At the conclusion of the experiments, mice were euthanized by CO2 asphyxiation. The cecum of each mouse was removed and washed in phosphate-buffered saline to remove the luminal contents. The cecal tip was then excised, bisected, and snap-frozen in liquid nitrogen prior to storage at −80°C. Genomic DNA was then extracted from cecal tip samples (25 to 100 mg) with the Qiagen DNeasy Blood & Tissue kit by using a modified protocol. These modifications included (i) adding a bead-beating step using UltraClean fecal DNA bead tubes (Mo Bio Laboratories, Inc.) that were shaken using a Mini-Beadbeater-8 (BioSpec Products, Inc.) at the “homogenize” setting for 1 min, (ii) increasing the amount of buffer ATL used in the initial steps of the protocol (from 180 μl to 360 μl), (iii) increasing the volume of proteinase K used (from 20 μl to 40 μl), and (iv) decreasing the amount of buffer AE used to elute the DNA at the end of the protocol (from 200 μl to 100 μl).
Sequencing and data analysis.
The data presented here are based on 39 PCR amplicon libraries sequenced in five 454 runs using the GS-FLX platform (454 Life Sciences, Roche Diagnostics Corp.). V6 tag sequence amplicon libraries were constructed as described previously (54). The primer sets corresponding to 967F and 1046R, used in the library preparation, and the permuted primer approach for sequencing multiple libraries within a single GS-FLX 454 run without use of a physical partition have been described by Huber et al. (21). Primers were trimmed off, and all sequences without an exact match to the forward primer, shorter than 50 nucleotides, or containing ambiguous base calls were removed as low-quality reads. Sequences were organized in a relational database, and OTUs were created by aligning with MUSCLE (14) and clustering with DOTUR (50) as described by Huber et al. (21). Taxonomic assignments were made by directly comparing tags to a reference database of close to 200,000 distinct V6 sequences and by using a consensus of the nearest tags in a global alignment of tags and reference sequences (22).
Quantitative PCR.
Quantitative PCRs were used to separately assay the quantity of rRNA operons in the DNA samples relative to a single-copy host gene (mouse tumor necrosis factor alpha [TNF-α]). A portion of the 16S rRNA gene from Helicobacter hepaticus 3B1 was cloned and used as a positive control (between positions 331 and 797, based on Escherichia coli numbering of the 16S rRNA gene). A 264-bp portion of the gene encoding TNF-α from Mus musculus was also cloned and used as a positive control for the host gene target (between positions 6455 and 6718 of the mouse gene encoding TNF-α; GenBank accession number Y00467). Plasmids were purified from each clone, and a 10-fold dilution series was used to determine the detection limits of the assay as well as to provide standard curves for absolute quantification in the quantitative PCRs (range, 101 to 107 copies per reaction). Assays used the LightCycler 480 Probes Master reaction mixture (Roche) at 1× concentration and appropriate primer-probe sets to increase the specificity of the signals detected from the sample DNA (100 ng). For detection of the bacterial signal, 100 nmol of each of the forward and reverse primers and the fluorogenic probe were included in the reaction mixtures. Sequences for the forward primer (5′-TCCTACGGGAGGCAGCAGT-3′), the reverse primer (5′-GGACTACCAGGGTATCTAATCCTGTT-3′), and the probe (5′-[6-carboxyfluorescein]-CGTATTACCGCGGCTGCTGGCAC-[6-carboxytetramethylrhodamine]-3′) were based on the work of Nadkarni et al. (39). Final assay volumes of 20 μl were dispensed in triplicate into 96-well plates. Signals were detected with a LightCycler 480 instrument (Roche). The reaction conditions for the amplification of DNA were 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Detection of the host signal used 200 nmol of the forward (TNFa_mu_se; 5′-GGCTTTCCGAATTCACTGGAG-3′) and reverse (TNFa_mu_as; 5′-CCCCGGCCTTCCAAATAAA-3′) primers and 100 nmol of the probe (TNFa_mu_probe; 5′-Cy5-ATGTCCATTCCTGAGTTCTGCAAAGGGA-Iowa Black RQ-3′) adapted from the work of Nitsche et al. (40). Amplification of the host signal began with incubation at 95°C for 10 min, followed by 45 cycles of 95°C for 20 s and 64°C for 30 s. Relative bacterial loads were compared via the ΔΔCT method by normalizing the 16S signal to the host signal (51).
RESULTS
Antibiotic administration alters the structure of the gut microbiota.
To characterize the impact of antibiotic administration on the composition of the gut microbiota, AMB was administered to C57BL/6 IL-10−/− mice via their chow for 10 days (Fig. 1). The C57BL/6 IL-10−/− strain was chosen because it is utilized as a model of inflammatory bowel disease that is responsive to antibiotic therapy (33). The microbial community in one group of mice was assessed immediately following the 10-day treatment, while a second group of mice was switched back to drug-free chow for 2 weeks before microbial community analysis. The microbiotas from mice in both groups were compared to those from a group of control mice that had been fed conventional chow for the duration of the study.
FIG. 1.

Schemata for antibiotic administration. (A) Fifteen C57BL/6 IL-10−/− mice received AMB in their chow for 10 days, while 10 animals remained on control chow. Mice were euthanized either immediately after antibiotic administration or after a 2-week period of recovery on nonmedicated chow. Control animals remained on nonmedicated chow for the entire experiment. (B) Five animals remained on sterile water, while 15 mice were treated with 0.5 mg/ml of cefoperazone in sterile drinking water for 10 days. The antibiotic-treated animals were subsequently divided into three groups. One group of three animals was immediately sacrificed. One group of six animals (divided into two cages) was returned to sterile water without antibiotics for a 6-week recovery period. A final group of six animals (also divided into two cages) was returned to water without antibiotics, and a nontreated control mouse was added to each cage for the 6-week recovery period. The ceca of all animals were harvested for microbial community analysis.
We used a massively parallel pyrosequencing strategy to retrieve sequences of the V6 hypervariable region of the small-subunit (SSU) rRNA gene (54) in order to determine the compositions of the microbial communities associated with the cecal mucosae of these mice. These SSU sequence tags are generated by PCR amplification and function as proxies for the presence of individual phylotypes in a given community. The use of pyrosequencing permitted characterization of a greater number of phylotypes than was previously practical via PCR amplification, cloning, and capillary sequencing of SSU genes. GAST (Global Alignment for Sequence Taxonomy) provided taxonomic assignments for each of the tag sequences (22).
We collected a total of 1,006,137 sequence tags generated from representative samples (from 9 control mice, 2 treated mice, and 10 mice that were treated and then allowed to recover). The vast majority of the sequence tags recovered from the cecal communities of control animals were affiliated with the phyla Bacteroidetes and Firmicutes, with only about 1% belonging to the Proteobacteria (Fig. 2; see also Table S1 in the supplemental material). In the antibiotic-treated animals, however, the majority of tags (73,010 of 102,822 [71%]) were Proteobacteria. Two specific sequences that were assigned to the family Enterobacteriaceae accounted for 67,717 (93%) of these Proteobacteria tags (see Table S2 in the supplemental material). In the control animals, these two sequences represented only 106 out of a total of 5,214 (2%) tags belonging to the Proteobacteria.
FIG. 2.

Comparison of microbial community composition in the ceca of antibiotic-treated mice. More than 1 million V6 sequence tags were retrieved from cecal DNA purified from untreated mice (control), animals that received AMB in chow for 10 days (antibiotic treated), and AMB-treated mice that were allowed to recover on plain chow for 2 weeks (recovery). The sequence tags were classified to the level of bacterial division (phylum). The pie charts show the distribution of the pooled tags recovered from each experimental group. Numbers are mean percentages of pooled tags in each experimental group assigned to particular phyla (± standard deviations).
In the animals whose gut microbial communities were allowed to recover via a 2-week antibiotic-free period, Firmicutes and Bacteroidetes returned to dominance (70% and 22% of the total number of tags, respectively). Proteobacteria decreased to 5.77% of the total, more than the 1.2% in the animals that never received antibiotics but much less than the 73% that they constituted at the end of the AMB treatment (Fig. 2). The relative increase in the proportion of Proteobacteria resulted from increases in the numbers of tags that mapped to several taxonomic groups within the phylum (see Table S1 in the supplemental material). The two Enterobacteriaceae tags that were dominant among the AMB-treated mice were encountered only 201 times out of a total of 26,964 (0.75%) Proteobacteria tags (see Table S2 in the supplemental material).
A global comparison of all of the gut microbial communities in each of the animals was performed by calculating the Bray-Curtis measure of community similarity (34). This index is based on the presence or absence and the relative abundance of each phylotype encountered in the mucosa-associated communities. We calculated the average Bray-Curtis similarity for each pairwise comparison of the control, treated, and recovered animals (Table 1). Analysis of variance of these Bray-Curtis values confirmed that the mucosa-associated microbiotas from antibiotic-treated animals differed significantly both from those of control animals and from those of recovered animals (P < 0.05). The average Bray-Curtis similarity between communities from antibiotic-treated animals and communities from the other two experimental groups was significantly lower than the values for all other pairwise comparisons.
TABLE 1.
Bray-Curtis similarities of microbial communities from control, AMB-treated, and recovered mice
| Group (n) | Bray-Curtis similarity (avg ± SD)a to the following group:
|
||
|---|---|---|---|
| Control | Recovery | Treatment | |
| Control (9) | 0.765 ± 0.014 A | ||
| Recovery (10) | 0.756 ± 0.008 A | 0.749 ± 0.011 A | |
| Treatment (2) | 0.212 ± 0.019 B | 0.205 ± 0.018 B | 0.845 ± 0.083 A |
Values followed by different letters are significantly different. Groups were compared by analysis of variance, with significance set at a P value of <0.05 by the Tukey-Kramer method.
Variability in the murine gut microbiota.
Despite the significant differences in the gut microbiota between control animals and animals that received the triple-antibiotic cocktail, interanimal variation was still noted within each experimental group. The animals in this experiment were selected from a breeding colony maintained at MSU over a period of approximately 5 months. The animals, therefore, came from several different litters born to separate mothers.
To determine the degree of similarity among animals that shared as many variables as possible, we sequenced and compared 48,594 V6 sequence tags from the mucosa-associated microbiotas located in the ceca of three age-matched, wild-type C57BL/6 mice purchased from a commercial vendor. Figure 3 depicts the results of taxonomic assignments and Bray-Curtis measures of community similarity based on presence or absence and relative phylotype abundance for the mucosa-associated community of each animal. As observed previously, Firmicutes and Bacteroidetes dominated the microbial communities in the ceca of each of the three animals. All three communities displayed similar phylotype distributions at all taxonomic levels, with Bray-Curtis similarities of >0.9 for all pairwise community comparisons. We recovered approximately 16,000 tags from each community (Fig. 3). Using an OTU assignment of 97% sequence similarity yielded ∼1,000 OTUs in each community. The nonparametric Chao1 estimator (8) suggests that for this sampling effort, there are ∼1,200 unique 97% OTUs in each mucosa-associated gut community.
FIG. 3.

Genus level diversity of the gut communities in the ceca of control animals. Approximately 16,000 V6 SSU hypervariable region tags were retrieved from the cecal mucosa-associated microbiota from each of three wild-type C57BL/6 mice. Pie charts show the distribution of the most prevalent taxonomically assigned tags, while the percentages for the 12 most common assignments are given below. Bray-Curtis similarities were calculated for each pairwise comparison. The nonparametric Chao1 diversity estimator was calculated for each community based on 97% sequence similarity. c.i., confidence interval.
Antibiotic administration can result in a prolonged decrease in the diversity of the gut microbiota.
In spite of the dramatic shifts in the composition of the gut microbiota following administration of the AMB cocktail, the community structure returned largely to the baseline state 2 weeks after discontinuation of the drugs. In an additional pilot experiment, the broad-spectrum cephalosporin antibiotic cefoperazone appeared to have a similar dramatic effect on the microbiotas of C57BL/6 IL-10−/− mice, but in this case there were significant long-term effects on the community structure, including lower overall diversity, after antibiotic recovery (data not shown).
To extend this initial observation, we undertook an additional antibiotic administration trial employing cefoperazone for 20 wild-type female C57BL/6 mice (Fig. 1B). Five mice were maintained in a single cage on standard mouse chow and sterile water (control group). The remaining 15 mice were switched to water supplemented with cefoperazone (0.5 mg/ml), and after 10 days, these antibiotic-treated mice were divided into three subsequent treatment groups. Three mice were immediately euthanized to enable observation of the effects of cefoperazone on the gut microbiota. Six mice, housed three animals per cage, were returned to sterile water for 6 weeks (isolated recovery), while another group of six mice (again divided into two cages) were housed with a control mouse added to each cage during the antibiotic-free period (donor recovery). The addition of the control mouse allowed reinoculation of the gut microbiota via natural coprophagic activity.
A total of 308,505 tag sequences were recovered from the 17 samples representing the animals in the three experimental groups (see Table S3 in the supplemental material). On average, ∼18,000 high-quality sequence tags (23) were recovered per sample. Amplification of sequence tags was not possible for the three cefoperazone-treated mice that were euthanized at the end of antibiotic treatment (without a drug-free recovery period). Real-time PCR targeting the 16S SSU gene was used to determine what effect the antibiotics had on the overall bacterial load (judged by the relative ratio of the 16S SSU signal to a genomic murine target gene) and whether this could explain the inability to amplify sequence tags from these animals. The bacterial loads in animals treated with antibiotics decreased by 3 orders of magnitude (average change, 4,300-fold) from those in control animals. Both groups of animals that were allowed to recover from antibiotic administration for 6 weeks (isolated recovery and donor recovery) had levels of bacteria comparable to those of control animals (average change, 0.91-fold for donor recovery animals and 1.20-fold for isolated-recovery animals).
Pairwise Bray-Curtis similarities are displayed in heat map format (Fig. 4) to allow visualization of all of the pairwise comparisons. The bacterial communities in cefoperazone-treated animals 6 weeks after discontinuation of the drug were distinct from those in control animals. However, the microbial communities in the animals that recovered from antibiotic administration in the presence of an untreated donor animal returned to a state very similar to that seen in the control animals.
FIG. 4.
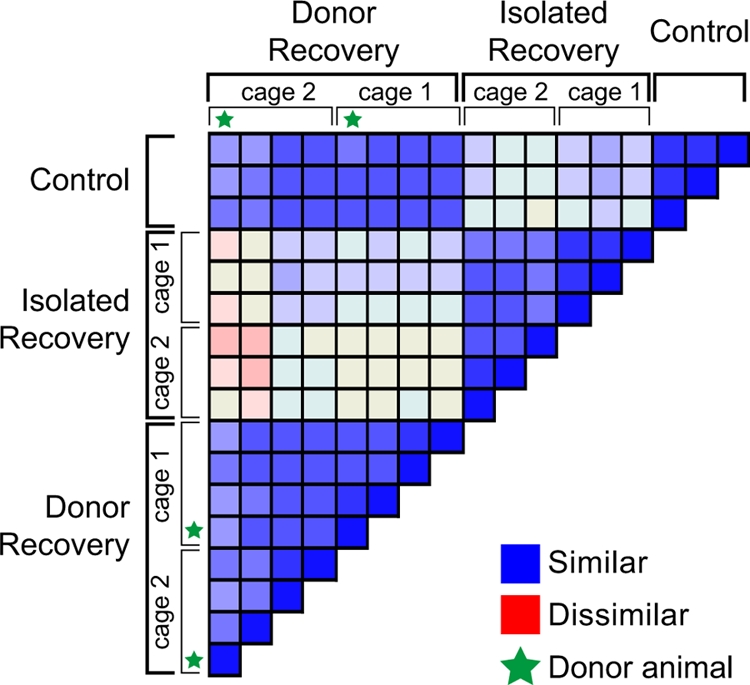
Comparison of microbial communities in cefoperazone-treated animals. More than 300,000 V6 sequence tags were retrieved from the ceca of cefoperazone-treated mice that recovered from drug treatment in the presence or absence of an untreated “donor” animal. Taxonomic assignments at the genus level were provided for the tags, and the pairwise Bray-Curtis distance was calculated for all possible comparisons. The Bray-Curtis values are presented in a heat map fashion as a color-coded distance matrix, with the most similar communities (Bray-Curtis similarity, 1.0) represented by blue and the most dissimilar communities (Bray-Curtis similarity, 0.0) represented by red. The housing of the animals is indicated, and each animal that served as a “donor” is marked with a star.
When we examined the composition of the phylotypes at the phylum level, the primary distinction was a reduction in Bacteroidetes diversity in the animals that recovered without any additional input of microbes. While phylotypes assigned to Bacteroidetes made up about 15% of the total community in control animals and in animals that recovered in the presence of a donor animal, they accounted for only 0.33% of the total community in animals that recovered in the absence of a donor.
At finer levels of taxonomic distinction, additional differences were noted between the animals that did not have a donor animal present during the recovery phase and either the control animals or those that recovered with a donor (Fig. 5; see also Table S3 in the supplemental material). At the genus level, the compositions of the microbial communities from controls and from animals that recovered in the presence of a donor were quite different from the compositions of the communities from animals that recovered without a donor. These data also suggest that the gut communities in animals that recovered without a donor comprised a decreased number of phylotypes.
FIG. 5.

Genus-level diversity of the gut communities from cefoperazone-treated animals. The taxonomic assignments of V6 tags from untreated animals (control), animals that recovered without an untreated animal (isolated recovery), and animals that recovered in the presence of an untreated animal (donor recovery) are shown. The pie charts show the most abundant genus level assigned tags for the pooled animals in each experimental group. The average (± standard deviation) percentage of tags belonging to each genus is given below the pie charts. ND, not detected; NA, not assignable with ≥67% confidence via GAST (22).
Rarefaction curves demonstrated that fewer phylotypes were present in the microbial communities from animals that recovered without a donor. Rarefaction analysis involves resampling of community survey data to generate idealized collector's curves, providing an indication of overall phylotype richness (17, 34). In addition, rarefaction can provide an estimate of the depth to which a complex community has been sampled. Rarefaction curves from the control animals and the animals that recovered in the presence of a donor overlapped, confirming that the overall levels of diversity (phylotype richness) in the two groups were similar (Fig. 6). Conversely, rarefaction analysis of the communities from the animals that recovered without a donor indicated that the diversity of these communities was lower than that for the other two experimental groups.
FIG. 6.
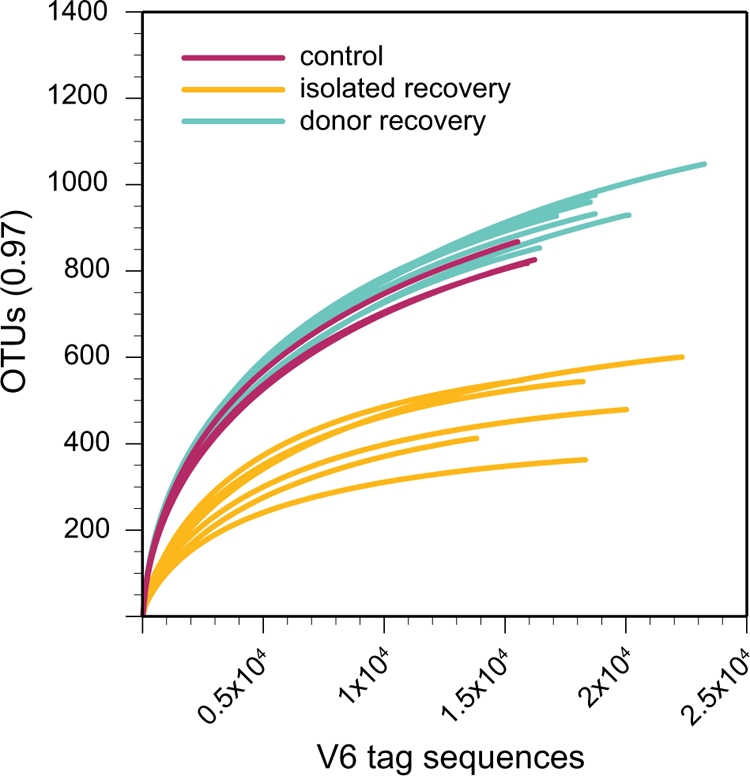
Rarefaction analysis of microbial communities from cefoperazone-treated animals. The number of assigned phylotypes is plotted as a function of the number of tags retrieved. The V6 tags from untreated animals (control), animals that recovered without an untreated animal (isolated recovery), and animals that recovered in the presence of an untreated animal (donor recovery) were used to construct rarefaction curves with an OTU definition of >97% sequence similarity.
DISCUSSION
The myriad interactions between the indigenous gastrointestinal microbiota and its mammalian host have been a focus of considerable recent scientific investigation. Studies of human subjects have the advantage of directly examining the natural community responsible for specific diseases. However, due to technical and ethical constraints on examining the human microbiota, a great deal of effort has been applied to studying model systems, in particular murine models.
Two main lines of research have provided insights about host-microbiota interactions in murine models. Studies with germ-free and gnotobiotic mice have demonstrated that gut bacteria can transmit signals that influence host responses (20, 44). However, these are highly simplified systems where community complexity is orders of magnitude lower than that of the naturally occurring murine microbiota. An alternative approach has been to study how ecological stressors shape complex communities in murine model systems. In many cases, antibiotics are employed to alter the indigenous microbiota, thus disturbing the normal, baseline host-microbe interactions. Such an approach has demonstrated a role for the microbiota in genetic models of murine inflammatory bowel disease (26, 33) and in the modulation of glucose tolerance in mouse models of insulin resistance (37). Antibiotic treatment studies have shown that antibiotic-resistant bacterial pathogens can exploit innate immune deficits triggered by antibiotic administration (5). Antibiotic regimens have been used to demonstrate a role for the indigenous microbiota in shaping physiological responses of the gut mucosa, including mediating protective responses to direct epithelial injury (43) and directing the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine (24). Antibiotic-treated mice demonstrate altered susceptibility to experimental infection with pathogenic bacteria. Treatment of mice with streptomycin increases susceptibility to oral infection with Salmonella enterica serovar Typhimurium (52). A recently described murine model of Clostridium difficile-associated colitis employed pretreatment with a mixture of five antibiotics followed by a single dose of clindamycin a day prior to oral challenge with C. difficile (9).
These studies have generally focused on the host response to the alteration in the indigenous microbiota. In most cases, the nature of the antibiotic-induced changes in the microbiota was not investigated. Some studies measured changes in total aerobic and anaerobic culturable bacteria following antibiotic administration, and in a few cases, limited culture-independent investigation of the microbiota was performed. An implicit assumption of these studies is that genetically identical mice harbor a consistent baseline microbiota. A further assumption is that the use of antibiotics would result in reproducible changes in the microbiota that would be responsible for the altered host responses observed. These critical assumptions have never been formally addressed in detail until the current study.
It has been proposed that an adult mammal harbors a stable “climax community” in each anatomic area of the gastrointestinal tract (48). Although there can be individual variation in the composition of shallow phylogenetic lineages within the gut microbiota, there are relatively few deep lineages, with Firmicutes and Bacteroidetes generally dominant in most surveys (11, 29). These observations most likely reflect the influence of a variety of ecological and evolutionary constraints on the gut microbial community (28, 29). Our results, demonstrating marked similarity between the gut microbiotas from individual animals, albeit among individuals with identical genetic backgrounds maintained in a tightly controlled environment, provide strong evidence that the gut microbial community represents a stable ecosystem. This high degree of similarity also provides evidence for the existence of community “assembly rules” that govern the establishment and stability of these microbial consortia.
Perhaps the more critical assumption in experiments that involve antibiotic manipulation of the indigenous gut microbiota is that drug treatment results in reproducible alterations of the microbial community structure. The relative stability of the indigenous microbiota has been debated. From an ecological standpoint, the term “stability” (also commonly referred to as “robustness”) encompasses a number of components (2, 31). One aspect is temporal stability, which is the constancy of community structure over time. In addition, the term “resistance” refers to the ability of a community to maintain a given structure in the setting of a perturbation, while “resilience” is the ability of a community to return to its baseline structure following a perturbation in community structure. In this regard, if a community exhibits temporal stability, this implies the presence of resistance and resilience in the community structure, since one assumes that most communities will experience ecological stress at some point.
A number of studies have indicated that an individual's gut microbiota can have a relatively stable community composition over a period of months to years (36, 49, 57, 62). These observations have led to the conclusion that the community of microbes in the gut is relatively resistant to perturbation by various ecological stressors. Subsequent environmental influences, including diet, host genetics, medication use, and exposure to infectious agents, can all influence the resultant microbial community (11). It has been reported that short-term administration of antibiotics could result in long-term changes in the structure of the fecal microbiota of humans (12, 25, 32). In all of these human studies, there was considerable individual baseline variation in the microbiotas, which made it difficult to make interindividual comparisons in the microbiota responses.
Although human studies such as these are important, one advantage of conducting murine experiments as described here is the ability to conduct true controlled, replicate experiments. Our replicate experiments allowed us to detect consistent shifts in the gut microbial community in response to antibiotic administration. The reproducibility of these changes indicates that even if the influences on microbial community structure are complex and numerous, the community will exhibit stereotypic responses if ecological stressors are consistently applied. We observed reproducible shifts in the community structure of the gut microbiota following antibiotic treatment, including significant alterations in both the richness and the distribution of 16S V6 phylotypes. The power of a deep survey of diversity allowed us to demonstrate that certain low-abundance phylotypes present at baseline could become dominant in response to the shift in environmental conditions brought about by antibiotic administration. In control animals, 16S V6 tag sequences corresponding to members of the family Enterobacteriaceae made up only a small fraction of the population (1%). During AMB administration, this group of organisms became the dominant phylotype, indicating that this antibiotic regimen created an environment that somehow favored this taxonomic group of organisms. Simple resistance to the antibiotics cannot entirely explain this observation, because other phylotypes were unchanged in relative abundance following AMB administration and did not undergo the remarkable relative expansion during drug treatment exhibited by the Enterobacteriaceae.
In this case, the gut microbial community exhibited resilience as the community structure shifted back toward the baseline state following cessation of the AMB treatment. However, the ability of this community to recover following antibiotic disturbance was not absolute. The administration of cefoperazone also caused dramatic shifts in community structure, but in this case, diversity did not recover even 6 weeks after the discontinuation of the drug. Rarefaction analysis revealed a persistent, significant decrease in overall species richness in the gut community following cefoperazone administration. However, the addition of an untreated mouse to cages of cefoperazone-treated animals during the recovery phase allowed complete restoration of diversity, presumably through natural coprophagic activity. This observation indicates that cefoperazone administration did not change host physiology, since exposure to a baseline microbiota resulted in normalization of the community structure. Additionally, we infer that the baseline community structure is “preferred,” since all four animals in the cage possessed a community that we cannot distinguish from that in untreated control animals. Since the donor animals were exposed to the altered communities present in the cefoperazone-treated animals, it is possible that the resultant communities would possess the antibiotic-altered community structure or an intermediate structure.
The reasons for the differences observed in community resilience are not entirely clear. The ecological disturbance mediated by cefoperazone appears to have overcome community resilience, potentially due to different spectra of antimicrobial activity. Regardless of the underlying reasons for the differences in observed community resilience, from an experimental standpoint it is important to understand that manipulation of the indigenous gut microbiota by various antibiotic regimens may result in altered community structures that persist even after the antibiotic is discontinued. Whether or not the gut community returns to the baseline state after perturbation can influence the conclusions that can be drawn from a particular experiment.
The implications of long-lasting changes in community diversity following antibiotic administration are severalfold. Even though the microbial composition of the animals that recovered from cefoperazone treatment remained altered from the baseline, overall bacterial biomass returned to the level observed in control mice. It has been postulated that functional redundancy in complex microbial communities can allow an altered community to perform ecosystem functions equivalent to those of the original community (2). Studies are ongoing to determine if the specific alterations in the gut microbiota result in any significant changes in gut ecosystem functioning.
One function of the gut microbiota that has captured our attention is “colonization resistance,” the ability of the indigenous microbiota to prevent the ingress of pathogens into the gut community (16, 18, 58). Antibiotic-associated colitis resulting from Clostridium difficile infection may result from a loss of the intrinsic colonization resistance of the gut microbiota (4, 38). Theoretically, the administration of antibiotics could disturb the indigenous microbiota, allowing C. difficile spores encountered in the environment to germinate and successfully colonize the gut (4, 60). Although C. difficile infection responds to the administration of specific antimicrobial therapy, including metronidazole or vancomycin, recurrence following the end of C. difficile therapy has become an increasing problem (35). In a previous study, we provided evidence that recurrent C. difficile infection is associated with a decrease in fecal microbial diversity (7). This observation is in line with the fact that the administration of stool from healthy individuals to patients with recurrent C. difficile can break the cycle of recurrence (1, 56). The data presented here indicate that antibiotic therapy of sufficient magnitude can result in an altered microbial community. It remains to be determined if this can be directly correlated with a loss of colonization resistance, but our findings provide evidence that antibiotic administration can result in long-term decreases in gut microbial diversity, which in turn are associated with recurrent C. difficile disease.
As we learn more about the intricate relationship between the gut microbiota and its host, we may find that unintended disturbance of this microbial community will have significant deleterious health effects. A more complete understanding of the ecological forces that determine the formation and maintenance of microbial community structures could lead to novel ways to prevent or treat diseases that result from disruptions of the gut microbiota.
Supplementary Material
Acknowledgments
We thank Nabeetha Nagalingam, Judith Opp, and Jason Pratt for assistance with animal experimentation and Katia Andreishcheva and Christina Holmes for assistance with 454 sequencing. Thanks to Courtney Robinson for critical reading of the manuscript.
The main projects were funded in whole with federal funds from the NIAID, NIH, Department of Health and Human Services, under contract N01-AI-30058. Additional funding was supplied via subcontracts from the Woods Hole Center for Oceans and Human Health from the National Institutes of Health and National Science Foundation (NIH/NIEHS 1 P50 ES012742-01 and NSF/OCE 0430724 [J. Stegeman, principal investigator] to H.G.M. and M.L.S. and R01 DK070875 to V.B.Y.) and by grants from the W. M. Keck Foundation and the G. Unger Vetlesen Foundation (to M.L.S.). D.A.A. was supported by the National Institutes of Health under a Ruth L. Kirschstein National Research Service Award (T32 HL07749).
This article's contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 23 March 2009.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Aas, J., C. E. Gessert, and J. S. Bakken. 2003. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin. Infect. Dis. 36580-585. [DOI] [PubMed] [Google Scholar]
- 2.Allison, S. D., and J. B. Martiny. 2008. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 105(Suppl. 1)11512-11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bäckhed, F., R. E. Ley, J. L. Sonnenburg, D. A. Peterson, and J. I. Gordon. 2005. Host-bacterial mutualism in the human intestine. Science 3071915-1920. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett, J. G. 2008. Historical perspectives on studies of Clostridium difficile and C. difficile infection. Clin. Infect. Dis. 46(Suppl. 1)S4-S11. [DOI] [PubMed] [Google Scholar]
- 5.Brandl, K., G. Plitas, C. N. Mihu, C. Ubeda, T. Jia, M. Fleisher, B. Schnabl, R. P. DeMatteo, and E. G. Pamer. 2008. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455804-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broekaert, I. J., and W. A. Walker. 2006. Probiotics and chronic disease. J. Clin. Gastroenterol. 40270-274. [DOI] [PubMed] [Google Scholar]
- 7.Chang, J. Y., D. A. Antonopoulos, A. Kalra, A. Tonelli, W. T. Khalife, T. M. Schmidt, and V. B. Young. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197435-438. [DOI] [PubMed] [Google Scholar]
- 8.Chao, A. 1984. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11265-270. [Google Scholar]
- 9.Chen, X., K. Katchar, J. D. Goldsmith, N. Nanthakumar, A. Cheknis, D. N. Gerding, and C. P. Kelly. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 1351984-1992. [DOI] [PubMed] [Google Scholar]
- 10.De La Cochetière, M. F., T. Durand, V. Lalande, J. C. Petit, G. Potel, and L. Beaugerie. 2008. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb. Ecol. 56395-402. [DOI] [PubMed] [Google Scholar]
- 11.Dethlefsen, L., P. B. Eckburg, E. M. Bik, and D. A. Relman. 2006. Assembly of the human intestinal microbiota. Trends Ecol. Evol. 21517-523. [DOI] [PubMed] [Google Scholar]
- 12.Dethlefsen, L., S. Huse, M. L. Sogin, and D. A. Relman. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckburg, P. B., E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman. 2005. Diversity of the human intestinal microbial flora. Science 3081635-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 321792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank, D. N., and N. R. Pace. 2008. Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 244-10. [DOI] [PubMed] [Google Scholar]
- 16.Freter, R. 1962. In vivo and in vitro antagonism of intestinal bacteria against Shigella flexneri. II. The inhibitory mechanism. J. Infect. Dis. 11038-46. [DOI] [PubMed] [Google Scholar]
- 17.Gotelli, N. J., and R. K. Colwell. 2001. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 4379-391. [Google Scholar]
- 18.Hentges, D. J., and R. Freter. 1962. In vivo and in vitro antagonism of intestinal bacteria against Shigella flexneri. I. Correlation between various tests. J. Infect. Dis. 11030-37. [DOI] [PubMed] [Google Scholar]
- 19.Holdeman, L. V., I. J. Good, and W. E. Moore. 1976. Human fecal flora: variation in bacterial composition within individuals and a possible effect of emotional stress. Appl. Environ. Microbiol. 31359-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hooper, L. V., J. Xu, P. G. Falk, T. Midtvedt, and J. I. Gordon. 1999. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc. Natl. Acad. Sci. USA 969833-9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huber, J. A., D. B. Mark Welch, H. G. Morrison, S. M. Huse, P. R. Neal, D. A. Butterfield, and M. L. Sogin. 2007. Microbial population structures in the deep marine biosphere. Science 31897-100. [DOI] [PubMed] [Google Scholar]
- 22.Huse, S. M., L. Dethlefsen, J. A. Huber, D. M. Welch, D. A. Relman, and M. L. Sogin. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4e1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huse, S. M., J. A. Huber, H. G. Morrison, M. L. Sogin, and D. M. Welch. 2007. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 8R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivanov, I. I., L. Frutos Rde, N. Manel, K. Yoshinaga, D. B. Rifkin, R. B. Sartor, B. B. Finlay, and D. R. Littman. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4337-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jernberg, C., S. Löfmark, C. Edlund, and J. K. Jansson. 2007. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 156-66. [DOI] [PubMed] [Google Scholar]
- 26.Kang, S. S., S. M. Bloom, L. A. Norian, M. J. Geske, R. A. Flavell, T. S. Stappenbeck, and P. M. Allen. 2008. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 5e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly, D., and S. Conway. 2005. Bacterial modulation of mucosal innate immunity. Mol. Immunol. 42895-901. [DOI] [PubMed] [Google Scholar]
- 28.Ley, R. E., M. Hamady, C. Lozupone, P. J. Turnbaugh, R. R. Ramey, J. S. Bircher, M. L. Schlegel, T. A. Tucker, M. D. Schrenzel, R. Knight, and J. I. Gordon. 2008. Evolution of mammals and their gut microbes. Science 3201647-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ley, R. E., D. A. Peterson, and J. I. Gordon. 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124837-848. [DOI] [PubMed] [Google Scholar]
- 30.Ley, R. E., P. J. Turnbaugh, S. Klein, and J. I. Gordon. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 4441022-1023. [DOI] [PubMed] [Google Scholar]
- 31.Little, A. E., C. J. Robinson, S. B. Peterson, K. F. Raffa, and J. Handelsman. 2008. Rules of engagement: interspecies interactions that regulate microbial communities. Annu. Rev. Microbiol. 62375-401. [DOI] [PubMed] [Google Scholar]
- 32.Löfmark, S., C. Jernberg, J. K. Jansson, and C. Edlund. 2006. Clindamycin-induced enrichment and long-term persistence of resistant Bacteroides spp. and resistance genes. J. Antimicrob. Chemother. 581160-1167. [DOI] [PubMed] [Google Scholar]
- 33.Madsen, K. L., J. S. Doyle, M. M. Tavernini, L. D. Jewell, R. P. Rennie, and R. N. Fedorak. 2000. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology 1181094-1105. [DOI] [PubMed] [Google Scholar]
- 34.Magurran, A. E. 2004. Measuring biological diversity. Blackwell Science Ltd., Oxford, United Kingdom.
- 35.Maroo, S., and J. T. Lamont. 2006. Recurrent Clostridium difficile. Gastroenterology 1301311-1316. [DOI] [PubMed] [Google Scholar]
- 36.Maukonen, J., R. Satokari, J. Matto, H. Soderlund, T. Mattila-Sandholm, and M. Saarela. 2006. Prevalence and temporal stability of selected clostridial groups in irritable bowel syndrome in relation to predominant faecal bacteria. J. Med. Microbiol. 55625-633. [DOI] [PubMed] [Google Scholar]
- 37.Membrez, M., F. Blancher, M. Jaquet, R. Bibiloni, P. D. Cani, R. G. Burcelin, I. Corthesy, K. Mace, and C. J. Chou. 2008. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 222416-2426. [DOI] [PubMed] [Google Scholar]
- 38.Mylonakis, E., E. T. Ryan, and S. B. Calderwood. 2001. Clostridium difficile-associated diarrhea: a review. Arch. Intern. Med. 161525-533. [DOI] [PubMed] [Google Scholar]
- 39.Nadkarni, M. A., F. E. Martin, N. A. Jacques, and N. Hunter. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148257-266. [DOI] [PubMed] [Google Scholar]
- 40.Nitsche, A., M. Becker, I. Junghahn, J. Aumann, O. Landt, I. Fichtner, B. Wittig, and W. Siegert. 2001. Quantification of human cells in NOD/SCID mice by duplex real-time polymerase-chain reaction. Haematologica 86693-699. [PubMed] [Google Scholar]
- 41.Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276734-740. [DOI] [PubMed] [Google Scholar]
- 42.Peterson, D. A., D. N. Frank, N. R. Pace, and J. I. Gordon. 2008. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 3417-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rakoff-Nahoum, S., J. Paglino, F. Eslami-Varzaneh, S. Edberg, and R. Medzhitov. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118229-241. [DOI] [PubMed] [Google Scholar]
- 44.Samuel, B. S., and J. I. Gordon. 2006. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc. Natl. Acad. Sci. USA 10310011-10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanders, M. E. 2008. Probiotics: definition, sources, selection, and uses. Clin. Infect. Dis. 46(Suppl. 2)S58-S61. [DOI] [PubMed] [Google Scholar]
- 46.Sansonetti, P. J., and J. P. Di Santo. 2007. Debugging how bacteria manipulate the immune response. Immunity 26149-161. [DOI] [PubMed] [Google Scholar]
- 47.Sartor, R. B. 2008. Microbial influences in inflammatory bowel diseases. Gastroenterology 134577-594. [DOI] [PubMed] [Google Scholar]
- 48.Savage, D. C. 1977. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 31107-133. [DOI] [PubMed] [Google Scholar]
- 49.Scanlan, P. D., F. Shanahan, C. O'Mahony, and J. R. Marchesi. 2006. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn's disease. J. Clin. Microbiol. 443980-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schloss, P. D., and J. Handelsman. 2005. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 711501-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmittgen, T. D., and K. J. Livak. 2008. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 31101-1108. [DOI] [PubMed] [Google Scholar]
- 52.Sekirov, I., N. M. Tam, M. Jogova, M. L. Robertson, Y. Li, C. Lupp, and B. B. Finlay. 2008. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect. Immun. 764726-4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shreiner, A., G. B. Huffnagle, and M. C. Noverr. 2008. The “microflora hypothesis” of allergic disease. Adv. Exp. Med. Biol. 635113-134. [DOI] [PubMed] [Google Scholar]
- 54.Sogin, M. L., H. G. Morrison, J. A. Huber, D. Mark Welch, S. M. Huse, P. R. Neal, J. M. Arrieta, and G. J. Herndl. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. USA 10312115-12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turnbaugh, P. J., R. E. Ley, M. A. Mahowald, V. Magrini, E. R. Mardis, and J. I. Gordon. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 4441027-1031. [DOI] [PubMed] [Google Scholar]
- 56.Tvede, M., and J. Rask-Madsen. 1989. Bacteriotherapy for chronic relapsing Clostridium difficile diarrhoea in six patients. Lancet i1156-1160. [DOI] [PubMed] [Google Scholar]
- 57.Vanhoutte, T., V. De Preter, E. De Brandt, K. Verbeke, J. Swings, and G. Huys. 2006. Molecular monitoring of the fecal microbiota of healthy human subjects during administration of lactulose and Saccharomyces boulardii. Appl. Environ. Microbiol. 725990-5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vollaard, E. J., and H. A. Clasener. 1994. Colonization resistance. Antimicrob. Agents Chemother. 38409-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walker, W. A. 2008. Mechanisms of action of probiotics. Clin. Infect. Dis. 46(Suppl. 2)S87-S91. [DOI] [PubMed] [Google Scholar]
- 60.Wilson, K. H. 1993. The microecology of Clostridium difficile. Clin. Infect. Dis. 16(Suppl. 4)S214-S218. [DOI] [PubMed] [Google Scholar]
- 61.Wilson, K. H., and R. B. Blitchington. 1996. Human colonic biota studied by ribosomal DNA sequence analysis. Appl. Environ. Microbiol. 622273-2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zoetendal, E. G., A. D. Akkermans, and W. M. De Vos. 1998. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl. Environ. Microbiol. 643854-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.