

Abstract
Group B streptococcus (GBS), the most frequent single isolate in neonatal sepsis and meningitis, potently activates inflammatory macrophage genes via myeloid differentiation antigen 88 (MyD88). However, events parallel to and downstream of MyD88 that instruct the macrophage response are incompletely understood. In this study, we found that only MyD88, not the Toll-like receptor (TLR) adapter proteins MAL/TIRAP, TRIF, and TRAM, essentially mediates the cytokine (tumor necrosis factor [TNF] and interleukin-6) and chemokine (RANTES) responses to whole GBS organisms, although MAL, TRIF, and TRAM have been shown to mediate the responses to substructures in other gram-positive and gram-negative bacteria. GBS-induced, MyD88-dependent phosphorylation of the mitogen-activated protein kinase p38 activated the transcription factor AP-1 and early growth response factor 1 (Egr-1) but not NF-κB. Furthermore, phosphorylation of Ets-like molecule 1 (Elk-1) was mediated by p38. However, in contrast to Egr-1 and AP-1, Elk-1 was dispensable for transcriptional activation of TNF by GBS organisms. Studies of macrophages from Elk-1-deficient mice revealed that Elk-1 was furthermore nonessential for the TNF responses to purified TLR2 and TLR4 agonists, which was in notable contrast to what was revealed in studies employing in vitro expression systems. In conclusion, MyD88, p38, and Egr-1, but not Elk-1, essentially mediate the inflammatory cytokine response to GBS organisms.
The advent of Toll-like receptors (TLRs) has revealed that the conventional synonymous use of “innate immune response” and “nonspecific immune response” is flawed. Specific receptor-based interactions between bacteria and immune cells result in the formation of distinct ecologic niches on the surface of the human body. Furthermore, bacteria are likely to express TLR ligands in an inducible fashion, therefore adapting to their specific immunological environment (12, 15). In other words, molecular variations among conserved structures might transform a ligand important for the recognition of one organism into a less important one in another species (10, 13). Accordingly, the interface between microbial particle and phagocyte is critical for the specificity of the host response. A further level of specific regulation is added by events downstream of TLRs that drive the phagocyte response toward transcriptional activation of inflammatory and/or directly antibacterial genes (8). The pivotal relay in uttermost proximity to the transmembrane TLRs is formed by a heteromer containing at least four well-defined intracellular proteins that share the so-called Toll-interleukin-1 (IL-1) resistance (TIR) domain with TLRs: (i) MyD88 (myeloid differentiation primary response gene 88), (ii) TRIF (TIR domain-containing adapter-inducing beta interferon), (iii) TRAM (TRIF-related adapter molecule), and (iv) MAL/TIRAP (MyD88 adapter-like/TIR domain-containing adapter protein). The role of a fifth adapter, SARM, is less well established. SARM appears to negatively regulate the TRIF-dependent cytokine response (3). Specific combinations of these proteins are engaged by substructures from distinct bacterial species, i.e., MyD88 and MAL are engaged by bacterial lipoproteins (35), TRIF is engaged by double-stranded RNA, and all four are engaged by Escherichia coli lipopolysaccharide (LPS) (7). With respect to group B streptococcus (GBS) and other streptococci, it is conceivable that optimal macrophage activation requires MAL and TRAM, since both have been reported as signaling intermediates for the responses to diacylated lipoproteins and lipoteichoic acid, which are putative TLR2/6 agonists in gram-positive organisms (27, 34). Subsequently, inflammatory cytokines (such as tumor necrosis factor [TNF], IL-6, chemokines, and type I interferons) are induced in a fashion depending on usage of individual adapter components (7). Downstream of the TLR adapter, the family of mitogen-activated protein kinases (MAPKs) comprises particularly attractive intermediates of TLR activation. MAPKs affect several (in part contrasting) macrophage properties, such as formation of inflammatory and anti-inflammatory cytokines, apoptosis, cell migration, and particle uptake (2, 11, 29, 37). Accordingly, the MAPKs c-Jun N-terminal protein kinase (JNK) and p38 have been found to be important for GBS-induced cytokine formation (19, 24, 31). However, the contributions of individual TLR adapter proteins other than MyD88 to MAPK and cytokine activation have not been assessed for GBS organisms or any other streptococcal species. This lack of knowledge sharply contrasts with what is known for E. coli, which has been shown to engage a variety of adapters (MyD88, TRIF, MAL/TIRAP, and TRAM) via its lipoproteins, LPS, flagellin, and DNA (6, 17, 18). Furthermore, events downstream of GBS-induced p38 activation eventually resulting in TNF formation remain largely elusive. It seems obvious that discrete knowledge of signaling events initiated by GBS is necessary for the development of adjunctive antisepsis strategies.
Here, we analyzed the roles of the individual components of the TLR adapter heteromer and the downstream MAPK axis in the cytokine response to GBS organisms. We found that MyD88 and p38 constitute a master regulator signaling pathway in the GBS-induced cytokine and chemokine responses via control of the transcription factors c-Fos/AP-1 and Egr-1.
MATERIALS AND METHODS
Reagents.
Reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless stated otherwise. Phosphate-buffered saline (PBS), Dulbecco's modified Eagle's medium (DMEM), G418, and trypsin-versene mixture were from Cambrex (Walkersville, MD). Low-endotoxin fetal bovine serum (FBS) was from HyClone (Erembodegem, Belgium). LPS derived from Escherichia coli strain 0111:B4 was purchased from Sigma-Aldrich and twice reextracted by phenol chloroform. SB203580 was from CalBiochem (San Diego, CA).
Generation of ethanol-fixed GBS.
GBS type III strain COH1, initially isolated from a newborn infant with sepsis, has previously been described (26). The strain was grown on blood agar plates (Remel, Lenexa, KS). Bacterial colonies were removed from the plates after overnight culture, washed three times in PBS, and then used to inoculate chemically defined medium prepared from endotoxin-free water (for GBS [25]) and grown to mid-log phase (A650 = 0.27 to 0.30). Subsequently, bacteria were harvested by centrifugation; fixed with ethanol (70%; 45 min on ice), heat (45 min at 70°C), or antibiotics (90 min in DMEM supplemented with ciprofloxacin); washed once with water; and resuspended in pyrogen-free water at a concentration of 20 mg/ml (corresponding to approximately 1 × 1010 organisms/ml, as determined by the number of CFU/ml before inactivation; accordingly, 200 μg/ml [dry weight] corresponds to 1 × 108 organisms/ml, as depicted in the figures). The entire procedure was performed under pyrogen-free conditions, resulting in preparations that were essentially free of endotoxin, as determined by a highly LPS-sensitive reporter system (CHO-CD14 with ELAM promoter-driven CD25; lower limit of detection, 10 to 100 pg/ml).
Macrophages.
Eight-week-old MyD88 −/− MAL/TIRAP −/− TRIF −/− TRAM −/− mice (engineered as described in reference 30), Elk−/− mice (4), and C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) were injected intraperitoneally with 2.5 ml of a 3% thioglycolate solution (Remel, Lenexa, KS). After 72 to 96 h, peritoneal exsudate cells were harvested exactly as described previously (14). Peritoneal macrophages were washed with medium, counted with a hemocytometer, and plated. After 2 h, nonadherent cells were removed by washing with medium and adherent cells were stimulated.
Bone marrow-derived macrophages were generated from bone marrow stem cells cultured on a 100-mm-diameter dish for 7 to 10 days in DMEM containing 10 μg/ml ciprofloxacin and supplemented with 10% FBS and 10 ng macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN) per ml. Differentiated bone marrow macrophages were counted, plated in 96-well plates, and stimulated as indicated. All cultures were kept at 37°C in a 5% CO2 (in air) atmosphere.
RAW 264.7 mouse macrophages were cultured in DMEM containing 10% FBS and 10 μg of ciprofloxacin/ml.
Transfection of RAW 264.7 macrophages and reporter constructs.
RAW 264.7 cells were seeded into 96-well tissue culture plates at a density of 105 cells per well. The following day, cells were transiently transfected with luciferase reporter constructs comprising Egr-1 (pEGR1.600.Luc, a kind gift from Dipika Gupta, Northwest Center for Medical Education, Indiana University School of Medicine, Gary, IN) (1) or NF-κB (κB3.Luc; Stratagene, La Jolla, CA) minimal promoter constructs or a human wild-type TNF promoter (TNF.WT.Luc, kindly provided by Nigel Mackman, Scripps Research Institute, La Jolla, CA), using Fugene (Roche, Basel, Switzerland) or Superfect (Qiagen, Hilden, Germany) per the manufacturers' recommendations. The following day, the cells were stimulated as indicated. After 4 to 6 h of stimulation, the cells were lysed in passive lysis buffer (Promega), and reporter gene activity was measured using a plate reader luminometer (MicroLumat Plus; Berthold Detection Systems, Pforzheim, Germany). In all cases, the data shown represent one of at least three separate but similar experiments and are presented as mean values ± standard deviations (SD) for triplicate samples.
Measurement of proinflammatory activity.
For determination of cytokine formation, macrophages were seeded at a density of 1 × 106 cells/ml in 96-well dishes in RPMI 1640 with 10% FBS plus 10 μg of ciprofloxacin/ml and incubated for 16 h at 37°C in a 5% humidified CO2 environment. Supernatants were processed directly for determination of released cytokines by an enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN) per the manufacturer's protocols.
Analysis of MAPK activation.
Phosphorylation of MAPK was evaluated according to standard protocols. Briefly, following stimulation, cell lysates were prepared, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and analyzed by Western immunoblotting using nitrocellulose membranes (HyClone, Erembodegem, Belgium). Primary antibodies directed the phosphorylated (activated) forms of MAPK p38 (Cell Signaling Technology, Beverly, MA), c-Jun (Cell Signaling Technology, Beverly, MA), Elk-1 (Cell Signaling Technology, Beverly, MA), and intracellular IκB (Santa Cruz Biotechnologies, Heidelberg, Germany). The identities of the bands were confirmed using molecular mass markers and positive controls.
RNA isolation.
Macrophages from MyD88-deficient or wild-type mice were seeded onto 24-well plates at a density of 300,000 cells per well. Cells were stimulated with heat-fixed GBS strain COH1 (107/ml) for 6 hours. Subsequently, cells were washed with ice-cold PBS and samples were frozen until RNA isolation.
Quantitative reverse transcription-PCR.
Total RNA was extracted from samples by using an RNeasy mini kit in accordance with the instruction manual (Qiagen). For quantitative two-step reverse transcription-PCR, 2 μg of total RNA was reverse transcribed to first-strand cDNA with an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Aliquots of 20 ng of first-strand cDNA were subsequently used as a template for quantitative PCR with gene-specific primers. The mouse-specific GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene served as a control for constitutive gene expression. GAPDH expression was consistent after bacterial inoculation in comparison with the amount of 18S rRNA. Amplifications were performed with 20 μl of SYBR green JumpStart Taq ReadyMix (Sigma-Aldrich, Munich, Germany) and 350 nM oligonucleotides, using an Eppendorf RealPlex thermal cycler (Eppendorf, Hamburg, Germany). After an initial activation step at 95°C for 7 min, 40 cycles (94°C for 30 s, 60°C for 30 s, 72°C for 30 s, and 82°C for 15 s) were performed, and a single fluorescence reading was obtained after the 82°C step of each cycle. A melting curve was determined at the end of cycling to ensure amplification of only a single PCR product. Threshold cycle values were determined with the Realplex version 1.5 software program, supplied with the instrument. Comparative expression levels (2−ΔΔCT) were calculated according to the method of Livak and Schmittgen (22a). The expression levels are relative to the level of GAPDH expression, which was constant in all RNA samples used and was set to 1. The values shown are representative of six samples from two biological experiments performed using quantitative PCR in triplicate. The oligonucleotide primers used were as follows: MoGAPDH (GenBank accession no. BC145812; 5′ACTCCACTCACGGCAAATTC3′ and 5′TCTCCATGGTGGTGAAGACA 3′), MoCfos (GenBank accession no. NM010234; 5′CCTGCCCCTTCTCAACGAC3′ and 5′GCTCCACGTTGCTGATGCT3′), and MoEGR1 (GenBank accession no. NM007913; 5′AGGAAGTTTGCCAGGAGTGA3′ and 5′TGGGTAGGAGGTAGCCACTG3′).
RESULTS
MyD88, but not MAL/TIRAP, TRIF, TRAM, or the IL-1 receptor, is essential for the formation of TNF and IL-6 in response to GBS.
Peritoneal macrophages from wild-type mice were potently activated by fixed GBS organisms to form both TNF and IL-6 (Fig. 1), starting at concentrations of dry bacterial mass as low as 200 ng/ml. Deletion of MAL/TIRAP had no effect on IL-6 and TNF formation, despite the putative role of MAL/TIRAP in the recognition of the cell wall constituent lipoteichoic acid and lipoproteins in gram-positive organisms. Similarly, TRIF and TRAM were dispensable for a robust cytokine response to GBS organisms (Fig. 1A to D). As previously reported, the TNF and IL-6 responses were abrogated in macrophages from mice with a targeted deletion of the MyD88 gene (Fig. 1B and D) (23). The essential role of MyD88 was independent of the mode of GBS fixation (ethanol, heat, or antibiotics) and was similarly observed in response to live bacteria (Fig. 1G). In contrast to GBS, all four tested adapter proteins were essential for the cytokine response to LPS in E. coli (Fig. 1E). In addition, inhibition of the IL-1-converting enzyme by N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD x fmk) had no effect on GBS-induced TNF formation (Fig. 1F). Furthermore, GBS-induced cytokine formation in IL-1 receptor 1-deficient macrophages was indistinguishable from cytokine formation in wild-type macrophages (data not shown). Accordingly, induction of IL-1/18 by GBS organisms cannot explain the role of MyD88 in this context, although MyD88 is an adapter for the IL-1 and IL-18 receptors.
FIG. 1.

MyD88, but not MAL/TIRAP, TRIF, or TRAM, is essential for GBS-induced TNF and IL-6 formation. Bone marrow-derived macrophages from mice with a targeted deletion of TRIF and TRAM (A and C) or MAL/TIRAP and MyD88 (B and D) (open symbols) or from wild-type (WT) C57B/J6 mice (closed symbols) were stimulated with escalating doses of ethanol-fixed GBS (strain COH1) or LPS (E) for 16 h. Then, TNF and IL-6 concentrations in the supernatants were determined by ELISA. Depicted are means ± SD of results from triplicate wells for one representative experiment out of three. (F) RAW 264.7 macrophages were preincubated with zVAD x fmk (20 μM) or a vehicle control and subsequently stimulated with GBS strain NEM 316 as indicated. (G) Macrophages from wild-type or MyD88- or TLR2-deficient mice were stimulated with ethanol-, heat-, or antibiotic-killed GBS or live bacteria. Then, TNF and IL-6 concentrations in the supernatants were determined by ELISA. Depicted are means ± SD for triplicate wells.
p38 is essential for the transcriptional activation of TNF by GBS organisms (Fig. 2).
FIG. 2.

GBS requires activation of MAPK p38 for proper formation of TNF. (A) Macrophages from mice with a targeted deletion of MyD88 or from wild-type mice were stimulated with GBS for 30 min. The cellular lysates were probed with antibodies specific for the phosphorylated form (Tyrx and Thrx) of p38 (pp38). (B and C) RAW 264.7 cells were stimulated with ethanol-fixed GBS in the presence of SB203580 (30 μM; open symbols) or a vehicle control (closed symbols) for 16 h. Then, TNF (B) and IL-6 (C) concentrations were determined by ELISA. Depicted are means ± SD of results from triplicate wells for one representative experiment out of three. (D) RAW 264.7 cells were transfected with the wild-type TNF luciferase reporter (TNF.WT.Luc) and stimulated with 108 GBS/ml in the presence of escalating dosages of the p38 inhibitor SB203580 for 5 h, and cellular lysates were analyzed by luminometry for reporter activity. Depicted are numbers of light units (means ± SD for triplicate wells) as n-fold activation over the background level (medium control).
GBS organisms robustly phosphorylated MAPK p38 in wild-type macrophages but not in MyD88-deficient macrophages (Fig. 2A). Activation of p38 was absolutely required for GBS-induced cytokine formation since inhibition of p38 by SB203580 (30 μM) abrogated the formation of TNF (Fig. 2B) and IL-6 (Fig. 2C). The effect of p38 on GBS-induced formation of TNF occurred at the transcriptional level, since SB203580 negatively affected the activation of a TNF promoter-driven luciferase reporter in a dose-dependent fashion (Fig. 2D).
p38 is essential for the MyD88-dependent induction of the transcription factor c-Fos/AP-1 by GBS but is dispensable for NF-κB activation (Fig. 3).
FIG. 3.

p38 mediates GBS-induced transcription of c-Fos/AP-1 but does not interfere with NF-κB activation. (A and B) RAW 264.7 cells were transfected with an NF-κB luciferase reporter construct. Then, cells were stimulated with or without fixed GBS (108/ml) plus an inhibitor for p38 (SB203580) (A) or JNK (SP600125) (B), and reporter activity was determined by luminometry. (C) RAW 264.7 cells were transfected with an AP-1 luciferase reporter construct (AP1.Luc). Then, cells were stimulated with a vehicle control or GBS (108/ml) plus an inhibitor for p38 (SB203580), and reporter activity was determined by luminometry. Depicted are results for one representative experiment out of three.
NF-κB has been shown to be essential for GBS-induced formation of TNF and to require the presence of MyD88 (14, 19, 31). Moreover, p38 has previously been shown to promote the transactivation of the NF-κB subunit p65 (16). Accordingly, we tested whether SB203580 inhibited GBS-induced transcriptional activation of NF-κB. However, in contrast to our expectations, SB203580 concentrations as high as 100 μM did not inhibit NF-κB activation in response to GBS organisms, clearly indicating the p38-independent nature of this pathway (Fig. 3A). In contrast, inhibition of the MAPK JNK by the anthrapyrazolone SP600125 led to inhibition of NF-κB-dependent transcriptional activation (Fig. 3B). Another signature substrate of p38 is c-Fos, which constitutes an important element of dimeric transcription factor activator protein 1 (AP-1). GBS-induced AP-1 activation was dependent on p38, since SB203580 inhibited an AP-1-dependent minimal promoter in a dose-dependent fashion (Fig. 3C). In conclusion, the GBS-induced MyD88-p38 pathway mediates TNF formation via c-Fos/AP-1 but not NF-κB activation.
GBS induces de novo synthesis of the transcription factor Egr-1 in a MyD88- and p38-dependent fashion (Fig. 4).
FIG. 4.

p38 mediates GBS-induced transcription of egr-1, which is essential for transcriptional activation of TNF. (A) Wild-type mouse macrophages were transfected with an egr-1 luciferase reporter construct (Egr-1.Luc). Then, cells were stimulated with GBS at the indicated concentrations, and reporter activity was determined by luminometry. (B) Wild-type (WT) or MyD88-deficient mouse macrophages were stimulated with the indicated concentrations of fixed GBS, and transcriptional activation of egr-1 was assessed by quantitative PCR. (C) RAW 264.7 cells were transfected with an egr-1 luciferase reporter construct (Egr-1.Luc). Then, cells were stimulated with GBS (108/ml) in the presence of escalating doses of an inhibitor for p38 (SB203580), and reporter activity was determined by luminometry. Depicted are results for one representative experiment out of three. (D) RAW 264.7 cells were transfected with either the wild-type TNF luciferase reporter construct or a TNF promoter construct with a mutation at the Egr-1 or AP-2 binding site. Subsequently, cells were stimulated as indicated and analyzed by luminometry.
Next to NF-κB and AP-1, Egr-1 is an essential transcription factor that triggers TNF production and has previously been associated with p38 activation. However, data on the role of MyD88 in Egr-1 induction by streptococci were not available. Accordingly, we wondered whether Egr-1 was a second MyD88- and p38-dependent branch of GBS-induced transcriptional activation of cytokine genes. First, we assessed whether egr-1 was induced by GBS organisms in macrophages and transcribed in a MyD88-dependent fashion. We found a strong dose-dependent egr-1 induction in wild-type macrophages (Fig. 4A). In contrast, egr-1 was not induced by GBS in MyD88-deficient mouse macrophages, as assessed by quantitative PCR (Fig. 4B). Furthermore, p38 was essential for egr-1 transcriptional activation since SB203580 inhibited GBS-induced egr-1 reporter activation (Fig. 4C). Next, we wondered whether Egr-1 was an essential transcription factor for GBS-induced TNF production. We found that a point mutation in the TNF promoter, which abolishes Egr-1 interaction with its canonical binding site at the TNF promoter, abrogates transcriptional activation of TNF by GBS organisms (Fig. 4D). In contrast, a mutation that abrogates the binding of AP-2 to the TNF promoter region did not alter the transcriptional activity induced by GBS on the TNF promoter. Accordingly, GBS organisms induce Egr-1 via p38 in a MyD88-dependent fashion. Egr-1 is essential for TNF induction in response to GBS organisms.
In contrast to TLR4 signaling, RANTES induction by GBS occurs in a MyD88- and p38-dependent fashion (Fig. 5).
FIG. 5.

GBS requires MyD88 and p38 activation for the formation of the chemokine RANTES. (A) Macrophages from mice with a targeted deletion of MyD88 or from wild-type (WT) mice were stimulated with fixed GBS for 10 h. (B) RAW 264.7 cells were stimulated with escalating concentrations of GBS in the presence or absence of the p38 inhibitor SB203580 (30 μM). RANTES concentrations in the macrophage supernatants were determined by ELISA. Depicted are means ± SD of results from triplicate wells for one representative experiment out of three.
Egr-1 has previously been linked to the chemokine RANTES (“regulated upon activation, normal T-cell expressed, and secreted”) mRNA. RANTES is a signature cytokine of the MyD88-independent pathway with respect to some TLRs (TLR3 and TLR4) (7). Data on RANTES induction by whole gram-positive bacteria were not available prior to our study. Accordingly, it seemed important to assess whether RANTES was induced by GBS organisms in a MyD88-dependent fashion. This was indeed the case, since GBS organisms induced large amounts of RANTES in wild-type macrophages but not in MyD88-deficient macrophages (Fig. 5A). In contrast, LPS in E. coli induced RANTES irrespective of MyD88 expression. Similar to what was found for TNF and IL-6, GBS-induced formation of RANTES was largely dependent on p38, since SB203580 potently reduced the response (Fig. 5B).
GBS-induced p38 mediates activation of Elk-1; however, Elk-1 is dispensable for TNF activation by GBS (Fig. 6).
FIG. 6.

MyD88 and p38 mediate GBS-induced activation of Elk-1, but Elk-1 is not essential for GBS-induced formation of TNF or IL-6. (A) RAW 264.7 cells were stimulated with 108 GBS/ml with or without SB203580 for the indicated time periods. Then, cellular lysates were subjected to Western blot analysis with antibodies against Phospho-Elk-1. (B and C) Bone marrow-derived macrophages from mice with a targeted deletion of elk-1 (open symbols) and from wild-type (WT) C57B/J6 mice (closed symbols) were stimulated with escalating doses of ethanol-fixed GBS (strain COH1) (B) or LPS (C) 16 h. Then, TNF and IL-6 concentrations in the supernatants were determined by ELISA. Depicted are means ± SD of results from triplicate wells for one representative experiment out of three.
The observation that p38 and Egr-1 were essential for GBS-induced transcriptional activation of TNF prompted us to analyze the putative role of Elk-1 in transcriptional activation of TNF. Elk-1 is a transcription factor for both the Egr-1 and TNF promoter; hence, Elk-1 might mediate GBS-induced cytokine formation through transcriptional activation of cytokine genes on several levels. First, we found that GBS organisms induced phosphorylation of the transcription factor Elk-1 in mouse macrophages in a dose-dependent fashion (data not shown). Elk-1 activation by GBS organisms was dependent on p38 activation, since inhibition of p38 prevented phosphorylation of Elk-1 by GBS (Fig. 6A). Interestingly, the kinetics of p38 and Elk-1 phosphorylation clearly differed. Whereas p38 was activated at 30 min only, Elk-1 was phosphorylated at 30 and 90 min, and both phosphorylations were prevented by inhibition of p38 (Fig. 6A and data not shown).
Since Elk-1 was activated by GBS organisms and had previously been implicated in the inflammatory response to TLRs, we addressed the specific role of Elk-1 in GBS-induced cytokine formation. To this end, we analyzed GBS-induced cytokine formation and GBS phagocytosis in wild-type and Elk-1-deficient macrophages. In contrast to our expectations, we did not observe a role for Elk-1 in GBS-induced cytokine formation (Fig. 6B) or phagocytosis of fluorescein isothiocyanate-labeled GBS (data not shown). Next, we assessed whether Elk-1 deficiency resulted in abrogation of the inflammatory response to LPS, since all available data generated by in vitro analysis of reporter expression indicated a role for Elk-1 downstream of TLR4. However, and in apparent contrast to what was found in previous studies with other cellular models (36), Elk-1 was not involved in LPS-induced cytokine formation (Fig. 6C). Accordingly, although Elk-1 is phosphorylated by GBS (via p38) and by LPS, Elk-1 is dispensable for the subsequent cytokine formation.
DISCUSSION
This study provides novel insights into the macrophage response to GBS organisms. First, MyD88 is the key TLR adapter protein for the formation of inflammatory cytokines and chemokines by macrophages, whereas the TLR adapter proteins MAL/TIRAP, TRIF, and TRAM are dispensable for this response. Second, downstream of MyD88, MAPK p38 controls at least two modes of transcriptional activation, c-Fos/AP-1 and EGR-1, which essentially mediate cytokine formation. In contrast, Elk-1, which had previously been suggested as an essential signaling intermediate in TLR signaling, is dispensable for GBS-induced cytokine formation.
The discrete delineation of TLR adapter usage by whole streptococci seems important, since available studies of the TLR adapters (other than MyD88) are largely limited to purified bacterial toxins. The absolute dependence of GBS inflammatory signaling on MyD88 but not any other well-defined TLR adapter protein is consistent with the notion that GBS organisms activate TNF and IL-6 in macrophages via a pathway clearly distinct from that engaged by TLR2 and TLR4 (14). With respect to the essential role of MyD88 alone, GBS mimics CpG DNA. Indeed, GBS has been reported to interact with the CpG receptor TLR9. However, the overall cytokine response to GBS is largely TLR9 independent, whereas it is entirely dependent on MyD88 (reference 14 and unpublished observations). It seems that this GBS-induced MyD88-dependent pathway, which does not involve any of the single TLRs commonly associated with gram-positive bacteria, is a common signaling route for many gram-positive bacteria, since recent data found similar modes of signal activation in response to Streptococcus pyogenes, Streptococcus pneumoniae, and Bacillus anthracis (9, 22).
Beyond the formation of inflammatory cytokines, MyD88 was essential for GBS-induced RANTES formation. This was intriguing, since, to the best of our knowledge, the role of MyD88 in the context of RANTES formation by gram-positive organisms had not been examined previously to this study. In contrast, RANTES formation in response to gram-negative bacteria occurs independently of MyD88 through engagement of TRIF and TRAM (7).
Why MyD88 functionally interacts with MAL/TIRAP, TRIF, and TRAM for some ligands but not others is currently not understood. Docking studies suggest that TLRs, MyD88, and MAL/TIRAP form heterotetrameric complexes. Specifically, MAL and MyD88 bind to different regions in TLRs and interact at a third, nonoverlapping site (8). It is unlikely that MyD88 can mediate TNF formation independently of other adapter proteins in the cases of GBS, flagellin, and CpG but requires interaction with one adapter protein (plus MAL/TIRAP) for the cytokine response to bacterial lipoproteins and four adapter proteins (plus MAL/TIRAP, TRIF, and TRAM) for the response to LPS. Thus, it is tempting to speculate on the existence of at least one further, currently unknown TLR adapter protein which serves as a signaling partner of MyD88 in the inflammatory response to GBS organisms. The current paradigm holds that, subsequently to TLR-MyD88 interaction, IL-1 receptor-associated kinase 1 (IRAK-1), IRAK-2, IRAK-4, and IRAK-M are recruited. Phosphorylation of IRAK-1 by IRAK-4 results in the dissociation of IRAK-1 from the receptor complex and interaction with TNF receptor-associated factor 6 (TRAF-6) through its COOH terminus. TRAF-6 activates the evolutionarily conserved signaling intermediate in Toll pathways (ECSIT), which in turn activates downstream MAPK cascades (20, 21). However, several technical issues, like the early lethality of the knockout mice, currently preclude absolute certainty as to the role of ECSIT between TRAF-6 and MAPK kinase kinases (33). MAPKs are a family of serine/threonine protein kinases. Among these, p38 can be regarded as a signaling relay that funnels various upstream signals and allows downstream diversification depending on the immediate cellular environment. p38 itself is activated through phosphorylation on both threonine and tyrosine residues at a Thr-Gly-Tyr dual phosphorylation motif in the kinase catalytic domain. Of the four known p38 isoforms α, β, γ, and δ, only the first two are significantly inhibited by SB203580, a pyridinyl-imidazole that exerts its relatively high kinase specificity by binding to the ATP binding pocket of p38 (28). Since SB203580 inhibits GBS-induced cytokine formation, p38 isoforms α and β are critical for GBS-induced inflammatory signaling, and the γ and δ isoforms cannot compensate for the inhibition of α and β.
Notably, the signaling pathways of p38 and other MAPKs are interrelated on several levels. As an example, Elk-1 can be phosphorylated by both p38 and JNK. However, our data indicate that, in the absence of p38 activity, JNK is not sufficient for GBS-induced Elk-1 phosphorylation. JNK and p38 signaling events furthermore converge on the level of AP-1 transcription factors, which are homo- or heterodimers of the Jun family, Fos proteins (c-Fos, FosB, Fra-1, and Fra-2), and members of subfamilies of the activating transcription factor family (ATF2, LRF1/ATF3, B-ATF, JDP1, and JDP2). While c-Jun proteins are mainly activated by the JNK, the Fos and ATF proteins are phosphorylated downstream of p38 and other MAPKs. In our hands, AP-1 activation is similarly reduced by the p38 inhibitor SB203580 and the JNK inhibitor SP600125 (Fig. 3A and unpublished observations).
Two transcription factor heteromers (c-Jun/c-Fos and c-Jun/ATF2) have previously been shown in more detail to have particularly high affinities for preferentially binding to the AP-1 binding site within the TNF promoter. Hence, c-Jun and therefore JNK activity is essential for the activation of AP-1 as a transcription factor for tnf. Cross-inhibition of JNK substrates by p38 inhibition is unlikely to be relevant in this context, since JNK (19) but not p38 (current study) mediates transactivation of NF-κB in response to GBS. It seems notable that GBS differs from IL-1β in this context, since the latter engages p38 for the transactivation of the NF-κB subunit p65 (through amino-terminal phosphorylation) (16). Accordingly, GBS engages a specific MyD88-dependent pathway that comprises the MAPKs p38 and JNK as essential inflammatory intermediates with overlapping but clearly distinct regulatory functions.
Several publications suggest that the microbial engagement of TLRs leads to the activation of Elk-1 (5, 32) and that this event in turn mediates transcriptional activation of TNF (36). However, conclusive data on the cytokine response to TLR agonists, such as data generated for Elk−/− mice, were not available. We report here that GBS and LPS induced normal amounts of TNF in Elk−/− macrophages. Accordingly, Elk-1 is dispensable for the responses to both stimuli. Furthermore, and in contrast to the upstream kinase p38, Elk-1 was not essential for GBS phagocytosis (19). We conclude that signaling events upstream of or parallel to Elk-1 mediate the p38 effect on transcriptional activation of tnf and phagocytosis.
Several lines of evidence suggest that, next to Elk-1, neither AP-1 nor NF-κB accounts for the antimicrobial effects of p38 activation. First, JNK mediates AP-1 activation, but its inhibition does not interfere with phagocytosis or bacterial killing. Furthermore, GBS-induced NF-κB activation is not dependent on p38 activation. Therefore, antimicrobial activity and TNF formation are independently regulated events downstream of p38. The Rab proteins are attractive intermediates in the p38-dependent antimicrobial pathway, since they modulate the endocytic traffic downstream of p38 (2).
In summary, MyD88 and p38 are parts of a distinct and functionally pivotal pathway activated by streptococci. AP-1 and Egr-1 inhabit key downstream positions in this highly inflammatory process (Fig. 7). Since p38 exerts desirable antimicrobial functions, interventional strategies that aim to target cytokine formation need to modulate molecules further downstream in the signaling cascade.
FIG. 7.
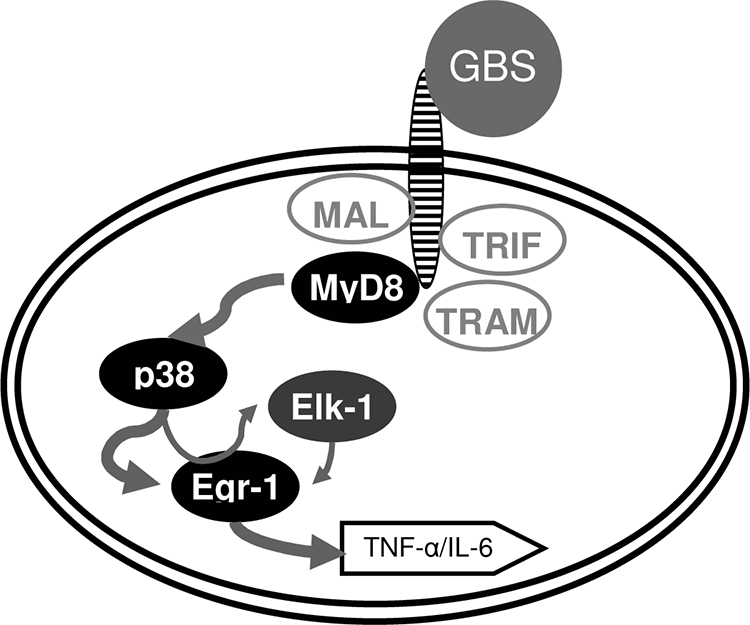
The signaling pathway MyD88 ⇒ p38 ⇒ EGR-1/AP-1 is essential for GBS-induced transcriptional activation of TNF. In response to GBS, MyD88 serves as a master signaling molecule, which essentially mediates p38 phosphorylation. In turn, p38 activates Egr-1 and AP-1, critical transcription factors for TNF. Similarly, Elk-1 is activated by p38; however, it is not required for TNF formation.
Acknowledgments
We are grateful to A. Nordheim for the provision of the Elk−/− mice and to Marie Charrel-Dennis for valuable discussions. We acknowledge Kristina Brückner and Anita Imm for expert technical assistance.
This work was supported by the Deutsche Forschungsgesellschaft (HE3127/2-3 and HE3127/3-1 to P.H.) and the National Institutes of Health (R01 AI052455-06 to D.T.G. and R01 AI067497-01 to K.A.F.).
Editor: J. N. Weiser
Footnotes
Published ahead of print on 30 March 2009.
REFERENCES
- 1.Bennett, B. L., D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J. C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, S. S. Bhagwat, A. M. Manning, and D. W. Anderson. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 9813681-13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blander, J. M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science 3041014-1018. [DOI] [PubMed] [Google Scholar]
- 3.Carty, M., R. Goodbody, M. Schroder, J. Stack, P. N. Moynagh, and A. G. Bowie. 2006. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 71074-1081. [DOI] [PubMed] [Google Scholar]
- 4.Cesari, F., V. Rennekampff, K. Vintersten, L. G. Vuong, J. Seibler, J. Bode, F. F. Wiebel, and A. Nordheim. 2004. Elk-1 knock-out mice engineered by Flp recombinase-mediated cassette exchange. Genesis 3887-92. [DOI] [PubMed] [Google Scholar]
- 5.Chow, J. C., D. W. Young, D. T. Golenbock, W. J. Christ, and F. Gusovsky. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 27410689-10692. [DOI] [PubMed] [Google Scholar]
- 6.Fischer, H., M. Yamamoto, S. Akira, B. Beutler, and C. Svanborg. 2006. Mechanism of pathogen-specific TLR4 activation in the mucosa: fimbriae, recognition receptors and adaptor protein selection. Eur. J. Immunol. 36267-277. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald, K. A., D. C. Rowe, B. J. Barnes, D. R. Caffrey, A. Visintin, E. Latz, B. Monks, P. M. Pitha, and D. T. Golenbock. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 1981043-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster, S. L., D. C. Hargreaves, and R. Medzhitov. 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447972-978. [DOI] [PubMed] [Google Scholar]
- 9.Glomski, I. J., J. H. Fritz, S. J. Keppler, V. Balloy, M. Chignard, M. Mock, and P. L. Goossens. 2007. Murine splenocytes produce inflammatory cytokines in a MyD88-dependent response to Bacillus anthracis spores. Cell. Microbiol. 9502-513. [DOI] [PubMed] [Google Scholar]
- 10.Hajjar, A. M., R. K. Ernst, J. H. Tsai, C. B. Wilson, and S. I. Miller. 2002. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 3354-359. [DOI] [PubMed] [Google Scholar]
- 11.Hazzalin, C. A., and L. C. Mahadevan. 2002. MAPK-Regulated transcription: a continuously variable gene switch? Nat. Rev. Mol. Cell Biol. 330-40. [DOI] [PubMed] [Google Scholar]
- 12.Henneke, P., and R. Berner. 2006. Interaction of neonatal phagocytes with group B streptococcus: recognition and response. Infect. Immun. 743085-3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henneke, P., S. Morath, U. Uematsu, S. Weichert, Pfitzenmaier., O. Takeuchi, A. Muller, C. Poyart, S. Akira, R. Berner, G. Teti, A. Geyer, T. Hartung, P. Trieu-Cuot, D. L. Kasper, and D. T. Golenbock. 2005. Role of lipoteichoic acid in the phagocyte response to group B streptococcus. J. Immunol. 1746449-6455. [DOI] [PubMed] [Google Scholar]
- 14.Henneke, P., O. Takeuchi, R. Malley, E. Lien, R. R. Ingalls, M. W. Freeman, T. Mayadas, V. Nizet, S. Akira, D. L. Kasper, and D. T. Golenbock. 2002. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 1693970-3977. [DOI] [PubMed] [Google Scholar]
- 15.Henneke, P., O. Takeuchi, J. A. van Strijp, H.-K. Guttormsen, J. A. Smith, A. B. Schromm, T. A. Espevik, S. Akira, V. Nizet, D. L. Kasper, and D. T. Golenbock. 2001. Novel engagement of CD14 and multiple Toll-like receptors by group B streptococci. J. Immunol. 1677069-7076. [DOI] [PubMed] [Google Scholar]
- 16.Jefferies, C. A., and L. A. J. O'Neill. 2000. Rac1 regulates interleukin 1-induced nuclear factor kappa B activation in an inhibitory protein kappa Balpha-independent manner by enhancing the ability of the p65 subunit to transactivate gene expression. J. Biol. Chem. 2753114-3120. doi: 10.1074/jbc.275.5.3114. [DOI] [PubMed] [Google Scholar]
- 17.Jeyaseelan, S., R. Manzer, S. K. Young, M. Yamamoto, S. Akira, R. J. Mason, and G. S. Worthen. 2005. Toll-IL-1 receptor domain-containing adaptor protein is critical for early lung immune responses against Escherichia coli lipopolysaccharide and viable Escherichia coli. J. Immunol. 1757484-7495. [DOI] [PubMed] [Google Scholar]
- 18.Jeyaseelan, S., S. K. Young, M. B. Fessler, Y. Liu, K. C. Malcolm, M. Yamamoto, S. Akira, and G. S. Worthen. 2007. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia. J. Immunol. 1783153-3160. [DOI] [PubMed] [Google Scholar]
- 19.Kenzel, S., G. Mancuso, R. Malley, G. Teti, D. T. Golenbock, and P. Henneke. 2006. c-Jun kinase is a critical signaling molecule in a neonatal model of group B streptococcal sepsis. J. Immunol. 1763181-3188. [DOI] [PubMed] [Google Scholar]
- 20.Kopp, E., R. Medzhitov, J. Carothers, C. Xiao, I. Douglas, C. A. Janeway, and S. Ghosh. 1999. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 132059-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopp, E. B., and R. Medzhitov. 1999. The Toll-receptor family and control of innate immunity. Curr. Opin. Immunol. 1113-18. [DOI] [PubMed] [Google Scholar]
- 22.Lee, K. S., C. A. Scanga, E. M. Bachelder, Q. Chen, and C. M. Snapper. 2007. TLR2 synergizes with both TLR4 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae. Cell. Immunol. 245103-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22a.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(ΔΔCT) method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 23.Mancuso, G., A. Midiri, C. Beninati, C. Biondo, R. Galbo, S. Akira, P. Henneke, D. Golenbock, and G. Teti. 2004. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J. Immunol. 1726324-6329. [DOI] [PubMed] [Google Scholar]
- 24.Mancuso, G., A. Midiri, C. Beninati, G. Piraino, A. Valenti, G. Nicocia, D. Teti, J. Cook, and G. Teti. 2002. Mitogen-activated protein kinases and NF-kappa B are involved in TNF-alpha responses to group B streptococci. J. Immunol. 1691401-1409. [DOI] [PubMed] [Google Scholar]
- 25.Paoletti, L. C., R. A. Ross, and K. D. Johnson. 1996. Cell growth rate regulates expression of group B Streptococcus type III capsular polysaccharide. Infect. Immun. 641220-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodewald, A. K., A. B. Onderdonk, H. B. Warren, and D. L. Kasper. 1992. Neonatal mouse model of group B streptococcal infection. J. Infect. Dis. 166635-639. [DOI] [PubMed] [Google Scholar]
- 27.Sacre, S. M., A. M. Lundberg, E. Andreakos, C. Taylor, M. Feldmann, and B. M. Foxwell. 2007. Selective use of TRAM in lipopolysaccharide (LPS) and lipoteichoic acid (LTA) induced NF-kappaB activation and cytokine production in primary human cells: TRAM is an adaptor for LPS and LTA signaling. J. Immunol. 1782148-2154. [DOI] [PubMed] [Google Scholar]
- 28.Shi, Y., and M. Gaestel. 2002. In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol. Chem. 3831519-1536. [DOI] [PubMed] [Google Scholar]
- 29.Shin, O. S., R. R. Isberg, S. Akira, S. Uematsu, A. K. Behera, and L. T. Hu. 2008. Distinct roles for MyD88 and Toll-like receptors 2, 5, and 9 in phagocytosis of Borrelia burgdorferi and cytokine induction. Infect. Immun. 762341-2351. doi: 10.1128/IAI.01600-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeuchi, O., K. Hoshino, and S. Akira. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 1655392-5396. [DOI] [PubMed] [Google Scholar]
- 31.Vallejo, J. G., P. Knuefermann, D. L. Mann, and N. Sivasubramanian. 2000. Group B Streptococcus induces TNF-alpha gene expression and activation of the transcription factors NF-kappa B and activator protein-1 in human cord blood monocytes. J. Immunol. 165419-425. [DOI] [PubMed] [Google Scholar]
- 32.Wu, W., R. D. Mosteller, and D. Broek. 2004. Sphingosine kinase protects lipopolysaccharide-activated macrophages from apoptosis. Mol. Cell. Biol. 247359-7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao, C., J. H. Shim, M. Kluppel, S. S. Zhang, C. Dong, R. A. Flavell, X. Y. Fu, J. L. Wrana, B. L. Hogan, and S. Ghosh. 2003. Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 172933-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto, M., S. Sato, H. Hemmi, H. Sanjo, S. Uematsu, T. Kaisho, K. Hoshino, O. Takeuchi, M. Kobayashi, T. Fujita, K. Takeda, and S. Akira. 2002. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420324-329. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-{beta} promoter in the Toll-like receptor signaling. J. Immunol. 1696668-6672. [DOI] [PubMed] [Google Scholar]
- 36.Yang, H., D. W. Young, F. Gusovsky, and J. C. Chow. 2000. Cellular events mediated by lipopolysaccharide-stimulated Toll-like receptor 4. MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J. Biol. Chem. 27520861-20866. doi: 10.1074/jbc.M002896200. [DOI] [PubMed] [Google Scholar]
- 37.Yates, R. M., and D. G. Russell. 2005. Phagosome maturation proceeds independently of stimulation of Toll-like receptors 2 and 4. Immunity 23409-417. [DOI] [PubMed] [Google Scholar]