

Abstract
We had shown earlier that the concentrations of circulating interleukin-18 (IL-18) are increased significantly in human immunodeficiency virus (HIV)-infected persons compared to HIV-seronegative healthy subjects. In the present study, we investigated the consequences of these elevated levels of IL-18 on natural killer (NK) cells and the immunopathogenesis of AIDS. We show here an inverse correlation between IL-18 concentrations and absolute numbers of various subsets of NK cells in infected persons. Recombinant human IL-18 caused increased death of a human NK cell line, as well as of primary human NK cells in vitro. The IL-18-mediated cell death was dependent upon Fas-FasL interactions and tumor necrosis factor alpha. IL-18 induced the expression of FasL on NK cells, increased the transcription from the human FasL promoter, reduced the expression of Bcl-XL in NK cells, and increased their sensitivity to FasL-mediated cell death. These results suggest that increased IL-18 concentrations present in the circulation of HIV-infected persons contribute to the immunopathogenesis of AIDS by altering NK cell homeostasis.
Interleukin-18 (IL-18), originally named as the gamma interferon (IFN-γ)-inducing factor, is a multifunctional and pleiotropic cytokine with proinflammatory properties (24, 25, 27; reviewed in reference 10). The cytokine is a member of the IL-1 family and is produced mainly by monocytes, macrophages, dendritic cells, keratinocytes, enterocytes, Kupffer cells, adrenal cortex, adipose tissues, and neurohypophysis in the human body. The cells and tissues constitutively produce IL-18 and enhance its production in response to stress, infection, lipopolysaccharide, cold, and stimulation via Toll-like receptors. Since IL-18 induces production of IFN-γ from T and natural killer (NK) cells, it was regarded as a cytokine that promotes Th1 type immune responses. However, the cytokine promotes Th2 type responses in the absence of IL-12 by inducing production of IL-4 from mast cells and eosinophils. Therefore, depending upon the context, the cytokine can promote both Th1 and Th2 type immune responses (reviewed in references 26 and 37). IL-18 exerts its biological effects by binding with a unique receptor called IL-18 receptor (IL-18R), which is a heterodimer of α and β chains, both of which carry Toll-IL-1 receptor domains in their intracytoplasmic regions. Given that IL-18 is a proinflammatory cytokine, it is not surprising that increased concentrations of this cytokine have been reported in many chronic inflammatory conditions in humans, e.g., rheumatoid arthritis, multiple sclerosis, Crohn's disease, graft-versus-host disease, atherosclerosis, etc. (10).
Human immunodeficiency virus type 1 (HIV-1) is the etiological agent of AIDS in humans. The infections with this virus are usually accompanied by changes in the production of several immunologically important cytokines, e.g., IL-15, tumor necrosis factor alpha (TNF-α), IL-4, IL-12, IL-10, transforming growth factor β1, etc. (1, 5, 8; reviewed in references 7, 16, and 20). These cytokine disturbances play an important role in the immunopathogenesis of AIDS in HIV-infected persons. Concerning IL-18, we and others have demonstrated increased concentrations of this cytokine in HIV-infected persons (3, 30, 33; see reference 32 for a review). The simian immunodeficiency virus (SIV), which is a close cousin of HIV-1, also induces IL-18 production in rhesus monkeys (12).
NK cells constitute an important cellular component of innate immunity. Not only do they kill virus-infected cells, they also kill these cells via antibody-dependent cell-mediated cytotoxicity (4, 16). Activated NK cells have been associated with protection from HIV infection, as well as with delaying its progression toward AIDS. However, NK cell functions become compromised in HIV-infected AIDS patients. Furthermore, the absolute numbers and percentages of various NK cell subsets are decreased overtime in these patients (reviewed in references 11 and 15). It has been demonstrated that increased serum concentrations of IL-18 correlate inversely with NK cell numbers in the patients suffering from chronic inflammatory conditions (22, 29). In the present study, we sought to determine whether such a correlation also existed between the serum concentrations of this cytokine and NK cell numbers in HIV-infected individuals. We show here a significant inverse correlation between IL-18 serum concentrations and NK cell numbers in HIV-infected AIDS patients. We also provide experimental evidence to show that IL-18 causes fratricidal cell death in human NK cells by inducing increased expression of FasL and TNF-α from these cells. The cytokine increases transcriptional activity of the human FasL gene promoter. Our study unravels the molecular mechanism underlying the inverse correlation between IL-18 levels and NK cell numbers and functions observed in chronic inflammatory conditions. Collectively, our studies demonstrate how increased IL-18 concentrations may be contributing to the immunopathogenesis of AIDS in HIV-infected persons by compromising NK cell responses.
MATERIALS AND METHODS
Cell culture.
All cells used in the present study were cultured at 37°C in 5% CO2 humidified atmosphere. An IL-2-dependent NK cell line, NK92, was used in the present study. The cell line was established from a patient with rapidly progressing non-Hodgkin's lymphoma (ATCC; catalogue no. CRL-2407). NK92 cells were maintained in α-MEM (Gibco, Burlington, Ontario, Canada) containing 12.5% fetal calf serum (FCS), 12.5% horse serum, 2 mM l-glutamate, 100 μg of penicillin/ml, and 100 μg of streptomycin/ml (all from Life Technologies, Burlington, Ontario, Canada) and supplemented with 100 U of IL-2 (Roche, Mississauga, Ontario, Canada)/ml. The erythroleukemia cell line K562 was maintained in the RPMI 1640 culture medium (Gibco) supplemented with 10% FCS, 2 mM l-glutamate, 100 μg of penicillin/ml, and 100 μg of streptomycin/ml (hereafter referred to as culture medium). These cells are frequently used as target cells in measuring NK cell-mediated cytotoxicity by human peripheral blood mononuclear cells (PBMC). The PBMC were obtained from the peripheral blood from HIV-infected AIDS patients and HIV-seronegative healthy subjects. For this purpose, blood was collected in heparinized vacuum tubes, and PBMC were isolated by centrifugation over Ficoll-Hypaque (Pharmacia, Montreal, Quebec, Canada) as described earlier (3). The buffy coat at the interface of Ficoll-Hypaque and plasma was collected, washed, and resuspended in RPM1 1640 medium containing 10% FCS, 2 mM l-glutamate, 100 μg of penicillin/ml, and 100 μg of streptomycin/ml as described previously (3).
Antibodies and recombinant cytokines.
The antibodies used in the present study were purchased: mouse anti-human FasL from BD Biosciences (Mississauga, Ontario, Canada); phycoerythrin (PE)-conjugated mouse anti-human FasL, mouse anti-human TNF-α, and its fluorescein isothiocyanate (FITC)-conjugated version from eBioscience (San Diego, CA); rabbit anti-human Bcl-XL from Cell Signaling Technology (Boston, MA); mouse anti-human Bcl-2 from Calbiochem (San Diego, CA); mouse anti-human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) from Ambion (Austin, TX); and allophycocyanin (APC)- or PE-conjugated mouse anti-human CD3, APC-conjugated mouse anti-CD56, FITC-conjugated mouse anti-human CD16 and PE-conjugated mouse anti-human TRAIL, and isotype-matched antibodies from Biolegend (San Diego, CA). The recombinant human IL-18 used for the present study was purchased from MBL (Woburn, MA).
Patients and sera collection.
Twenty-six HIV-infected viremic AIDS patients and an equal number of age-matched HIV-seronegative healthy subjects were recruited for the present study. The study was approved by the Institutional Ethics Committee. Peripheral blood samples were obtained from the study participants after we obtained their written informed consent. All of the patients had one or more AIDS-defining conditions and were receiving highly active antiretroviral therapy that comprised at least one protease inhibitor (saquinavir, ritonavir, or indinavir) and one or two reverse transcriptase inhibitors (stavudine, lamivudine, zidovudine, or didanosine). Detailed clinical and virological parameters of the patients are shown in Table 1. Serum was obtained from each blood sample after it was allowed to clot at room temperature for 2 to 3 h. Each serum sample was divided into aliquots and stored at −80°C until used.
TABLE 1.
Characteristics of 26 patients in this study
| Patient | CD4 (no. of cells/μl)a | Viral load (log10)b | Clinical stagec | Durationd |
|---|---|---|---|---|
| 1 | 334 | 2.70 | C2 | >4Y |
| 2 | 172 | 3.59 | C3 | >3Y |
| 3 | 497 | 2.70 | C1 | ND |
| 4 | 297 | 3.47 | C2 | 4Y 6M |
| 5 | 187 | 4.65 | C1 | 5Y 7M |
| 6 | 190 | 2.70 | C3 | ND |
| 7 | 254 | 4.18 | C3 | ND |
| 8 | 242 | 3.80 | C1 | >6Y |
| 9 | 299 | 2.70 | B1 | 6Y |
| 10 | 16 | 4.55 | C3 | 3Y |
| 11 | 165 | 2.70 | C1 | ND |
| 12 | 476 | 2.70 | B2 | 8Y 8M |
| 13 | 119 | 3.29 | C1 | ND |
| 14 | 171 | 4.79 | C3 | 4Y |
| 15 | 746 | 2.80 | C1 | ND |
| 16 | 26 | 3.80 | C3 | 4Y |
| 17 | 578 | 4.11 | C1 | >3Y |
| 18 | 164 | 2.80 | C1 | >3Y |
| 19 | 489 | 3.20 | C1 | 5Y |
| 20 | 519 | 2.70 | C1 | >5Y |
| 21 | 299 | 4.54 | C2 | 6Y |
| 22 | 176 | 2.70 | C1 | 6Y |
| 23 | 55 | 4.76 | C1 | ND |
| 24 | 38 | 5.34 | C3 | 3Y |
| 25 | 72 | 3.58 | C3 | 3Y |
| 26 | 643 | 2.80 | C1 | ND |
Mean ± standard error, 278 ± 203; median, 216; range, 16 to 746.
Mean ± standard error, 3.53 ± 0.85; median, 3.38; range, 2.70 to 5.34.
The clinical stages are as described in reference 19.
Y, year(s); M, month(s); ND, not determined.
Measuring cytokine concentrations.
The concentrations of IL-18 were determined in serum samples by using commercial enzyme-linked immunosorbent assay (ELISA) kits from Bender Medsystems (Burlingame, CA). The detection limit for the kit was 12 pg/ml. Soluble TNF-α in the culture supernatants was measured by a commercial ELISA kit (eBioscience, San Diego, CA) with minimum detection limit of 4 pg/ml.
NK cell microcultures.
For NK cell microculture experiments, 105 NK92 or primary NK cells were resuspended in 200 μl of the culture medium containing 5 U of IL-2/ml in triplicate in the wells of a round-bottom 96-well plate. NK cells were obtained from PBMC by negative selection using a kit from Stem Cell Technology (Vancouver, British Columbia, Canada). Recombinant human IL-18 was added to the microcultures at different concentrations with or without different neutralizing antibodies (described for each experiment individually). At different time points, the cells were washed with phosphate-buffered saline (PBS; pH 7.2) and were analyzed for different parameters.
Western blotting.
The expression of different proteins was analyzed by Western blotting as described in our earlier publications (3). Briefly, 5 × 106 NK cells were incubated in the culture medium with or without treatment as detailed in individual experiments. At different time points after the incubation, the cells were washed with PBS and lysed in a lysis buffer containing Tris-HCl (pH 6.8; 50 mM), sodium dodecyl sulfate (SDS; 2%), leupeptin (1 mg/ml), phenylmethylsulfonyl fluoride (1 mM), and pepstatin (1 mg/ml). The lysates were clarified by centrifugation at 14,000 × g for 15 min. Protein concentrations were determined in the lysates by using a commercial kit (Pierce, Inc., Nepean, Ontario, Canada). Portions (40 μg) of the lysate proteins were mixed with 2× SDS-polyacrylamide gel electrophoresis sample loading buffer containing 1 mM dithiothreitol, boiled, run on SDS-12% polyacrylamide gel electrophoresis gels, and electroblotted onto polyvinylidene difluoride membranes (Immobilon-P; Millipore, Ontario, Canada). After blockage of the membranes in 1% casein for 2 h at room temperature, they were incubated on a shaker with human protein-specific antibodies generated in mice, i.e., anti-FasL, anti-Bcl-XL, anti-Bcl-2, or anti-GAPDH, at 4°C overnight. The protein bands were revealed by autoradiography by using biotinylated goat anti-mouse antibodies and a commercial chemiluminescent kit (Vectastain ABC-AmP; Vector Laboratories, Burlington, CA). Individual bands on the X-ray films were quantified by densitometry.
Flow cytometry.
For immunostaining, 106 cells were incubated with fluorochrome-conjugated antibodies for 30 min on ice and washed three times with PBS containing 0.05% bovine serum albumin and 0.002% sodium azide. The washed cells were resuspended in 2% paraformaldehyde and analyzed by flow cytometry using FACSCalibur (BD Biosciences).
Determination of NK cell cytotoxicity.
NK cell cytotoxicity was determined with a standard 51Cr-release assay as described in our earlier publications (2, 4). Briefly, 104 51Cr-labeled K562 cells were dispended in 100 μl of the culture medium in triplicate in the wells of a V-bottom 96-well plate. Then, 2 × 105 PBMC obtained from HIV-infected/AIDS patients were added in 100 μl of the culture medium to each well. This gave a target-to-effector cell ratio of 1:20. The microcultures were incubated for 16 h at 37°C in a humidified 5% CO2 atmosphere, after which 100 μl of the culture supernatants were collected from each well. The radioactivity released into these supernatants was counted by an automatic gamma counter (Clini Gamma; LKB Wallac, Finland). The percentage lysis of the target cells was determined as described earlier (2).
Detection of cell death.
Dead cells were counted in microcultures by trypan blue exclusion. Apoptotic cells were detected by annexin V and propidium iodide (PI) staining using a commercial kit (BD Biosciences). Briefly, 105 cells were washed in PBS and resuspended in 100 μl of 1× annexin V binding buffer. Cells were then stained with 5 μl of FITC-conjugated annexin V and 5 μl of PI for 15 min at room temperature in the dark. Cells were then diluted with 400 μl of 1× binding buffer and analyzed within 1 h after staining by flow cytometry using FACSCalibur (BD Biosciences).
Transfection and FasL reporter gene assay.
Five million NK92 cells were transfected with a human FasL promoter-reporter gene construct. The construct (−511FasL pGL-3) contains a 511-bp region upstream the FasL gene start codon fused with the firefly luciferase gene in the plasmid pGL-3 basic (Promega) as described previously (23). The construct (5 μg of the DNA) was transfected transiently into the cells by using an Amaxa transfection system according to the accompanying protocol (Amaxa, Gaithersburg, MD). The transfected cells were divided into two equal aliquots at 12 h posttransfection. The cells were incubated in the culture medium for another 12 h with or without the addition of rhIL-18. The cells were washed and processed for luminescence detection using a luciferase reporter assay kit (Promega). The luminescence was analyzed by using a Mithras LB940 instrument (Berthold Technologies, Germany).
Statistical analysis.
Group means were compared using Student t test and correlation between two parameters were determined by Pearson correlations using the software Prism (GraphPad, San Diego, CA). The multivariate analysis using partial Pearson correlations were determined with the software SPSS (SPSS, Inc., Chicago, IL). Differences and correlations were deemed significant at P ≤ 0.05.
RESULTS
Levels of circulating IL-18 inversely correlate with NK cell numbers.
Increased IL-18 concentrations in the sera of patients suffering from chronic inflammatory conditions have been shown to be associated with loss of NK cell numbers and functionality (10, 22, 29). It is noteworthy that decreased NK cell numbers, as well as their decreased functionality, have been reported in HIV-infected AIDS patients. In order to determine whether increases in serum IL-18 levels were also associated with NK cell number and/or their cytolytic potential in HIV-infected persons, we measured IL-18 in the sera of HIV-infected persons by a commercial ELISA kit as described above and determined the absolute numbers of CD3− CD56+, CD3− CD16+, and CD56+ CD16+ NK cells present in their peripheral blood by flow cytometry. The staining and gating of the different NK cell subsets is shown in Fig. 1A. Furthermore, we measured spontaneous NK cell activities of freshly isolated PBMC of these patients against K562 cells in 51Cr-release assays as detailed in Materials and Methods. As shown in Fig. 1B to D, serum concentrations of IL-18 correlated negatively with absolute numbers of CD3− CD56+ (P = 0.04; r = −0.40), CD3− CD16+ (P = 0.008; r = −0.52), and CD16+ CD56+ NK cells (P = 0.006; r = −0.52). We determined whether the inverse correlation observed between IL-18 concentrations and NK cell numbers was influenced by viral loads of the patients. For this purpose, we performed a multivariate analysis between these three factors. Partial Pearson correlations were determined between IL-18 levels and NK cell numbers in controlling for viral loads. As shown in Table 2, the relationship between IL-18 concentrations and NK cell numbers in our cohort was independent of viral load.
FIG. 1.

Correlations of serum IL-18 levels with NK cell numbers and cytotoxicity. IL-18 concentrations in the sera of 26 HIV-infected persons were measured by a commercial ELISA kit, and the absolute numbers of different NK cells subsets present in their peripheral blood after gating on the viable lymphocyte population were determined by flow cytometry. (A) Representative flow cytometry data showing the gates used to identify the three different subsets of NK cells. The panel on the right shows results for CD3− CD16+ CD56+ NK cells. (B to D) Pearson correlations between serum concentrations of IL-18 and their absolute numbers of CD3− CD56+ (B), CD56+ CD16+ (C), and CD3− CD16+ (D) NK cells. The two parameters showed a statistically significant negative correlations (P = 0.04; r = −0.40 for panel B), (P = 0.006; r = −0.52 for panel C) and (P = 0.008; r = −0.52 for panel D). (E) Spontaneous NK cell activities of freshly isolated PBMC from these patients against K562 cells in a standard 51Cr-release assay. The serum IL-18 concentrations were plotted against NK cell-mediated cytotoxicities of the PBMC from HIV/AIDS patients. A bimodal effect of the serum IL-18 concentrations on NK cell-mediated cytotoxicities is evident.
TABLE 2.
Multivariate analysis between NK cell absolute numbers, IL-18 concentrations, and patient viral loads
| Control variable | Analysisa | Correlation between:
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CD3- CD56+ NK cells and IL-18 concn
|
CD56+ CD16+ NK cells and IL-18 concn
|
CD56+ CD16+ NK cells and IL-18 concn
|
||||||||
| NK cells | IL-18 | Viral load | NK cells | IL-18 | Viral load | NK cells | IL-18 | Viral load | ||
| None | ||||||||||
| NK cell no. | Correlation | 1.000 | -0.404 | -0.070 | 1.000 | -0.523 | -0.012 | 1.000 | -0.507 | -0.0169 |
| Significance | 0.041 | 0.734 | 0.006 | 0.955 | 0.008 | 0.408 | ||||
| df | 0 | 24 | 24 | 0 | 24 | 24 | 0 | 24 | 24 | |
| IL-18 concn | Correlation | -0.404 | 1.000 | -0.036 | -0.523 | 1.000 | -0.036 | -0.507 | 1.000 | -0.036 |
| Significance | 0.041 | 0.863 | 0.006 | 0.863 | 0.008 | 0.863 | ||||
| df | 24 | 0 | 24 | 24 | 0 | 24 | 24 | 0 | 24 | |
| Viral load | Correlation | -0.070 | -0.036 | 1.000 | -0.012 | -0.036 | 1.000 | -0.0169 | -0.036 | 1.000 |
| Significance | 0.734 | 0.863 | 0.955 | 0.863 | 0.408 | 0.863 | ||||
| df | 24 | 24 | 0 | 24 | 24 | 0 | 24 | 24 | 0 | |
| Viral load | ||||||||||
| NK cell no. | Correlation | 1,000 | -0.407 | 1.000 | -0.524 | 1.000 | -0.521 | |||
| Significance | 0.043 | 0.007 | 0.008 | |||||||
| df | 0 | 23 | 0 | 23 | 0 | 23 | ||||
| IL-18 concn | Correlation | -0.407 | 1.000 | -0.524 | 1.000 | -0.521 | 1.000 | |||
| Significance | 0.043 | 0.007 | 0.008 | |||||||
| df | 23 | 0 | 23 | 0 | 23 | 0 | ||||
df, Degree of freedom. Significance refers to bilateral significance. Correlations are Pearson correlations.
Interestingly, the cytokine concentrations did not show a significant correlation with NK cell cytotoxicity. However, when we plotted the serum IL-18 concentrations against NK cell-mediated cytotoxicities of the PBMC, a bimodal effect of the cytokine was observed on the cytotoxic activities of the PBMC obtained from the patients (Fig. 1E). These data also suggest that at higher concentrations, IL-18 causes decreased NK cell-mediated cytotoxicity of the PBMC in HIV-infected patients. Collectively, these results show that, similar to other chronic inflammatory diseases, increased serum IL-18 concentrations are associated with diminished NK cell numbers. The findings also suggest that NK cell cytotoxic activities also start declining progressively as the cytokine concentration increases beyond 400 pg/ml.
IL-18 treatment induces fratricidal killing of NK cells.
In order to investigate the molecular mechanism(s) involved in this IL-18-mediated NK cell loss, we cultured the NK cell line NK92 in the presence of various concentrations of recombinant human IL-18. It is noteworthy that NK92 cells are in phase III clinical trials as therapeutic tools of cell therapy for different human disease conditions (6, 31). Using the trypan blue exclusion assay, we found that addition of the rhIL-18 resulted in significant death of NK cells compared to the cells cultured in the absence of the cytokine. These experiments were repeated several times. The results of a representative experiment are shown in Fig. 2A. Remarkably, the death was dose dependent. Since 10 ng of the cytokine/ml gave consistently more than 50% cell death (Fig. 2A and data not shown), we used this concentration in subsequent experiments. In order to determine whether the IL-18-mediated cell death was due to apoptosis, we stained the cells with FITC-conjugated annexin V and PI and analyzed them by flow cytometry. Again, the results of a representative of three experiments are shown in Fig. 2C. The culture of NK92 cells in the presence of rhIL-18 induced their increased binding to FITC-annexin V, which was reduced by an anti-FasL antibody.
FIG. 2.

IL-18 induces cell death in NK cells. (A) NK92 cells were cultured for 24 h with or without increasing concentrations of rhIL-18. The IL-18-mediated cell death was measured using a trypan blue exclusion assay. (B) NK92 cells were cultured for 24 h with or without rhIL-18 at a concentration of 10 ng/ml. Simultaneously, FasL-, TNF-α-, or TRAIL-neutralizing antibodies were added to the wells. (C) IL-18-mediated cell death was measured by using a trypan blue exclusion assay, as well as by staining with FITC-conjugated annexin V and PI.
In order to determine the molecular mechanism(s) underlying this IL-18-mediated apoptosis of NK cells, we performed experiments to determine whether this occurred via Fas/FasL, TNF-α/ΤΝFR, and/or the TRAIL (TNF-related apoptosis-inducing ligand) pathways. For this purpose, we added anti-FasL antibodies, TNF-α neutralizing antibodies, and anti-TRAIL antagonist antibodies to NK92 cell cultures and determined their effects on the IL-18-mediated cell death. In these experiments, anti-FasL and anti-TNF-α antibodies significantly and consistently reduced apoptosis in the IL-18-added cultures compared to control antibodies. The results from a typical experiment are shown in Fig. 2B and C. Note a reduction by anti-FasL and anti-TNF-α to 20 and 25%;, respectively, versus 80% with control antibodies). Simultaneous addition of the anti-FasL and anti-TNF-α antibodies only slightly enhanced effect in the reduction of apoptosis (15% versus 20 and 25%). These results suggest that Fas-FasL and TNF-α-TNFR interactions were involved in the IL-18-mediated cell death in NK cell cultures. No reduction in the NK cells apoptosis was seen when anti-TRAIL antibodies were added in the NK cell cultures in the presence of rhIL-18 (data not shown). These results suggest that IL-18-mediated NK cell death does not involve TRAIL. This conclusion is further supported by the fact that NK92 cells do not express TRAIL and IL-18 does not induce its expression on these cells (data not shown).
IL-18 induces cell surface expression of FasL on NK cells.
Since IL-18-mediated cell death involved Fas-FasL interactions, we sought to determine whether this cytokine induced expression of Fas and/or FasL on NK92 cells. For this purpose, we determined the expression of FasL by flow cytometry on NK92 cells after their cultures for 20 h with or without IL-18. In repeated experiments, we found that this NK cell line expresses little FasL on the cell surface constitutively and that IL-18 induces its expression on the cell surface (Fig. 3A). We also verified this increased expression of FasL by Western blots. As shown in Fig. 3B, the cell line expresses FasL constitutively intracellularly and rhIL-18 increases this expression after 20 h of treatment. Since FasL is known to be cleaved by certain metalloproteases into culture supernatants of FasL-expressing cells (18), we sought to determine whether IL-18-treated NK92 cells were secreting FasL in the culture media. For this purpose, we used a commercial ELISA kit to measure soluble FasL (sFasL) in the culture supernatants of these cells. No sFasL could be detected in the supernatants of NK92 cells cultured in the presence or absence of rIL-18 (data not shown). This result supports our unpublished data that the supernatants taken from IL-18-treated cells do not cause the death of NK92 cells. We next wanted to know whether IL-18 was inducing the expression of FasL at the gene expression level. For this purpose, we used a human IL-18 promoter-reporter gene construct as described in Materials and Methods. We transfected the construct into NK92 cells and determined the effect of IL-18 on the reporter gene expression. Our data (Fig. 3C) show that the FasL promoter activity is significantly increased in IL-18-treated cells compared to control untreated cells (12,850 ± 650 versus 7,250 ± 510 relative luciferase activity, respectively; P < 0.01). These data suggest that IL-18 increases FasL gene expression in NK92 cells.
FIG. 3.

IL-18 induces cell surface expression of FasL in NK cells. (A) NK92 cells were cultured for 20 h with or without rhIL-18 (10 ng/ml), stained for the surface-expressed FasL, and analyzed by flow cytometry. The black histogram show the signal obtained with the isotype-matched control antibody. (B) Cell lysates from NK92 cells cultured in medium (lane 1) or medium supplemented with 10 ng of rhIL-18/ml (lane 2) for 20 h were analyzed by Western blotting with a monoclonal anti-FasL antibody. (C) NK92 cells were transfected with the IL-18 promoter-firefly luciferase reporter gene construct. Twelve hours later, the transfected cells were divided into aliquots, followed by incubation in culture medium for another 12 h with or without the addition of rhIL-18. The expression of the reporter gene (luminescence) was measured in arbitrary units as described in Materials and Methods. Consistently similar results were obtained with three different transfections. Promoter-luc and Basic-luc represent positive and negative controls, respectively. The bars labeled Medium and IL-18 refer to the activity with the vector −511FasL pGL-3.
IL-18-rich sera from AIDS patients induces cell death and FasL expression in NK92 cells.
In an attempt to reproduce the in vivo observation that increased IL-18 concentrations in the sera are associated with decreased NK cell numbers in AIDS patients under in vitro conditions, we incubated NK92 cells in sera from AIDS patients and control subjects. These sera had been previously tested for their IL-18 contents. In repeated experiments, we found that only the sera containing more than 500 pg of the cytokine/ml caused significant cell death (P < 0.001). The results from four representative sera are shown in Fig. 4A. The sera containing less than this concentration of the cytokine, irrespective of the fact whether they were from healthy or HIV-infected persons, did not cause significant cell death (data not shown). The addition of anti-IL-18 neutralizing antibodies in these cultures reduced cell death to the level of control sera. Furthermore, treatment of NK92 cells with these IL-18-rich sera induced the cell surface expression of FasL compared to sera from control subjects, and the presence of neutralizing anti-IL-18 antibodies in the culture abrogated the cell surface expression of FasL (Fig. 4B). Due to limited amounts of the sera containing more than 500 pg of IL-18/ml, the experiments were conducted with two samples. Taken together, these results confirm our earlier data obtained with the recombinant human IL-18.
FIG. 4.

Effect of IL-18-rich sera from AIDS patients on NK92 cells (A) NK92 cells were incubated for 24 h with medium only (M) or with IL-18-rich sera from AIDS patients (P1 and P2) and control donors (D1 and D2). These sera had been previously tested for their IL-18 contents (570 and 900 pg/ml for patients 1 and 2, respectively, and 73 and 63 pg/ml for donors 1 and 2, respectively). Simultaneously, neutralizing anti-IL-18 antibodies or control antibodies (mouse immunoglobulin G1) were added to the wells. After 24 h, the IL-18-mediated cell death was measured by counting dead cells using a trypan blue exclusion assay. (B) NK92 cells were incubated as described in panel A and stained for their surface expression of FasL by flow cytometry. Col Abody, control antibodies.
IL-18 induces TNF-α in the culture supernatants of NK92 cells.
Since anti-TNF-α antibodies inhibited IL-18-mediated cell death in NK92 cells, we determined whether IL-18 induced the secretion of TNF-α from these cells. For this purpose, we measured the concentration of TNF-α in the culture supernatants of the NK cells with or without treatment with IL-18. Figure 5 shows results from one of the two independent experiments. NK92 secreted low levels of this cytokine, and IL-18 treatment significantly (P < 0.0001) increased the amount of its secretion.
FIG. 5.
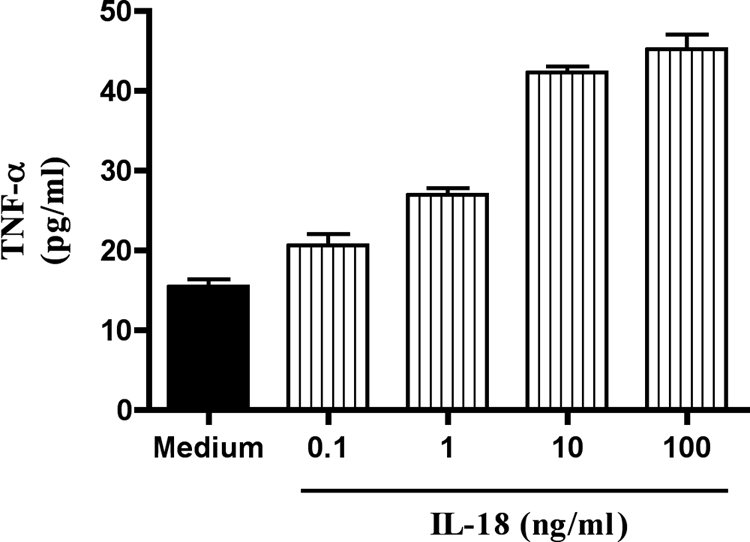
IL-18 induces TNF-α in the culture supernatants of NK92 cells. NK92 cells were incubated for 24 h with increasing concentrations (0.1, 1, 10, and 100 ng/ml) of rhIL-18. After 24 h, the concentrations of TNF-α in the culture supernatants were measured by a commercial ELISA kit.
IL-18 enhances susceptibility of NK92 cells to Fas/FasL-mediated apoptosis by downregulating Bcl-XL expression.
The supernatants from IL-18-treated cells were unable to induce cell death when added to NK92 cell cultures despite containing increased amounts of TNF-α. This suggested that IL-18 may not only kill NK92 cells by increasing the expression of FasL and TNF-α but also have increased the sensitivity of these cells to death-inducing stimuli. To investigate this possibility, we treated these cells with rhIL-18 for 12 h, washed them with the culture medium, and incubated them with equal amounts of anti-Fas agonist antibody. As shown in the Fig. 6A, the anti-Fas agonist causes more cell death in IL-18-treated NK92 cells than in cells that were not treated with the cytokine (P = 0.0343). These results are representative of three independent experiments. It is noteworthy that NK92 cells constitutively express Fas on their surface and rhIL-18 does not cause significant changes in the expression of this antigen in these cells (our unpublished data). Therefore, we used the same amount of anti-Fas agonist antibody in these experiments to provide equal degrees of death-inducing stimuli. The results could be interpreted by assuming that IL-18 enhances the susceptibility of these cells to Fas/FasL-mediated cell death. The IL-18-induced susceptibility depended upon the dose of IL-18 used: the higher the dose, the more susceptible the cells become. We reasoned that one way of increasing this susceptibility could be by modulating the expression of one or more apoptosis-regulating proteins belonging to the Bcl-2 family in the IL-18-treated cells. In order to determine whether IL-18 treatment modulated the expression of any of these proteins, we performed Western blots for Bcl-2 and Bcl-XL on IL-18-treated and untreated NK92 cells. In repeated experiments, the cytokine downregulated the expression of Bcl-XL, but not of Bcl-2, in this cell line. The data from a representative experiment are shown in shown in Fig. 6B.
FIG. 6.

IL-18 enhances susceptibility of NK92 cells to Fas/FasL-mediated apoptosis. (A) NK92 cells were cultured with the medium (lane A) or with the medium supplemented with 10 ng of rhIL-18/ml (lane B) for 12 h, washed, and incubated for another 12 h with 1 μg of anti-Fas agonistic antibody/ml. The dead cells were counted by using a trypan blue exclusion assay. The results are representative of two independent experiments. (B) Cell lysates from NK92 cells cultured in medium alone (lane 1) or in medium supplemented with 10 ng of rhIL-18/ml (lane 2) for 24 h were analyzed by Western blotting with anti-Bcl-2 monoclonal antibody and Bcl-XL. A decrease in the expression of the latter protein in the IL-18-treated cell lysates is quite noticeable. The results are representative of three independent experiments.
Recombinant human IL-18-induced changes in NK92 cells are recapitulated in primary human NK cells.
Finally, we wanted to know whether IL-18-mediated cell death in NK92 cells also occurs in primary human NK cells. For this purpose, we isolated NK cells from the peripheral blood of three donors and incubated them in the culture medium with different concentrations of rhIL-18. As shown in Fig. 7A, IL-18 also caused increased cell death in these cells in a dose-dependent manner (P < 0.0001). Furthermore, the death was prevented by both FasL and TNF-α neutralizing antibodies (Fig. 7B and C). In separate experiments, we determined the effects of the cytokine treatment on the expression of Fas and FasL on freshly isolated human NK cells. As shown in Fig. 8, the primary human NK cells, like NK92 cells, expressed little FasL on their surface, and IL-18 treatment induced this expression (Fig. 8A). Furthermore, the cells constitutively expressed Fas on their surface, and the cytokine treatment did not affect this expression in any significant manner (Fig. 8B). We also investigated whether IL-18 increased the susceptibility of primary NK cells to Fas-mediated apoptosis. For this purpose, we treated NK cells with IL-18 (10 ng/ml) from two different donors for 12 h separately and then added equal amounts of anti-Fas agonist antibodies. As shown in the Fig. 9A, the Fas agonistic antibodies caused significantly more (P < 0.05) death in IL-18-pretreated cells than in mock-treated cells in both donors. These data suggest that IL-18 also increases the susceptibility of primary human NK cells to Fas/FasL-induced death. Finally, we investigated the effects of IL-18 on the expression of Bcl-2 and Bcl-XL in primary human NK cells taken from two different donors. As shown in Fig. 9B, IL-18 treatment caused a decreased expression of Bcl-XL, but not of Bcl-2, in primary human NK cells. Collectively, these results demonstrate that recombinant human IL-18-induced changes in the NK92 cell line are recapitulated in primary human NK cells.
FIG. 7.

IL-18 causes death in freshly isolated human NK cells. (A) Isolated primary NK cells from a healthy donor were cultured for 24 h with or without increasing concentrations of rhIL-18. IL-18-mediated cell death was measured by counting dead cells using a trypan blue exclusion assay. (B) Isolated human NK cells were cultured for 24 h in the presence of rhIL-18 (10 ng/ml) with or without the addition of anti-FasL antibodies or TNF-α- or TRAIL-neutralizing antibodies (1 μg/ml each). IL-18-mediated cell death was measured by counting dead cells using a trypan blue exclusion assay. (C) Human NK cells were treated as described above and stained with FITC-conjugated annexin V and PI and analyzed by flow cytometry. The results are representative of three independent experiments done on three different donors.
FIG. 8.

Effect of rhIL-18 on cell surface expression of Fas and FasL on primary NK cells. Isolated human NK cells were incubated for 24 h with or without 10 ng of rhIL-18/ml, stained for the expression of FasL (A) and Fas (B) on their surfaces, and measured by flow cytometry. The results are representative of three independent experiments with three different donors.
FIG. 9.

IL-18 enhances the susceptibility of primary NK cells to Fas/FasL-mediated apoptosis. (A) Isolated primary NK cells from two donors were cultured with the medium (lane A) or with the medium supplemented with 10 ng of rhIL-18/ml (lane B) for 12 h, washed, and incubated for another 12 h with 1 μg of anti-Fas agonistic antibody/ml. The dead cells were counted by using a trypan blue exclusion assay. (B) Cell lysates from primary NK cells coming from two different donors were cultured in medium alone (lane 1) or in medium supplemented with 10 ng of rhIL-18/ml (lane 2) for 24 h and then analyzed by Western blotting with anti-Bcl-2 monoclonal antibody and Bcl-XL. A decrease in the expression of the latter protein in the IL-18-treated cell lysates is notable.
DISCUSSION
Our results show that, like other chronic inflammatory disease conditions (22, 29), there exists a negative correlation between NK cell numbers and the levels of the circulating IL-18 in HIV-infected persons. All major subsets of NK cells—i.e., CD3− CD16+, CD3− CD56+, and CD16+ CD56+ NK cells—showed almost equally strong inverse correlations with serum IL-18 levels in these patients. These data suggest that IL-18 is implicated in the loss of NK cell numbers in HIV-infected persons (see below), and all of these subsets of NK cells are equally susceptible to this adverse effect of this cytokine. Our results also shed light on the molecular mechanism involved in the IL-18-mediated loss of NK cells. Using an established human NK cell line, as well as primary human NK cells, we demonstrate that IL-18 induces fratricidal cell death in NK cells via two mechanisms: by inducing the expression of FasL and by increasing the production of TNF-α from NK cells. Our unpublished data show that NK92 cells, as well as freshly isolated primary human NK cells, constitutively express Fas on the cell surface, and IL-18 treatment does not increase Fas expression on these cells. However, the cytokine induces expression of FasL on the cell surface and TNF-α production from these cells. The two molecules induce cell death in NK cell cultures. Despite an increase in the expression of FasL, we were not able to detect soluble FasL from the NK cell supernatants cultured in the presence of rhIL-18. Furthermore, these supernatants were not able to induce cell death when added to NK cell cultures (our unpublished data). We also noted that rhIL-18 caused an increase in the sensitivity of NK cells to apoptotic stimuli by decreasing the expression of the antiapoptotic protein, Bcl-XL, in both NK92 and human NK cells. This may explain why the supernatants from the NK cells cultured in the presence of rhIL-18 (which are supposed to contain TNF-α) were not able to induce apoptosis of NK cells. A decreased expression of this antiapoptotic protein in rhIL-18-treated cells may be crucial for their apoptosis via FasL. To the best of our knowledge, this is the first report implicating IL-18 in causing the downregulation of an antiapoptotic protein in human NK cells. It will be interesting to determine whether the cytokine induces a similar downregulation of this protein in other immune cells, e.g., T cells.
We observed that rhIL-18 caused cell death in NK92 cells, as well as in primary human NK cells, in a dose-dependent manner. At concentrations as low as 100 pg/ml, the cytokine consistently caused significant cell death. However, when we tested sera from healthy and HIV-infected persons with known concentrations of IL-18, the sera containing more than 500 pg of IL-18/ml caused cell death. The sera containing lower concentrations of the cytokine, whether from HIV-infected or healthy persons, did not cause significant cell death. We believe that many other factors in the sera, e.g., IL-18BP, cytokines, growth factors, etc., may explain these differential effects.
It is noteworthy that this IL-18-mediated NK cell killing may represent a negative-feedback mechanism to control and terminate a proinflammatory immune response. It was demonstrated previously that combinations of two cytokines, IL-12 and IL-15, as well as IL-2 and IL-12, also cause death of human NK cells, which could be blocked by TNF-α neutralizing antibodies (28). The authors of that study did not implicate Fas-FasL interactions in this cytokine-mediated cell death. It is noteworthy that IL-18, but not IL-12 or IL-15, has been shown to increase FasL-mediated cell death by murine NK cells and Th1 T-cell clones (9, 13, 36). However, the expression of FasL on NK cells was not measured in these studies. We show here for the first time that IL-18 induces FasL expression in a human NK cell line, as well as in freshly isolated primary human NK cells. Constitutively, these cells express very little of this antigen on the cell surface. We provide experimental evidence to show that in vitro the cytokine increases the transcription of the FasL gene via stimulating its promoter. It is quite conceivable that the cytokine-induced expression of FasL would result in upregulation of the FasL-mediated cytotoxicity of NK cells. However, these cells could also cause tissue destruction by killing Fas-positive body cells and tissues. Indeed, IL-18 has been implicated in FasL- and TNF-α-mediated liver damage by endotoxin in mice (34; reviewed in reference 35). It is noteworthy that cytokines such as IL-2, IL-12, IL-15, and IL-18 have the potential of enhancing innate and adaptive immunity against cancer and infectious diseases and are being used or considered both as adjuvants and as prophylactic and therapeutic tools. Our results suggest that careful consideration should be given to the in vivo use of these cytokines either alone or in different combinations due to their negative-feedback mechanisms and consequently their suppressive effects on immune responses.
Interestingly, one study (14) has shown that IL-18 prevents apoptosis of NK cells during their killing of target cells. The authors of that study ascribed this IL-18-mediated protection to enhanced expression of TRAF-1 and c-IAP. Another study (21) has shown that IL-18 induces expression of CD83, CCR7, and CD25 on NK cells and increases their migration to lymph nodes and interaction with other immunocytes (so-called helper function). It is noteworthy that our study examines NK cell death when they interact with each other after IL-18 treatment and does not contradict the results of these two studies. IL-18 increases FasL expression on NK cells and therefore IL-18-treated NK cells would kill Fas-positive target cells more efficiently. It is noteworthy that in our hands IL-18-treated NK92 did not cause enhanced killing of K562 cells, which lack Fas expression. Our results contradict those of Kalina et al. (17), who reported increased killing of these target cells by IL-18-treated NK92 cells. These differences may be attributed to different techniques used in the two studies: we used standard 51Cr-release assays, while the other study relied upon cell-staining dye PKH and PI intake. Furthermore, we cannot exclude the differential expression of Fas on the target cells.
It can be argued that our results may not be relevant since we used concentrations of IL-18 in several of our experiments that are much higher that those found in the circulation of healthy or HIV-infected persons. In fact, the concentrations of cytokines in the circulation represent a spillover from the cells and tissues that produce them. The precise concentrations of the cytokines in tissues where the cytokine-producing cells reside are not known. These concentrations are most likely to be much higher than those found in the circulation, the so-called “physiological” concentrations. Therefore, in the course of investigating basic biology of a cytokine, it is not inappropriate or “unphysiological” to perform experiments with cytokine concentrations higher than those found in the circulation. There are several examples in which researchers have used IL-18 concentrations in nanograms per milliliter (see, for example, references 17 and 21) and discovered certain unknown aspects of the cytokine biology.
In summary, the present study advances our understanding of the immunobiology of IL-18 and sheds light on how increased concentrations of this cytokine found in the circulation of HIV-infected patients may be contributing to AIDS pathogenesis.
Acknowledgments
We thank Jonathan Ashwell of the Immune Cell Biology Laboratory at NCI/NIH, Bethesda, MD, for providing us with the plasmid −511FasLpGL-3. We thank Mario Legault for administrative coordination for obtaining blood specimens from HIV-infected patients.
We also thank the Canadian Institutes for Health Research for support and the Fonds de Recherche en Santé du Québec for a doctoral research award to I.A.
Footnotes
Published ahead of print on 1 April 2009.
REFERENCES
- 1.Ahmad, A., R. Ahmad, E. Toma, R. Morisset, and J. Menezes. 2000. Impaired induction of IL-15 in response to herpes simplex virus type 1 infection in peripheral blood mononuclear cells of HIV-infected patients. AIDS 14744-746. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad, A., R. Morisset, R. Thomas, and J. Menezes. 1994. Evidence for a defect of antibody-dependent cellular cytotoxic (ADCC) effector function and anti-HIV gp120/41-specific ADCC-mediating antibody titres in HIV-infected individuals. J. Acquir. Immune Defic. Syndr. 7428-437. [PubMed] [Google Scholar]
- 3.Ahmad, R., S. T. A. Sindhu, E. Toma, R. Morisset, and A. Ahmad. 2002. Elevated levels of circulating interleukin-18 in human immunodeficiency virus-infected individuals: role of peripheral blood mononuclear cells and implications for AIDS pathogenesis. J. Virol. 7612448-12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad, R., S. T. A. K. Sindhu, E. Toma, R. Morisset, J. Vincelette, J. Menezes, and A. Ahmad. 2001. Evidence for a correlation between antibody-dependent cellular cytotoxicity-mediating anti-HIV-1 antibodies and prognostic predictors of HIV infection. J. Clin. Immunol. 21227-233. [DOI] [PubMed] [Google Scholar]
- 5.Allen, J., H. Wong, P. Guyre, G. Simon, and S. Wahl. 1991. Association of circulating receptor Fcγ RIII-positive monocytes in AIDS patients with elevated levels of transforming growth factor-beta. J. Clin. Investig. 871773-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arai, S., R. Meagher, M. Swearingen, H. Myint, E. Rich, J. Martinson, and H. Klingemann. 2008. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 10625-632. [DOI] [PubMed] [Google Scholar]
- 7.Cease, K. B., and J. A. Berzofsky. 1994. Toward a vaccine for AIDS: the emergence of immunobiology-based vaccine development. Annu. Rev. Immunol. 12923-989. [DOI] [PubMed] [Google Scholar]
- 8.Chehimi, J., S. Starr, I. Frank, A. D'Andrea, X. Ma, R. MacGregor, J. Sennelier, and G. Trinchieri. 1994. Impaired interleukin 12 production in human immunodeficiency virus-infected patients. J. Exp. Med. 1791361-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dao, T., K. Ohashi, T. Kayano, M. Kurimoto, and H. Okamura. 1996. Interferon-gamma-inducing factor, a novel cytokine, enhances Fas ligand-mediated cytotoxicity of murine T helper 1 cells. Cell. Immunol. 173230-235. [DOI] [PubMed] [Google Scholar]
- 10.Dinarello, C. 2007. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin. Nephrol. 2798-114. [DOI] [PubMed] [Google Scholar]
- 11.Fauci, A. S., D. Mavilio, and S. Kottilil. 2005. NK cells in HIV infection: paradigm for protection or targets for ambush. Nat. Rev. Immunol. 5835-843. [DOI] [PubMed] [Google Scholar]
- 12.Giavedoni, L. D., M. C. Velasquillo, L. M. Parodi, G. B. Hubbard, and V. L. Hodara. 2000. Cytokine expression, natural killer cell activation, and phenotypic changes in lymphoid cells from rhesus macaques during acute infection with pathogenic simian immunodeficiency virus. J. Virol. 741648-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto, W., T. Osaki, H. Okamura, P. D. Robbins, M. Kurimoto, S. Nagata, M. T. Lotze, and H. Tahara. 1999. Differential antitumor effects of administration of recombinant IL-18 or recombinant IL-12 are mediated primarily by Fas-Fas ligand- and perforin-induced tumor apoptosis, respectively. J. Immunol. 163583-589. [PubMed] [Google Scholar]
- 14.Hodge, D. L., J. J. Subleski, D. A. Reynolds, M. D. Buschman, W. B. Schill, M. W. Burkett, A. M. Malyguine, and H. A. Young. 2006. The proinflammatory cytokine interleukin-18 alters multiple signaling pathways to inhibit natural killer cell death. J. Interferon Cytokine Res. 26706-718. [DOI] [PubMed] [Google Scholar]
- 15.Iannello, A., O. Debbeche, S. Samarani, and A. Ahmad. 2008. Antiviral NK cell responses in HIV infection: I. NK cell receptor genes as determinants of HIV resistance and progression to AIDS. J. Leukoc. Biol. 841-26. [DOI] [PubMed] [Google Scholar]
- 16.Iannello, A., O. Debbeche, S. Samarani, and A. Ahmad. 2008. Antiviral NK cell responses in HIV infection. II. viral strategies for evasion and lessons for immunotherapy and vaccination. J. Leukoc. Biol. 8427-49. [DOI] [PubMed] [Google Scholar]
- 17.Kalina, U., D. Kauschat, N. Koyama, H. Nuernberger, K. Ballas, S. Koschmieder, G. Bug, W.-K. Hofmann, D. Hoelzer, and O. G. Ottmann. 2000. IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN-γ production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk-21. J. Immunol. 1651307-1313. [DOI] [PubMed] [Google Scholar]
- 18.Kayagaki, N., A. Kawasaki, T. Ebata, H. Ohmoto, S. Ikeda, S. Inoue, K. Yoshino, K. Okumura, and H. Yagita. 1995. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 1821777-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy, J. 1998. HIV and the pathogenesis of AIDS, p. 397. American Society for Microbiology, Washington, DC.
- 20.Lotz, M., and P. Seth. 1993. TGF beta and HIV infection. Ann. N. Y. Acad. Sci. 685501-511. [DOI] [PubMed] [Google Scholar]
- 21.Mailliard, R. B., S. M. Alber, H. Shen, S. C. Watkins, J. M. Kirkwood, R. B. Herberman, and P. Kalinski. 2005. IL-18-induced CD83+ CCR7+ NK helper cells. J. Exp. Med. 202941-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazodier, K., V. Marin, D. Novick, C. Farnarier, S. Robitail, N. Schleinitz, V. Veit, P. Paul, M. Rubinstein, C. A. Dinarello, J.-R. Harle, and G. Kaplanski. 2005. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 1063483-3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittelstadt, P. R., and J. D. Ashwell. 1998. Cyclosporin A-sensitive transcription factor Egr-3 regulates Fas ligand expression. Mol. Cell. Biol. 183744-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura, K., H. Okamura, K. Nagata, T. Komatsu, and T. Tamura. 1993. Purification of a factor which provides a costimulatory signal for gamma interferon production. Infect. Immun. 6164-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura, K., H. Okamura, M. Wada, K. Nagata, and T. Tamura. 1989. Endotoxin-induced serum factor that stimulates gamma interferon production. Infect. Immun. 57590-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakanishi, K., T. Yoshimoto, H. Tsutsui, and H. Okamura. 2001. Interleukin-18 regulates both TH1 and TH2 responses. Annu. Rev. Immunol. 19423-474. [DOI] [PubMed] [Google Scholar]
- 27.Okamura, H., H. Tsutsui, T. Komatsu, M. Yutsudo, A. Hakura, T. Tanimoto, K. Torigoe, T. Okura, Y. Nukada, K. Hattori, K. Akita, M. Namba, F. Tanabe, K. Konishi, S. Fukuda, and M. Kurimoto. 1995. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature 37888-91. [DOI] [PubMed] [Google Scholar]
- 28.Ross, M. E., and M. A. Caligiuri. 1997. Cytokine-induced apoptosis of human natural killer cells identifies a novel mechanism to regulate the innate immune response. Blood 89910-918. [PubMed] [Google Scholar]
- 29.Shibatomi, K., K. Ida, S. Yamasaki, T. Nakashima, T. Origuchi, A. Kawakami, K. Migita, Y. Kawabe, M. Tsujihata, P. Anderson, and K. Eguchi. 2001. A novel role for interleukin-18 in human natural killer cell death: high serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheum. 44884-892. [DOI] [PubMed] [Google Scholar]
- 30.Song, W., C. M. Wilson, S. Allen, C. Wang, Y. Li, R. A. Kaslow, and J. Tang. 2006. Interleukin 18 and human immunodeficiency virus type I infection in adolescents and adults. Clin. Exp. Immunol. 144117-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tam, Y., J. Martinson, K. Doligosa, and H. Klingemann. 2003. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy 5259-272. [DOI] [PubMed] [Google Scholar]
- 32.Torre, D., and A. Pugliese. 2006. Interleukin-18: a proinflammatory cytokine in HIV-1 infection. Curr. HIV Res. 4423-430. [DOI] [PubMed] [Google Scholar]
- 33.Torre, D., F. Speranza, R. Martegani, A. Pugliese, F. Castelli, C. Basilico, and G. Biondi. 2000. Circulating levels of IL-18 in adult and paediatric patients with HIV-1 infection. AIDS 142211-2212. [DOI] [PubMed] [Google Scholar]
- 34.Tsutsui, H., K. Matsui, N. Kawada, Y. Hyodo, N. Hayashi, H. Okamura, K. Higashino, and K. Nakanishi. 1997. IL-18 accounts for both TNF-alpha- and Fas ligand-mediated hepatotoxic pathways in endotoxin-induced liver injury in mice. J. Immunol. 1593961-3967. [PubMed] [Google Scholar]
- 35.Tsutsui, H., K. Matsui, H. Okamura, and K. Nakanishi. 2000. Pathophysiological roles of interleukin-18 in inflammatory liver diseases. Immunol. Rev. 174192-209. [DOI] [PubMed] [Google Scholar]
- 36.Tsutsui, H., K. Nakanishi, K. Matsui, K. Higashino, H. Okamura, Y. Miyazawa, and K. Kaneda. 1996. IFN-gamma-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol. 1573967-3973. [PubMed] [Google Scholar]
- 37.Yoshimoto, T., and K. Nakanishi. 2006. Roles of IL-18 in basophils and mast cells. Allergy Int. 55105-113. [DOI] [PubMed] [Google Scholar]