

Abstract
The virological synapse (VS) is a specialized molecular structure that facilitates the transfer of certain lymphotropic viruses into uninfected T cells. However, the role of the VS in the transfer of nonlymphotropic viruses into T cells is unknown. Herpes simplex virus (HSV) has been shown in vitro to infect T cells and modulate T-cell receptor function, thereby suppressing T-cell antiviral function. However, whether such infection of T cells occurs in vivo is unknown. Here, we examined whether T-cell infection could be observed in human HSV disease and investigated the mechanism of HSV entry into T cells. We found that HSV-infected T cells were readily detectable during human disease, suggesting that infection and modulation of T-cell function plays a role in human immunopathology. HSV infection of both CD4+ and CD8+ T cells occurred much more efficiently via direct cell-to-cell spread from infected fibroblasts than by cell-free virus. Activation of T cells increased their permissivity to HSV infection. Cell-to-cell spread to T cells did not require HSV glycoproteins E and I (gE and gI), which are critical for cell-to-cell spread between epithelial cells. Transfer of HSV to T cells required gD, and the four known entry receptors appear to be contributing to viral entry, with a dominant role for the herpesvirus entry mediator and nectin-1. VS-like structures enriched in activated lymphocyte function-associated antigen 1 (LFA-1) were observed at the point of contact between HSV-infected fibroblasts and T cells. Consistent with spread occurring via the VS, transfer of HSV was increased by activation of LFA-1, and cell-to-cell spread could be inhibited by antibodies to LFA-1 or gD. Taken together, these results constitute the first demonstration of VS-dependent cell-to-cell spread for a predominantly nonlymphotropic virus. Furthermore, they support an important role for infection and immunomodulation of T cells in clinical human disease. Targeting of the VS might allow selective immunopotentiation during infections with HSV or other nonlymphotropic viruses.
The virological synapse (VS) is a specialized molecular structure that facilitates the transfer of certain lymphotropic viruses, such as human immunodeficiency virus (HIV) and human T-cell leukemia virus type 1 (HTLV-1), into uninfected T cells (22, 28, 38). Entry and infection of T cells by HIV or HTLV-1 via the VS is far more efficient than infection by cell-free virus, and thus this structure plays a critical role in the pathogenesis of these viruses. The organization of the VS is in many respects similar to the immunological synapse (IS), in particular, to the immature IS. The VS is highly enriched in the adhesion molecule lymphocyte function-associated antigen 1 (LFA-1) and its ligands intercellular adhesion molecule 1 (ICAM-1) and ICAM-3 (29); however, it does not possess the CD3-enriched central region associated with the mature IS (28, 47). While the VS is critical to the pathogenesis of HIV and HTLV-1, it remains an unanswered question whether the VS is also involved in T-cell infection by other viruses, especially those not typically considered lymphotropic.
Herpes simplex virus (HSV) is a remarkably successful human pathogen that establishes lifelong latency in neurons of the dorsal root ganglia. HSV can efficiently reactivate from the latent state and transmit to new hosts despite the presence of preformed immunity. HSV is thought to achieve this feat by employing a number of sophisticated immune evasion mechanisms (33), many of which are directed at the cellular arm of the immune response. In one such potential mechanism, HSV has evolved the ability to enter and infect T cells. Although T cells do not support efficient viral replication (25), infection by HSV profoundly modulates T-cell receptor (TCR) signaling, which prevents T-cell cytotoxic function (55) and alters cytokine production profiles toward an interleukin-10-dominated immunosuppressive phenotype (54). However, it is unknown whether and to what extent HSV infection of T cells occurs during human HSV disease. Furthermore, the dominant mechanisms by which HSV might gain access to lesion-infiltrating T cells have not been elucidated.
Here, we evaluated T-cell infection during human HSV infections, the mechanisms by which HSV enters T cells, the relative involvement of cell-cell spread versus cell-free virus in T-cell infection, and the role of the VS in the infection of T cells by HSV. The demonstration of infection of T cells in human HSV disease and of a dominant role for the VS in entry of HSV into T cells suggests that the VS is important in the pathogenesis of nonlymphotropic as well as lymphotropic viruses. Thus, the VS may be a unique pharmacologic target to allow improved immune control of a wide variety of viral infections.
MATERIALS AND METHODS
Cells and viruses.
Vero cells, maintained in 10% fetal bovine serum (FBS) in Dulbecco's modified Eagle's medium (10% FBS-DMEM), and Jurkat T cells, maintained at between 2 × 105 and 1 × 106 cells/ml in RPMI 1640 medium supplemented with 4 mM HEPES, 5 mM l-glutamine, 5 mM sodium pyruvate, and 10% FBS (10% FBS-RPMI medium), were obtained from ATCC. VD60 (35), VB38, and VL303, derived from Vero cells, are complementing cell lines expressing the glycoprotein D (gD), gB, and gL, respectively. B78H1-A10 is a mouse melanoma cell line expressing HSV entry mediator (HVEM) (39), and 42H9-9 (also designated H-gD-1) is an HEp-2-derived cell line expressing gD from HSV-1(KOS) (27). The CD8+ T-cell clone SKH13 was a kind gift of E. H. Warren (5). Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Hypaque separation from blood obtained from healthy donors. Primary human fibroblasts were maintained in 10% FBS-DMEM up to passage 14.
The following viruses have been used in this study. The HSV-1(F) wild-type strain was obtained from J. Blaho, and the HSV-2(HG52) wild-type strain was obtained from J. Vieira. FΔgE-GFP and FΔgI-GFP, recombinant viruses in which green fluorescent protein (GFP) sequences coupled to the cytomegalovirus promoter replace gE and gI coding sequences, respectively, were obtained from D. Johnson (16). The HSV-1(KOS) mutants with a deletion of gB (K082), originally obtained from N. A. DeLuca (University of Pittsburgh School of Medicine, Pittsburgh, PA) (7); gL (gL86), in which the Escherichia coli lacZ gene with the cytomegalovirus promoter replaces part of the gL open reading frame (40); and gC (ΔgC2-3) (21) have been previously described. The gD mutant virus VRR1097, which has much of the gD coding sequences replaced by GFP sequences (50), was obtained from R. Roller. The gD mutant virus D30P (63), Δ7-21 virus(64) (along with the parental virus FRT-gD; FRT is Flp recognition target), and KOSRid1 virus (12) (along with the parental viruses KOS and KOStk12) have been described previously. D30P virus has a mutation in gD that abolishes the interaction of gD with the known HSV entry receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. The mutant forms of gD in the Δ7-21 and KOSRid1 mutants are unable to interact with HVEM but can still interact with nectin-1 and, to some extent, with nectin-2 and 3-O-sulfated heparan sulfate (63, 64). Viruses A3CY38C and Y38R, which have mutant gD alleles flanked by FRT sites as previously described (59), were derived from the parental virus KOStk12 and retain the wild-type ability to bind HVEM but not nectin-1, nectin-2, or 3-O-sulfated heparan sulfate (M. Yoon and P. Spear, unpublished observations). K26GFP is a mutant virus that carries the GFP gene fused in frame with the UL35 open reading frame coding for the capsid protein VP26. This recombinant virus produces a VP26-GFP fusion protein and was obtained from P. Desai (13). Except as indicated below, the viruses were grown and titers were determined on Vero cells. For VRR1097, K082, and gL86, VD60 (gD-expressing cells), VB38 (gB-expressing cells), and VL303 (gL-expressing cells) cells, respectively, were used for preparation of viral stocks and titering. The resulting virus stocks were named gD−/+, gB−/+, and gL−/+ to designate viruses with a deletion of either the gD, gB, or gL coding sequence but carrying gD, gB, or gL protein (expressed by the corresponding complementing cells) incorporated in the virion. A3CY38C and Y38R viruses were grown and titers were determined on B78H1-HVEM cells; KOSRid1 virus was grown on 42H9-9 cells, and titers were determined on Vero cells.
Antibodies.
The polyclonal rabbit anti-gD (R8) (23) and anti-gHt-gL (R137; 1 mg/ml) (44) antibodies as well as the monoclonal antibodies (MAbs; 1 mg/ml) anti-gD (HD1) (41, 45), DL11 (10, 41), DL6 (15, 23), 1D3 (18), anti-nectin-1 (CK41) (34), and anti-gB (SS10) (3) were kindly provided by G. H. Cohen and R. J. Eisenberg. The rabbit polyclonal anti-HVEM was described previously (40). Antibodies to T-cell markers CD3, CD4, CD8, and CD25, conjugated to either fluorescein isothiocyanate (FITC), phycoerythrin, allophycocyanin, peridinin chlorophyll protein, or peridinin chlorophyll protein-Cy5.5, were from Becton Dickinson. Anti-CD3 labeled with energy-coupled dye was also used (Beckman Coulter). Anti-HSV-2 rabbit polyclonal antibody was purchased from Dako Cytomation, and rabbit immunoglobulin G (IgG) was from R&D Systems. Anti-LFA-1 antibodies used were anti-CD18 (activation epitope; clone MEM-148) for the immunofluorescence staining (AbD Serotec) and anti-CD11a (clone MEM-25) for the blocking experiment and immunofluorescence staining (Invitrogen). FITC- and allophycocyanin-conjugated donkey anti-rabbit and Cy3-conjugated donkey anti-mouse were used as secondary antibodies (Jackson ImmunoResearch).
Specimen collection and processing.
Collection of vesicle fluid and lesion biopsies was approved by the University of Washington Institutional Review Board. Vesicle fluids were collected in 10% human serum-RPMI medium containing acyclovir (50 μM) as described previously (32) and immunostained for T-cell markers and HSV antigens, followed by flow cytometry analysis. One quarter of a punch biopsy (3 mm) from lesions (labia majora or buttock) and from normal contralateral skin was obtained. Single-cell suspensions were made by mechanical dissociation and enzymatic digest as described by Radoja et al. (49), followed by immunostaining for T-cell markers and HSV antigens and flow cytometry analysis. Virus from each of the lesions was typed as HSV-2 by the University of Washington Virology Laboratory.
T-cell infections.
Infection of T cells with cell-free viruses was performed at a multiplicity of infection (MOI) of 10 (or as otherwise indicated in the figure legends) by addition of viruses to the cell-containing medium, followed by incubation at 37°C overnight until further analysis. Infection of T cells through cell-cell spread was done by infecting monolayers of primary human fibroblasts at an MOI of 10 for 16 h or the indicated time at 37°C. The virus inoculum was then removed from the cell monolayer, T-cell-containing medium was added, and cells were incubated at 37°C overnight or the time indicated, until further analysis.
Flow cytometry analysis.
The staining for T-cell markers as well as HSV antigens was done as follows. After infection, the cells were collected by centrifugation, washed in 1% bovine serum albumin-containing phosphate-buffered saline (1% BSA-PBS), and stained for T-cell markers and HSV antigens in 0.1% Tween containing 1% BSA-PBS with the antibodies indicated in the figure legends for 30 min at room temperature. At the end of the incubation period, the cells were washed with 1% BSA-PBS, resuspended in 1% paraformaldehyde-containing PBS, and stored at 4°C until analysis on a FACSCalibur flow cytometer (Becton Dickinson). The data collected were analyzed using FlowJo software.
T-cell activation.
Activation was performed by incubating PBMC in 10% FBS-RPMI medium supplemented with either Staphylococcus enterotoxin B (SEB; 5 μg/ml; Sigma) or phytohemagglutinin ([PHA] 200 ng/ml; Sigma) for 2 to 5 days. Phorbol myristate acetate ([PMA] 50 ng/ml; Sigma) was also used for T-cell activation.
Blocking/increase of HSV cell-cell spread from fibroblasts to T cells.
PBMC were washed with PBS, resuspended in HEPES buffer (20 mM HEPES buffer, 2 mg/ml glucose, and 140 mM NaCl, pH 7.4) alone or supplemented with 5 mM MgCl2, 1 mM EGTA, 50 ng/ml PMA or 50 ng/ml PMA, and 1 mM CaCl2 for 20 min; PBMC were added to infected fibroblasts for 1 h at 37°C, removed, washed with citrate buffer (40 mM citric acid, 135 mM NaCl, 10 mM KCl, pH 3) and PBS, and then further incubated at 37°C overnight in 10% FBS-RPMI medium before being stained for flow cytometry analysis. Cell-cell spread inhibition using anti-LFA-1, anti-HVEM, anti-nectin-1, and antibodies directed against HSV glycoproteins were performed as follows. LFA-1, HVEM, and nectin-1 blocking experiments were done using 2 × 105 T cells or PBMC that were preincubated with or without either anti-LFA-1 blocking antibody (anti-CD11a), anti-HVEM, or anti-nectin-1 (CK41) at the concentration indicated in the figure or legend for 20 min at 37°C and then added to HSV-1(F) fibroblasts infected at an MOI of 10 for 16 h. After a 2-h coincubation at 37°C, the PBMC were collected, washed with citrate buffer and PBS, further incubated overnight at 37°C in 10% FBS-RPMI medium, and stained for flow cytometry analysis. Blocking experiments with antibodies directed against HSV glycoproteins were done by coincubating 2 ×105 T cells or PBMC with HSV-1(F)-infected fibroblasts (MOI of 10 for 16 h) that had been preincubated with the anti-HSV glycoprotein antibody added at the indicated dilution 20 to 30 min prior to the coincubation period. After 2 h at 37°C, the T cells or PBMC were collected, washed with citrate buffer and PBS, further incubated overnight at 37°C in 10% FBS-RPMI medium, and stained for flow cytometry analysis. Blocking of cell-free virus infection was performed by adding the indicated dilution of anti-gD or blocking anti-LFA-1 to 2 × 105 T cells or PBMC for 20 to 30 min at 37°C; cell-free virus was added at the indicated MOI for 2 h at 37°C, followed by washes with citrate buffer and PBS, with further incubation overnight at 37°C in 10% FBS-RPMI medium and staining for flow cytometry analysis.
Immunofluorescence.
Monolayers of fibroblasts grown overnight at 37°C on glass coverslips were infected with either HSV-1(F) or K26GFP at an MOI of 2 for 16 h at 37°C. After the culture medium was removed and cells were washed with PBS, 1 × 106 to 2 × 106 PBMC stimulated with PHA (5 μg/ml) for 2 days at 37°C were added to the HSV-infected fibroblasts. Flow cytometry analysis was performed on stimulated cells and confirmed that greater than 90% of CD3+ T cells were positive for the CD25 activation marker (data not shown). After a 1-h incubation at 37°C, the cells were fixed with 2% methanol-free formaldehyde (Polysciences Inc.) for 20 min at room temperature, washed with PBS, incubated overnight in blocking solution (10% donkey normal serum and 1% BSA-containing PBS). Cells were stained at room temperature in blocking solution. For HSV-1(F) infection, the following combinations of antibodies were used: anti-gD (1:1,000) and either anti-activated LFA-1 (CD18act; 1:1,000) or anti-total LFA-1 (CD11a; 1:1,000), followed by FITC-conjugated anti-rabbit, Cy3-conjugated anti-mouse, and Hoechst 333422 (10 μg/ml). For K26GFP infection, the cells were stained with anti-activated LFA-1 (CD18act; 1:1,000) or anti-total LFA-1 (CD11a; 1:1,000), followed by Cy3-conjugated anti-mouse and Hoechst 333422 (10 μg/ml). After PBS washes, the coverslips were mounted on slides in 0.1% Mowiol (antifade agent; Sigma) and 2.5% 1,4-diazabicyclo[2,2,2]-octane (antioxidant; Sigma) and sealed with clear nail polish. The slides were left at least overnight at 4°C before being viewed using a DeltaVision microscope system (Fred Hutchinson Cancer Research Center scientific imaging shared resources). Stacks of optical sections were acquired at 0.2-μm spacing with an Olympus 60× oil immersion objective (1.4 numerical aperture), with an additional magnification achieved through use of a 1.5× tube lens, on an Applied Precision DeltaVision RT microscope system (Applied Precision, Issaquah, WA). Stacks were deconvolved using a constrained iterative algorithm with DeltaVision SoftWorx software. The images presented in Fig. 6 are the projection of the high-fluorescence intensity signals from five optical sections.
FIG. 6.

Detection of LFA-1 at the site of contact between HSV-infected fibroblasts and T cells. (A to C) PBMC stimulated with PHA (5 μg/ml for 2 days at 37°C) were coincubated for 1 h at 37°C with fibroblasts infected with HSV-1(F) at an MOI of 2 for 16 h, fixed, and stained for either active LFA-1 (red) and gD (green) (A and B) or total LFA-1 (red) and gD (green) (C). (D to F) PBMC stimulated with PHA as above were coincubated for 1 h at 37°C with fibroblasts infected with HSV-1(K26GFP) at an MOI of 2 for 16 h, fixed, and stained for either active LFA-1 (red) (D and E) or total LFA-1 (red) (F). K26GFP produces and incorporates into the virion the capsid protein VP26 fused to GFP (VP26-GFP). (G and H) PBMC stimulated with PHA as above were coincubated for 1 h at 37°C with uninfected fibroblasts, fixed, and stained for either active LFA-1 (red) (G) or total LFA-1 (red) (H). In all panels, the nuclei were stained with Hoechst.
RESULTS
HSV-infected T cells can be detected in human HSV lesions.
In vitro, HSV infection of T cells leads to profound changes in TCR signaling and T-cell function, suggesting that HSV infection of T cells might be a critical determinant of HSV immune evasion (53-55). To determine whether such a mechanism might be operative in vivo, we obtained biopsies or lesion fluid from volunteers with HSV lesions. HSV lesions are markedly infiltrated by CD4+ and CD8+ T cells (11), and we were readily able to isolate T cells from both the biopsies and the vesicle fluid. Consistent with the idea that our in vitro observations are relevant to clinical human disease, we detected HSV antigens in approximately 12% of T cells from the vesicle fluid and 6 to 27% of T cells from the lesion biopsies (Fig. 1).
FIG. 1.
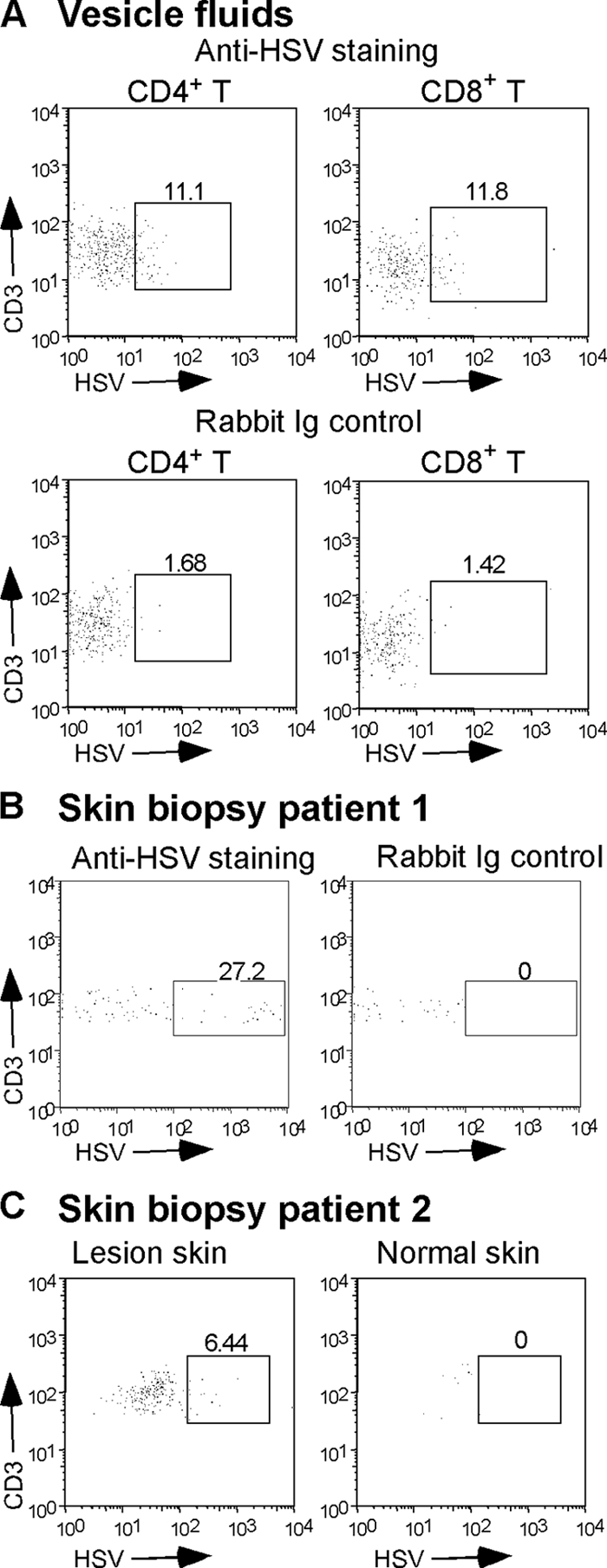
HSV is detectable in T cells from human herpes lesions. (A) Vesicle fluid collected from a herpetic lesion was stained and analyzed by flow cytometry for the presence of infected T cells using MAbs to CD3, CD4, and CD8 and rabbit polyclonal anti-HSV-2. Staining control was performed using rabbit Ig control instead of anti-HSV-2. The box in each dot plot contains the T cells positive for HSV antigens; numbers represent the percentage of T cells expressing HSV antigen. (B) A biopsy of a herpetic lesion from a second patient was stained with anti-CD3 and rabbit polyclonal anti-HSV-2 or rabbit Ig control. (C) Biopsies of a herpetic lesion (lesion skin) or control uninfected skin (normal skin) from a third patient were stained for the presence of infected T cells using anti-CD3 and rabbit polyclonal anti-HSV-2.
Infection of T cells by HSV is much more efficient via cell-cell spread than by cell-free virus.
We have previously shown that the Jurkat T-cell line, a leukemic T-cell line, is readily infected by HSV (25). On the other hand, primary T cells have been reported to be only poorly susceptible to HSV infection and/or permissive for HSV replication (62). Since we have previously reported that HSV can induce signaling alterations in primary T cells, we investigated the extent to which HSV could infect these cells. In agreement with our previous results, Jurkat T cells were efficiently infected with cell-free HSV (Fig. 2A). However, when either primary CD8+ T cells (clone SKH13) or PBMC were exposed to cell-free HSV, very few cells became infected (Fig. 2B and C). We then asked whether coincubation of T cells with infected fully permissive cells might allow more efficient infection of T cells. Indeed, while Jurkat T cells were easily infected with cell-free viruses (Fig. 2A), an even higher fraction of Jurkat T cells became infected after exposure to HSV-infected human fibroblasts for the same length of time (Fig. 2D). Even more strikingly, after coincubation of primary T cells or PBMC with HSV-infected fibroblasts, a large fraction of T cells became infected with HSV (Fig. 2E and F). In PBMC, CD4+ and CD8+ T cells were infected by HSV with similar efficiencies (Fig. 2F, inset). HSV-1 and HSV-2 both readily infected T cells from PBMC, and both viruses infected CD4+ and CD8+ T cells with equal efficiencies (Fig. 2F).
FIG. 2.

HSV infection of human T cells occurs preferentially via cell-cell spread. The Jurkat T cell line (A and D), a CD8+ T-cell clone (B and E), or PBMC (C and F) were either infected with cell-free HSV-1(F) at an MOI of 10 at 37°C (A to C) or coincubated at 37°C with human fibroblasts infected with HSV-1(F) or HSV-2(HG52) at an MOI of 10 for 5 h at 37°C (D to F). At 16 to 18 h postinfection or exposure, cells were collected, stained for T-cell markers and HSV antigens, and analyzed by flow cytometry. The percentage of HSV-positive T cells (CD3+ HSV positive) is indicated below each window. For PBMC infected by coincubation with HSV-infected fibroblasts, the inset shows the percentage of CD4+ (upper left quadrant) and CD8+ (lower right quadrant) cells among infected CD3+ T cells.
Activation of T cells is required for high permissivity to HSV infection.
It has previously been reported that T-cell permissiveness for HSV replication is increased in activated T cells (4). To investigate the effect of T-cell activation on permissivity to HSV via the cell-free virus and cell-to-cell spread methods of infection, we activated T cells in PBMC using PHA or SEB. The activation status of the T cells was confirmed by staining for CD25 (Fig. 3A, inset) and CD38 (data not shown). Activation with either SEB or PHA increased the efficiency with which T cells in PBMC could be infected with cell-free virus although the percentage of infected CD4+ and CD8+ T cells after infection at an MOI of 10 (∼10%) was still far less than that of Jurkat T cells (∼80%) (Fig. 3A). The poor infectivity of activated primary T cells could be overcome to some degree by dramatically increasing the MOI; however, even at an MOI of 100, infectivity remained less than 50% (Fig. 3B). Activation of T cells from PBMC dramatically increased their ability to be infected via cell-to-cell spread (Fig. 3C). In fact, T cells from PBMC activated using PHA were as infectible as Jurkat T cells (∼80%).
FIG. 3.

Activation of T cells increases the efficiency of HSV infection. (A) Jurkat T cells, PBMC, or PBMC activated for 4 days at 37°C with either SEB (200 ng/ml) or PHA (5 μg/ml) were infected with cell-free HSV-1(F) at an MOI of 10 for 16 to 18 h at 37°C, stained for T-cell markers (CD3, CD4, and CD8) and HSV antigens, and analyzed by flow cytometry. Bars indicate the percentage of T cells (CD3+) positive for HSV antigens. The inset shows flow cytometry analysis of T cells (CD3+) for CD25 prior to infection of either untreated (no Tx) PBMC or SEB- or PHA-activated PBMC. (B) Jurkat T cells, PBMC, or PBMC activated for 2 days with PHA (5 μg/ml) were infected with cell-free HSV-1(F) at the indicated MOI for 24 h at 37°C, stained for CD3 and HSV antigens, and analyzed by flow cytometry. Bars indicate the percentage of T cells (CD3+) positive for HSV antigens. (C) Jurkat T cells, PBMC, or PBMC activated for 4 days at 37°C with either SEB (200 ng/ml) or PHA (5 μg/ml) were coincubated for 16 to 18 h at 37°C with human fibroblasts infected with HSV-1(F) at an MOI of 10 for 5 h at 37°C, stained for T-cell markers (CD3, CD4, and CD8) and HSV antigens, and analyzed by flow cytometry. Bars represent the percentage of T cells (CD3+ cells) positive for HSV antigens. Panels A and C show the results from one representative experiment, and panel B shows the mean ± standard deviation of triplicate wells for each condition from one experiment.
Cell-cell spread of HSV to T cells requires gD, gB, and gH/gL.
Several HSV proteins are known to be required for infection of cells. HSV gB, gD, gH, and gL are all required for infection by either the cell-free or cell-to-cell spread routes of infection (8). Additionally, gE and gI are required for efficient cell-to-cell spread of virus between epithelial cells (14). We therefore evaluated the role of the HSV glycoproteins in the cell-cell spread of HSV to T cells. Consistent with the known role of gD, infection by cell-free virus could not occur in the absence of gD (data not shown), and deletion of this protein abrogated the ability of HSV to spread from infected fibroblasts to T cells (Fig. 4A to E). Similarly, deletion of gB or gL (which forms an obligate heterodimer with gH for HSV entry) also abrogated the ability of HSV to spread to T cells (Fig. 5E). However, deletion of gE or gI had no effect on the ability of virus to spread to T cells by either route (Fig. 4A to D and data not shown), suggesting that cell-cell spread to T cells is mechanistically different than spread between epithelial cells.
FIG. 4.

gD, but not gE or gI, is required for cell-cell spread of HSV to T cells. Jurkat T cells (A), unstimulated PBMC (B), or PBMC activated with either SEB (C) or PHA (D, E, and F) were coincubated at 37°C for either 16 to 18 h with uninfected human fibroblasts (no virus) or fibroblasts infected at an MOI of 10 for 5 h at 37°C with the indicated viruses (A to D); alternatively, cells were coincubated for 2 h with uninfected human fibroblasts (no virus) or fibroblasts infected at an MOI of 10 for 16 h at 37°C with the indicated viruses, removed, washed with citrate buffer and PBS, and further incubated in culture medium at 37°C for 24 h (E and F). After collection, the T cells were stained either for both HSV antigens and CD3 (A, E, and F) or for HSV antigens, CD3, CD4, and CD8 (B to D), followed by flow cytometry analysis. The graphs show the percentage of CD3+ T cells positive for HSV antigens (A, E, and F) or CD3+ CD4+ (black bars) and CD3+ CD8+ (gray bars) T cells positive for HSV antigens (B to D). In panels E and F, additional wells of fibroblasts were infected in parallel with the wells of infected fibroblasts used for the cell spread experiment; fibroblasts were collected at the end of the coincubation period of the cell spread experiment and stained for HSV antigens and analyzed by flow cytometry. Black bars indicate the percent infection of fibroblasts; gray bars indicate the percent infection of T cells. The table indicates the ability (+) or inability (−) of mutant viruses to use the known HSV entry receptors. HSV(F) is the parental virus for FgE− (deletion of gE), FgI− (deletion of gI), and gD−/+; FRT-gD is the parental virus for D30P and Δ7-21; KOS is the parental virus for A3CY38C, Y38R, and the gB, gC, and gL mutants in panel E. Panels A and B show the average of two independent experiments, panel C or D shows the results from one experiment, and panels E and F show the results of one representative experiment with duplicate wells.
FIG. 5.

Blocking of HSV spread with either antibody directed against known HSV entry receptors or HSV glycoproteins. (A) PBMC stimulated with PHA (1 μg/ml for 2 days at 37°C) were coincubated for 2 h at 37°C with fibroblasts infected with HSV-1(F) at an MOI of 10 for 16 h in the presence of various dilutions of antibody. For anti-HVEM and anti-nectin-1, the diluted antibodies were added to the PBMC and incubated for 20 min at 37°C before coincubation with the infected fibroblasts. For anti-gB, anti-gD, and anti-gH-gL, the diluted antibodies were added to the infected fibroblasts and incubated for 20 min at 37°C before coincubation with the PBMC. After the 2-h coincubation, the T cells were removed, washed with citrate buffer and PBS, further incubated at 37°C in culture medium for 24 h, stained for CD3 and HSV antigens, and analyzed by flow cytometry. The table indicates the reactivity of the different antibodies used in the experiment. Antibody binding and the ability to block infection were confirmed by direct infection of control HEp-2 cells in the presence of antibody and ranged from ∼90% to ∼40% inhibition of infection relative to cells in the absence of antibody (R8 > SS10 > R137 ≈ CK41 ≈ HVEM) (data not shown). (B) PBMC stimulated with PHA as described in panel A were coincubated for 2 h at 37°C with fibroblasts infected with HSV-1(F) at an MOI of 10 for 16 h in the presence of MAb dilutions directed against different gD epitopes. The diluted antibodies were added to the infected fibroblasts and incubated for 20 min at 37°C before coincubation with the PBMC. After the 2-h coincubation, the T cells were removed, washed with citrate buffer and PBS, further incubated at 37°C in culture medium for 24 h, stained for CD3 and HSV antigens, and analyzed by flow cytometry. The table indicates the ability of the different MAbs to block gD binding to HVEM and/or nectin-1. Antibody binding and the ability to block infection were confirmed by performing direct infection of control HEp-2 cells in the presence of antibody and ranged from ∼90% to ∼20% inhibition of infection (DL11 > 1D3 ≈ HD1 ≈ DL6) (data not shown). Both panels show results from duplicate wells from one representative of two independent experiments.
HSV gD is known to bind at least four cellular receptors, HVEM, nectin-1, nectin-2, and 3-O-S heparan sulfate (57). HVEM is expressed on the surface of T cells, including Jurkat T cells, and is involved in the regulation of T-cell function (19). Specifically, HVEM mediates costimulation of T cells via its interaction with LIGHT, a tumor necrosis factor superfamily member expressed mainly on lymphoid cells (19, 37), and can also negatively regulate T-cell proliferation and activation through interactions with members of the Ig superfamily (31). We therefore evaluated whether the interaction of gD with HVEM was required for the cell-cell spread of HSV to T cells. For these experiments, we used HSV variants containing mutations in gD altering their receptor tropism. Wild-type HSV-1 efficiently uses nectin-1 or HVEM for entry into cells and inefficiently uses also either nectin-2 or 3-O-sulfated heparan sulfate. The HSV mutant D30P fails to bind HVEM, nectin-2, or 3-O-sulfated heparan sulfate but retains wild-type binding to nectin-1 (63). The D30P mutant was severely defective in its ability to spread to T cells (Fig. 4A to D and F), suggesting that the interaction of gD with nectin-1 is not sufficient for efficient cell-cell spread of virus to T cells. Two additional HSV mutants, D7-21 and Rid1, also fail to bind HVEM and have wild-type binding to nectin-1 and 3-O-sulfated heparan sulfate. However, Rid1 has significantly enhanced ability to use nectin-2 for cell entry (64). D7-21 and Rid1 were more efficient than D30P in cell-cell spread of virus to T cells (Fig. 4A to D and F), suggesting that nectin-2 or 3-O-sulfated heparan sulfate may contribute to cell-cell spread of HSV to T cells. The HSV mutants A3CY38C and Y38R, which can bind HVEM but not nectin-1, nectin-2, or 3-O-sulfated heparan sulfate, also had reduced but not absent ability for cell-cell spread to T cells (Fig. 4F). Taken together, these results suggest that each of the known gD receptors may contribute to cell-cell spread of HSV to T cells but that for fully efficient transfer the binding of gD to multiple cellular receptors is required.
To confirm the roles of the HSV glycoproteins and their cellular receptors in cell-cell spread to T cells, we used a panel of antibodies previously shown to inhibit the interaction between these glycoproteins and their receptors. The interaction of each of the essential glycoproteins with its receptor could be inhibited by antibody although gD was more readily inhibited than gB or gH/gL, which showed inhibition only at the highest antibody concentrations (Fig. 5A). Taken together with the lack of T-cell infection by viruses with a deletion of gB, gD, or gH/gL (Fig. 4E), these results confirm the essential role of these glycoproteins. Neither antibody to HVEM nor antibody to nectin-1 could block cell-cell spread of HSV to T cells, possibly due to rapid recycling of these receptors or high receptor density on T cells.
To confirm the importance of gD binding to HVEM and nectin-1, we used antibodies to gD known to specifically block these interactions. While antibody HD1 blocking the gD-nectin-1 interaction had only a minor effect on cell-cell spread of HSV to T cells, blockade of the gD-HVEM interaction with antibodies 1D3 and DL11 had a more dramatic effect (Fig. 5B). The most effective inhibitory effect was seen with antibody DL11, which blocks the interaction of gD with both HVEM and nectin-1. These data support the previous findings that multiple cellular receptors facilitate the cell-cell spread of HSV to T cells.
Cell-cell spread of HSV to T cells occurs via the VS.
Infection of T cells by HIV occurs efficiently via cell-to-cell spread (46). Transmission of virus has been reported to occur at a structured area of contact between the T cell and the infected cell, which has been termed the VS (28). LFA-1 is central to the formation of the VS, and the intermediate and/or active conformations of LFA-1 promote synapse formation and HIV infection (1). In the case of adhesion between lymphocytes and target cells, Neeson et al. demonstrated the important role of the active form of LFA-1 on the lymphocyte cell surface for conjugate formation with the ICAM-1-expressing cell (42). To determine whether such a structure might account for the highly efficient cell-to-cell spread of HSV into T cells, we first evaluated the localization of LFA-1 and HSV components during cell-to-cell spread of the virus to T cells. Activated LFA-1 was found to be enriched at the site of contact between the infected fibroblast and the T cell (Fig. 6A and D) while total LFA-1 was more evenly distributed over the T cell (Fig. 6C and F). The distribution of active and total LFA-1 on T cells interacting with HSV-infected fibroblasts was similar to that of T cells interacting with uninfected fibroblasts (Fig. 6G and H). HSV gD was present over the entire surface of the infected fibroblast, including at the site where the two cell types (fibroblasts and T cells) were in contact (Fig. 6A). Consistent with the formation of a virological synapse, the HSV protein VP26, a marker of viral nucleocapsids, was observed in some T-cell-fibroblast conjugates to be localized to the area of cell-cell contact, suggesting that this structure might be involved in the efficient transfer of virus from infected fibroblasts to T cells (Fig. 6D). However, localization of VP26 to the area of contact was not observed in all conjugates, suggesting that polarization of VP26 is either incomplete or occurs only during a portion of the total duration of T-cell-fibroblast interaction. The role of the synapse in HSV transfer to T cells is supported by the observation of gD and VP26 within T cells having intimate contact with infected fibroblasts (Fig. 6B and E).
To confirm the role of VS-like structures in cell-cell spread of HSV from infected fibroblasts to T cells, we first used the anti-LFA-1 antibody MEM25. This antibody antagonizes the interaction between LFA-1 and ICAM-1 and has previously been shown to inhibit the infection of T cells by ICAM-1-bearing HIV virions (60). Consistent with a role for the VS in transfer of HSV to lymphocytes, anti-LFA-1 effectively inhibited the infection of cloned CD8+ T cells after contact with infected fibroblasts while antibody to the irrelevant lymphocyte surface marker CD28 had no effect (Fig. 7A). In contrast, anti-LFA-1 had no effect on the direct infection of T cells expanded from PBMC (data not shown), suggesting that the role of LFA-1 is limited to HSV spread via the VS. Like all other known means of HSV entry, spread via the VS required gD since antibody to gD strongly inhibited cell-cell spread to lymphocytes (Fig. 7B). A similar effect was seen in T cells from PBMC as either anti-gD or anti-LFA could block cell-cell spread of HSV from infected fibroblasts to PMA-activated PBMC (Fig. 7C). Importantly, while cell-cell spread appeared to be the nearly exclusive mechanism of infection of primary T cells, direct infection was quite efficient in Jurkat T cells (Fig. 7D), and thus anti-LFA-1 had little inhibitory effect on the infection of Jurkat T cells after coincubation with infected fibroblasts (data not shown). These data imply that Jurkat T cells are not an appropriate model for mechanistic studies of HSV entry into T cells.
FIG. 7.

Role of the VS during HSV cell-cell spread to T cells. (A) A CD8+ T-cell clone was preincubated for 20 min at 37°C with either anti-LFA-1 blocking antibody (CD11a) or anti-CD28 antibody at the indicated concentrations before addition to uninfected fibroblasts (no virus) or fibroblasts infected with HSV-1(F) at an MOI of 10 for 16 h at 37°C. After 2 h at 37°C, the T cells were removed, washed with citrate buffer, further incubated at 37°C for 16 h, stained for CD3 and HSV antigens, and then analyzed by flow cytometry. (B) A CD8+ T-cell clone was coincubated at 37°C with mock-infected fibroblasts or fibroblasts infected with HSV-1(F) at an MOI of 10 for 16 h at 37°C in the absence or presence of neutralizing anti-gD antibody at the indicated dilutions. After 2 h, the T cells were removed, washed with citrate buffer, further incubated at 37°C for 16 h, stained for CD3 and HSV antigens, and then analyzed by flow cytometry. (C) PBMC were incubated for 20 min at 37°C with either culture medium (untreated; no Tx), culture medium with PMA (50 ng/ml), or culture medium with PMA and anti-LFA-1 blocking antibody (PMA+anti-LFA-1), and then added for 2 h to fibroblasts infected with HSV-1(F) at an MOI of 10 for 16 h. In one group the HSV-infected fibroblasts were preincubated with neutralizing anti-gD antibody (1:50 dilution) for 20 min prior to the addition of PMA-stimulated PBMC (PMA+anti-gD). After the 2-h coincubation, the T cells were removed, washed with citrate buffer and PBS, further incubated at 37°C in culture medium for 24 h, stained for CD3 and HSV antigens, and analyzed by flow cytometry. (D) Jurkat T cells, unstimulated PBMC, and PHA-activated PBMC (5 μg/ml 2 days at 37°C) were exposed for 2 h at 37°C to either fibroblasts that were infected with HSV-1(F) at an MOI of 10 for 16 h at 37°C and washed with PBS prior to the addition of T cells (Cell) or the medium (16 to 18 h postinfection) collected from fibroblasts that were infected with HSV-1(F) at an MOI of 10 for 16 h, washed with PBS, and incubated in medium for 2 h (16 to 18 h postinfection) (Supernatant). In both cases after a 2-h incubation, T cells were removed, spun, washed with citrate buffer and PBS, further incubated at 37°C for 24 h, stained for CD3 and HSV antigens, and analyzed by flow cytometry. (E) PBMC were resuspended for 30 min in either HEPES buffer, HEPES buffer plus Mg2+-EGTA, HEPES buffer plus PMA-Ca2+, or HEPES buffer plus PMA and then incubated for 1 h at 37°C with fibroblasts infected with HSV-1(F) at an MOI of 10 for 16 h. At the end of the coincubation, the PBMC were removed, washed with citrate buffer and PBS, further incubated in culture medium for 24 h, stained for CD3 and HSV antigens, and analyzed by flow cytometry. (F) PHA-activated PBMC (5 μg/ml 2 days at 37°C) were added for 2 h at 37°C to either uninfected fibroblasts (24 h; Mock) or HSV-infected fibroblasts (MOI of 10) at different times postinfection in the absence (no Ab) or presence of anti-LFA-1 blocking antibody (+LFA-1); PBMC were then removed, washed with citrate buffer and PBS, further incubated at 37°C for 20 to 25 h, stained for CD3 and HSV antigens, and then analyzed by flow cytometry. hpi, hours postinfection.
We next asked whether the increased susceptibility of activated T cells to cell-cell spread of virus might relate to the activation state of LFA-1. T-cell activation is known to promote the conformational change of LFA-1 into an active form, manifest by clustering of LFA-1 on the cell surface and increased avidity to its ligand (58). Pretreatment of PBMC with Mg2+ in the absence of Ca2+ increases the activation of LFA-1, resulting in enhanced interaction with its ligand (6, 58). Consistent with LFA-1 being a major determinant of the susceptibility of T cells to cell-cell spread of HSV, treatment with Mg2+ increased the infectivity by severalfold (Fig. 7E).
Finally, we sought to determine when during the HSV replication cycle infected fibroblasts are most efficient at transmitting virus to T cells and whether the VS accounted for the major route of transmission at all times. Consistent with the known kinetics of viral replication and the formation of infectious virions, infectivity began to increase at about 9 h postinfection and reached a maximum at about 16 h postinfection (Fig. 7F). Treatment with anti-LFA-1 effectively blocked infection of T cells at all time points, suggesting that cell-cell spread is the dominant mechanism of T-cell infection throughout the viral life cycle.
DISCUSSION
Understanding the mechanisms by which chronic viral infections evade the host immune response is key for developing new and more effective vaccine and therapeutic approaches. In clinical HSV disease, the failure of T cells to adequately control reactivating virus is a defining characteristic. It is likely that such mechanisms also explain the failure of vaccination efforts directed at HSV. It is in this context that we demonstrate that HSV-infected T cells are present in human HSV lesions. Moreover, we show that HSV can efficiently infect both CD4+ and CD8+ T cells via direct transfer from infected somatic cells by means of the virological synapse. This mechanism of spread is notably distinct from the previously described cell-cell spread between epithelial cells. Together with the prior observation that entry of HSV into T cells remodels TCR signaling (resulting in reduced cytotoxicity and an altered cytokine synthesis profile dominated by the immunosuppressive cytokine interleukin-10), this likely constitutes a new and important means of immune modulation by HSV and is likely to play an important role in the pathogenesis of human HSV disease.
Viruses have evolved a plethora of mechanisms to deal with the human immune response (17). Since the cellular immune response is uniquely designed to detect and deal with intracellular pathogens such as viruses, many of these immune evasion mechanisms are directed at T cells. Many viruses inhibit peptide loading or otherwise downregulate major histocompatibility complex (MHC) class I, thus reducing the recognition of infected cells by T cells. As is common for the host-pathogen interaction, the host typically has its own countermeasures, in this case often via the action of interferon, which overcomes the viral inhibitors and upregulates MHC class I expression. Once infected cells are recognized by T cells, a second tier of viral immune evasion mechanisms comes to the fore. Expression of viral antiapoptotic proteins can protect infected cells from T-cell-induced apoptosis (2, 26). In addition, certain viruses can modulate TCR signaling, either to prepare the T cell to itself support viral replication or to disarm antiviral mechanisms, thus ameliorating the threat posed by virus-reactive T cells (24).
Despite the in vitro demonstration of numerous mechanisms by which viruses could affect the efficacy of the immune response, it has proven difficult to demonstrate that these mechanisms are operative in vivo. For human viruses, this has been especially problematic because certain immune evasion mechanisms are highly species dependent. For example, while the HSV protein ICP47 efficiently prevents peptide loading of MHC class I in humans, it is poorly functional in mouse cells (61). Only recently have HSV mutants been engineered that inhibit MHC class I loading in mice; such mutants are more pathogenic in mice than those unable to block MHC class I loading (43). For viral inhibitors of apoptosis, it has been difficult in model systems to separate the immune-modulatory effects of apoptosis inhibition from the other effects such as increased production of progeny virus from protected cells. Furthermore, some antiapoptotic gene products are multifunctional and play critical roles in other aspects of viral replication (48). In the human situation, direct evidence for the relevance of viral immune evasion is even scarcer. Viruses are inherently efficient with their genetic material, and it seems unlikely that the powerful effects observed in vitro are irrelevant to viral biology in vivo. In this context, we predicted that infection of T cells might be observable in human HSV lesions, and, indeed, this proved to be the case. While the human system does not allow us to evaluate the extent to which this infection contributes to immune evasion by HSV, our evidence supports the contention that the signaling alterations we have previously described (53-55) are likely to be relevant to human disease.
A large number of viruses are known to infect T cells. For lymphotropic viruses, T cells constitute an important cell type for viral replication or for the establishment of latency or persistence. For nonlymphotropic viruses, T cells may become infected but do not efficiently support viral replication. Superficially, such infection might seem unimportant. However, for many viruses of both the lymphotropic and nonlymphotropic groups, emerging evidence shows that infection of T cells can disrupt or otherwise alter T-cell function (24). In the specific case of HSV infection, T cells very poorly support viral replication, but infection of T cells leads to inhibition of cytotoxic function and the skewing of cytokine synthesis toward an immunosuppressive phenotype.
In this report we demonstrate that the infection of T cells by HSV occurs during human HSV disease. Since we could not control the microscopic location of the T cells we analyzed and since bulk biopsies of clinical lesions contain many T cells residing in the deep dermis, which presumably have not contacted HSV-infected cells, our observations may underestimate the infection rate of T cells infiltrating into areas of active epithelial cell infection. Given the profound effect HSV infection has on signaling pathways and, thus, the effector function of T cells, we believe it is likely that the effects we describe here significantly influence the ability of the immune system to control recurrent HSV disease.
One important aspect of the cell-cell spread of HSV into T cells described here is that it appears to be mechanistically distinct from previous examples of cell-cell spread by HSV. While cell-cell spread between epithelial cells requires HSV gE and gI, entry into T cells is distinct in that neither gE nor gI is required. On the other hand, each of the four HSV glycoproteins known to play an essential role in HSV entry, by either cell-free virus or cell-cell spread between epithelial cells (gB, gD, gH, and gL), is required for cell-cell spread to T cells. Furthermore, each of the known receptors for gD appears to contribute to HSV infection of T cells. Viruses containing mutant forms of gD defective in the ability to bind either HVEM or nectin-1 show impaired infection of T cells via cell-cell spread but retain some ability compared to gD null viruses. The gD-HVEM interaction appears to play a dominant role as disruption of this interaction via mutagenesis or antibody blocking had the strongest effect on the ability of the virus to infect T cells, while disruption of the gD-nectin-1 interaction had a somewhat lesser effect. Disruption of gD interactions with nectin-2 and 3-O-sulfated heparan sulfate, in the context of a disrupted gD-HVEM interaction, led to a nearly complete abrogation of cell-cell spread to T cells (equivalent to gD null virus), suggesting that these receptors also play a role. Interestingly, pooled antisera from HSV-1-seropositive individuals, but not control HSV-1-seronegative individuals, could reduce inactivation of T-cell effector function by about 50% (D. D. Sloan and K. R. Jerome, unpublished observations). Since T-cell inactivation requires virus entry into T cells (53-55), this suggests that at least some infected persons mount an antibody response that can partially block HSV infection of T cells. Precisely defining the requirements for HSV infection of T cells may therefore allow therapeutic vaccination approaches tailored to produce effective blocking antibodies, thereby facilitating efficient T-cell control of recurrent lesions.
An emerging body of work has implicated the virological synapse as a critical structure for the transfer of HIV from infected antigen presenting cells (APCs) to uninfected T cells (9, 20, 28, 47, 56). The virological synapse formed during HIV transfer is reminiscent of the immunological synapse formed between APCs and T cells. The mature immunological synapse is characterized by a central region, the central supramolecular activation cluster (SMAC), containing the TCR and associated signaling molecules (51). Surrounding the central SMAC is the peripheral SMAC, composed predominantly of LFA-1, which mediates adhesion of T cells with APCs bearing ICAM-1. A similar LFA-1-containing synapse forms between HIV-infected APCs and T cells, and viral proteins such as Gag and Env can be visualized at the synapse (28). The role of the VS in the cell-cell transmission of HIV is supported by studies using fluorescent virus, which suggest that transfer of virus to T cells via the VS is up to 18,000-fold more efficient than infection by cell-free virus (9).
These studies raised questions of whether the VS might be utilized by viruses that are not classically considered lymphotropic and whether the VS would be limited to the interaction between T cells and professional APCs. In addition to the synapses formed between T cells and professional APCs such as dendritic cells or B cells, T cells can also form synapses with other cell types, notably during the interaction of cytotoxic T lymphocytes with potential targets (59). These synapses are more transient than those with professional APCs and form as T cells scan the surface of cells with which they come in contact. The initial formation of the immunological synapse by cytotoxic T lymphocytes, including the clustering of LFA-1, is antigen independent (36). Since we have previously shown that the cell-cell spread of HSV to T cells was antigen independent (55), it was reasonable to ask whether a synapse-like structure might facilitate the transfer of virus. Our data confirm that this is the case in that HSV utilizes an LFA-1-dependent structure to transfer from infected fibroblasts to T cells. In fact, our data suggest that this is likely the dominant mechanism of T-cell infection in vivo since the direct infection of T cells by cell-free HSV is extremely inefficient. Our results thus point to a general role for the VS in both lymphotropic and nonlymphotropic virus infection of T cells.
While the VS as utilized by HSV has similarity to the VS utilized by retroviruses such as HIV or HTLV-I, there are also areas of distinction. First, HSV appears to utilize a synapse normally formed as T cells scan potential target cells since the distributions of total and activated LFA-1 were similar in T cells during contact with infected versus uninfected cells. This contrasts with the VS used by HIV during spread from infected T cells to uninfected T cells, in which the formation of T cell-T cell conjugates and the VS appears to be actively driven by the virus (28) since stable contacts between uninfected CD4+ T cells are rare (30). On the other hand, active virus induction of the VS is not always the case for HIV. For example, clustering and synapse formation between T cells and dendritic cells occur normally under uninfected conditions, and thus active viral induction of the VS between these cell types is unnecessary. We believe this latter condition more closely approximates the situation with HSV. Second, we observed only minor enrichment of HSV gD at the point of contact with infected fibroblasts, in contrast with the much greater enrichment of HIV Env protein reported at the point of contact with infected cells (52). However, we observed greater polarization of the nucleocapsid protein VP26 toward the VS, and both gD and VP26 could be observed within T cells after contact with HSV-infected fibroblasts. Thus, we argue for an inclusive definition of the VS, with the defining characteristic being viral transfer via a synapse formed between the T cell and a potential target cell or professional APC. Whether other nonlymphotropic viruses utilize the VS and the degree to which VS-mediated infection is mechanistically distinct from the other pathways of entry by these viruses remain unanswered questions. Emerging evidence suggests that T cells can be infected by many nonlymphotropic viruses, and this infection can have profound influences on the function of T cells (24). If the VS proves to be widely used by many viruses, pharmacological targeting of the VS may provide a unique target to minimize T-cell infection and the concomitant immune modulatory effects.
Acknowledgments
We thank Julio Vazquez and David McDonald from the Fred Hutchinson Cancer Research Center scientific imaging shared resource for assistance with microscopy and image analysis and Lawrence Corey and Michael Lagunoff for careful review of the manuscript.
This work was supported by NIH grants R56 AI 65956 (K.R.J.), R37 AI 36293 (P.G.S.), and R01 CA 21776 (P.G.S.).
Footnotes
Published ahead of print on 1 April 2009.
REFERENCES
- 1.Arthos, J., C. Cicala, E. Martinelli, K. Macleod, D. Van Ryk, D. Wei, Z. Xiao, T. D. Veenstra, T. P. Conrad, R. A. Lempicki, S. McLaughlin, M. Pascuccio, R. Gopaul, J. McNally, C. C. Cruz, N. Censoplano, E. Chung, K. N. Reitano, S. Kottilil, D. J. Goode, and A. S. Fauci. 2008. HIV-1 envelope protein binds to and signals through integrin α4β7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 9301-309. [DOI] [PubMed] [Google Scholar]
- 2.Aubert, M., E. M. Krantz, and K. R. Jerome. 2006. Herpes simplex virus genes Us3, Us5, and Us12 differentially regulate cytotoxic T lymphocyte-induced cytotoxicity. Viral Immunol. 19391-408. [DOI] [PubMed] [Google Scholar]
- 3.Bender, F. C., J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 7911588-11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouayyad, A., and J. Menezes. 1990. Comparative study of herpes simplex virus receptor expression on human lymphoid cells. Virology 179905-910. [DOI] [PubMed] [Google Scholar]
- 5.Brickner, A. G., E. H. Warren, J. A. Caldwell, Y. Akatsuka, T. N. Golovina, A. L. Zarling, J. Shabanowitz, L. C. Eisenlohr, D. F. Hunt, V. H. Engelhard, and S. R. Riddell. 2001. The immunogenicity of a new human minor histocompatibility antigen results from differential antigen processing. J. Exp. Med. 193195-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabanas, C., and N. Hogg. 1991. Lymphocyte-fibroblast adhesion. A useful model for analysis of the interaction of the leucocyte integrin LFA-1 with ICAM-1. FEBS Lett. 292284-288. [DOI] [PubMed] [Google Scholar]
- 7.Cai, W. Z., S. Person, S. C. Warner, J. H. Zhou, and N. A. DeLuca. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 61714-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campadelli-Fiume, G., M. Amasio, E. Avitabile, A. Cerretani, C. Forghieri, T. Gianni, and L. Menotti. 2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 17313-326. [DOI] [PubMed] [Google Scholar]
- 9.Chen, P., W. Hubner, M. A. Spinelli, and B. K. Chen. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 8112582-12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen, G. H., V. J. Isola, J. Kuhns, P. W. Berman, and R. J. Eisenberg. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunningham, A. L., R. R. Turner, A. C. Miller, M. F. Para, and T. C. Merigan. 1985. Evolution of recurrent herpes simplex lesions. An immunohistologic study. J. Clin. Investig. 75226-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dean, H. J., S. S. Terhune, M. T. Shieh, N. Susmarski, and P. G. Spear. 1994. Single amino acid substitutions in gD of herpes simplex virus 1 confer resistance to gD-mediated interference and cause cell-type-dependent alterations in infectivity. Virology 19967-80. [DOI] [PubMed] [Google Scholar]
- 13.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 727563-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dingwell, K. S., C. R. Brunetti, R. L. Hendricks, Q. Tang, M. Tang, A. J. Rainbow, and D. C. Johnson. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68834-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenberg, R. J., D. Long, M. Ponce de Leon, J. T. Matthews, P. G. Spear, M. G. Gibson, L. A. Lasky, P. Berman, E. Golub, and G. H. Cohen. 1985. Localization of epitopes of herpes simplex virus type 1 glycoprotein D. J. Virol. 53634-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farnsworth, A., K. Goldsmith, and D. C. Johnson. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 778481-8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finlay, B. B., and G. McFadden. 2006. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124767-782. [DOI] [PubMed] [Google Scholar]
- 18.Friedman, H. M., G. H. Cohen, R. J. Eisenberg, C. A. Seidel, and D. B. Cines. 1984. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 309633-635. [DOI] [PubMed] [Google Scholar]
- 19.Granger, S. W., and S. Rickert. 2003. LIGHT-HVEM signaling and the regulation of T cell-mediated immunity. Cytokine Growth Factor Rev. 14289-296. [DOI] [PubMed] [Google Scholar]
- 20.Groot, F., S. Welsch, and Q. J. Sattentau. 2008. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 1114660-4663. [DOI] [PubMed] [Google Scholar]
- 21.Herold, B. C., R. J. Visalli, N. Susmarski, C. R. Brandt, and P. G. Spear. 1994. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 751211-1222. [DOI] [PubMed] [Google Scholar]
- 22.Igakura, T., J. C. Stinchcombe, P. K. Goon, G. P. Taylor, J. N. Weber, G. M. Griffiths, Y. Tanaka, M. Osame, and C. R. Bangham. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2991713-1716. [DOI] [PubMed] [Google Scholar]
- 23.Isola, V. J., R. J. Eisenberg, G. R. Siebert, C. J. Heilman, W. C. Wilcox, and G. H. Cohen. 1989. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 632325-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jerome, K. R. 2008. Viral modulation of T-cell receptor signaling. J. Virol. 824194-4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jerome, K. R., R. Fox, Z. Chen, A. E. Sears, H.-Y. Lee, and L. Corey. 1999. Herpes simplex virus inhibits apoptosis through the action of two genes, US5 and US3. J. Virol. 738950-8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jerome, K. R., J. F. Tait, D. M. Koelle, and L. Corey. 1998. Herpes simplex virus type 1 renders infected cells resistant to cytotoxic T-lymphocyte-induced apoptosis. J. Virol. 72436-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson, R. M., and P. G. Spear. 1989. Herpes simplex virus glycoprotein D mediates interference with herpes simplex virus infection. J. Virol. 63819-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolly, C., K. Kashefi, M. Hollinshead, and Q. J. Sattentau. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199283-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolly, C., I. Mitar, and Q. J. Sattentau. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 8113916-13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jolly, C., and Q. J. Sattentau. 2004. Retroviral spread by induction of virological synapses. Traffic 5643-650. [DOI] [PubMed] [Google Scholar]
- 31.Kaye, J. 2008. CD160 and BTLA: LIGHTs out for CD4+ T cells. Nat. Immunol. 9122-124. [DOI] [PubMed] [Google Scholar]
- 32.Koelle, D. M., H. Abbo, A. Peck, K. Ziegweid, and L. Corey. 1994. Direct recovery of herpes simplex virus (HSV)-specific T lymphocyte clones from recurrent genital HSV-2 lesions. J. Infect. Dis. 169956-961. [DOI] [PubMed] [Google Scholar]
- 33.Koelle, D. M., and L. Corey. 2008. Herpes simplex: insights on pathogenesis and possible vaccines. Annu. Rev. Med. 59381-395. [DOI] [PubMed] [Google Scholar]
- 34.Krummenacher, C., I. Baribaud, M. Ponce de Leon, J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2000. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J. Virol. 7410863-10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ligas, M. W., and D. C. Johnson. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 621486-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marwali, M. R., M. A. MacLeod, D. N. Muzia, and F. Takei. 2004. Lipid rafts mediate association of LFA-1 and CD3 and formation of the immunological synapse of CTL. J. Immunol. 1732960-2967. [DOI] [PubMed] [Google Scholar]
- 37.Mauri, D. N., R. Ebner, R. I. Montgomery, K. D. Kochel, T. C. Cheung, G. L. Yu, S. Ruben, M. Murphy, R. J. Eisenberg, G. H. Cohen, P. G. Spear, and C. F. Ware. 1998. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 821-30. [DOI] [PubMed] [Google Scholar]
- 38.McDonald, D., L. Wu, S. M. Bohks, V. N. KewalRamani, D. Unutmaz, and T. J. Hope. 2003. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 3001295-1297. [DOI] [PubMed] [Google Scholar]
- 39.Miller, C. G., C. Krummenacher, R. J. Eisenberg, G. H. Cohen, and N. W. Fraser. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3160-168. [DOI] [PubMed] [Google Scholar]
- 40.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87427-436. [DOI] [PubMed] [Google Scholar]
- 41.Muggeridge, M. I., V. J. Isola, R. A. Byrn, T. J. Tucker, A. C. Minson, J. C. Glorioso, G. H. Cohen, and R. J. Eisenberg. 1988. Antigenic analysis of a major neutralization site of herpes simplex virus glycoprotein D, using deletion mutants and monoclonal antibody-resistant mutants. J. Virol. 623274-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neeson, P. J., P. J. Thurlow, and G. P. Jamieson. 2000. Characterization of activated lymphocyte-tumor cell adhesion. J. Leukoc. Biol. 67847-855. [DOI] [PubMed] [Google Scholar]
- 43.Orr, M. T., K. H. Edelmann, J. Vieira, L. Corey, D. H. Raulet, and C. B. Wilson. 2005. Inhibition of MHC class I is a virulence factor in herpes simplex virus infection of mice. PLoS Pathog. 1e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng, T., M. Ponce-de-Leon, H. Jiang, G. Dubin, J. M. Lubinski, R. J. Eisenberg, and G. H. Cohen. 1998. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 7265-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pereira, L., T. Klassen, and J. R. Baringer. 1980. Type-common and type-specific monoclonal antibody to herpes simplex virus type 1. Infect. Immun. 29724-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips, D. M. 1994. The role of cell-to-cell transmission in HIV infection. AIDS 8719-731. [DOI] [PubMed] [Google Scholar]
- 47.Piguet, V., and Q. Sattentau. 2004. Dangerous liaisons at the virological synapse. J. Clin. Investig. 114605-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poon, A. P., and B. Roizman. 2007. Mapping of key functions of the herpes simplex virus 1 US3 protein kinase: the US3 protein can form functional heteromultimeric structures derived from overlapping truncated polypeptides. J. Virol. 811980-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radoja, S., M. Saio, and A. B. Frey. 2001. CD8+ tumor-infiltrating lymphocytes are primed for Fas-mediated activation-induced cell death but are not apoptotic in situ. J. Immunol. 1666074-6083. [DOI] [PubMed] [Google Scholar]
- 50.Rauch, D. A., N. Rodriguez, and R. J. Roller. 2000. Mutations in herpes simplex virus glycoprotein D distinguish entry of free virus from cell-cell spread. J. Virol. 7411437-11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito, T., and T. Yokosuka. 2006. Immunological synapse and microclusters: the site for recognition and activation of T cells. Curr. Opin. Immunol. 18305-313. [DOI] [PubMed] [Google Scholar]
- 52.Sattentau, Q. J., and J. P. Moore. 1993. The role of CD4 in HIV binding and entry. Philos. Trans. R. Soc. Lond. B 34259-66. [DOI] [PubMed] [Google Scholar]
- 53.Sloan, D. D., J. Y. Han, T. K. Sandifer, M. Stewart, A. J. Hinz, M. Yoon, D. C. Johnson, P. G. Spear, and K. R. Jerome. 2006. Inhibition of TCR signaling by herpes simplex virus. J. Immunol. 1761825-1833. [DOI] [PubMed] [Google Scholar]
- 54.Sloan, D. D., and K. R. Jerome. 2007. Herpes simplex virus remodels T-cell receptor signaling, resulting in p38-dependent selective synthesis of interleukin-10. J. Virol. 8112504-12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sloan, D. D., G. Zahariadis, C. M. Posavad, N. T. Pate, S. J. Kussick, and K. R. Jerome. 2003. CTL are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J. Immunol. 1716733-6741. [DOI] [PubMed] [Google Scholar]
- 56.Sourisseau, M., N. Sol-Foulon, F. Porrot, F. Blanchet, and O. Schwartz. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 811000-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spear, P. G., S. Manoj, M. Yoon, C. R. Jogger, A. Zago, and D. Myscofski. 2006. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 34417-24. [DOI] [PubMed] [Google Scholar]
- 58.Stewart, M. P., C. Cabanas, and N. Hogg. 1996. T cell adhesion to intercellular adhesion molecule-1 (ICAM-1) is controlled by cell spreading and the activation of integrin LFA-1. J. Immunol. 1561810-1817. [PubMed] [Google Scholar]
- 59.Stinchcombe, J. C., G. Bossi, S. Booth, and G. M. Griffiths. 2001. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 15751-761. [DOI] [PubMed] [Google Scholar]
- 60.Tardif, M. R., and M. J. Tremblay. 2005. LFA-1 is a key determinant for preferential infection of memory CD4+ T cells by human immunodeficiency virus type 1. J. Virol. 7913714-13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tomazin, R., N. E. van Schoot, K. Goldsmith, P. Jugovic, P. Sempe, K. Fruh, and D. C. Johnson. 1998. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J. Virol. 722560-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westmoreland, D. 1978. Herpes simplex virus type-1 and human lymphocytes: virus expression and the response to infection of adult and foetal cells. J. Gen. Virol. 40559-575. [DOI] [PubMed] [Google Scholar]
- 63.Yoon, M., and P. G. Spear. 2004. Random mutagenesis of the gene encoding a viral ligand for multiple cell entry receptors to obtain viral mutants altered for receptor usage. Proc. Natl. Acad. Sci. USA 10117252-17257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoon, M., A. Zago, D. Shukla, and P. G. Spear. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 779221-9231. [DOI] [PMC free article] [PubMed] [Google Scholar]