

Abstract
Type 3 (T3) reovirus strains induce apoptotic neuronal cell death and lethal encephalitis in infected mice. T3 strain Dearing (T3D)-induced apoptosis in primary neuronal cultures occurs by a Fas-mediated mechanism and requires the activation of caspase 8. We now show that Fas mRNA is upregulated in the brains of mice infected with encephalitic reovirus T3D and T3 strain Abney (T3A) but not following infection with nonencephalitic reovirus type 1 strain Lang. Fas is upregulated in regions of the brain that are injured during infection with T3 reovirus strains and colocalizes with virus antigen in individual neurons. In contrast, levels of FasL mRNA induced by encephalitic and nonencephalitic reovirus strains do not differ significantly. Caspase 8, the initiator caspase associated with Fas-mediated apoptosis, is activated in the cortex and hippocampal regions of both T3D- and T3A-infected mice. Furthermore, Bid cleavage and the activation of caspase 9 in the brains of T3D-infected mice suggest that the caspase 8-dependent activation of mitochondrial apoptotic signaling contributes to virus-induced apoptosis. We have previously shown that the inhibition of c-Jun N-terminal kinase (JNK) signaling blocks T3D-induced apoptosis and improves the outcome of virus-induced encephalitis. We now show that the reovirus-induced upregulation of Fas requires JNK signaling, thereby providing a link between reovirus-induced death receptor signaling and mitogen-activated protein kinase pathways and a potential mechanism for the therapeutic action of JNK inhibition.
Reovirus infection of epithelial cell lines, primary neurons, and the mouse brain provides a model for virus-induced encephalitis that allows a detailed examination of cell signaling pathways that play a critical role in viral pathogenesis. Reovirus infection causes cell death, tissue injury, and disease by triggering apoptosis (11, 38). We and others have shown that apoptotic signaling in reovirus-infected epithelial cell lines involves both the extrinsic (7, 29) and intrinsic (29, 30, 47) apoptotic pathways (see below). In primary neurons, the extrinsic apoptotic pathway, mediated by Fas, also appears to be required for reovirus-induced apoptosis (45). Other signaling molecules have also been shown to play a role in reovirus-induced apoptosis in epithelial cell lines and primary neuronal cultures in vitro and in target organs in vivo. These molecules include nuclear factor κB (NF-κB) (8, 12, 21, 37) and c-Jun N-terminal kinase (JNK) (3, 9, 10). Defining how these pathways are activated and interactions between these pathways in reovirus-infected cells is critical to our understanding of the mechanisms involved in virus-induced apoptosis in vitro and disease progression in vivo.
Extrinsic apoptotic signaling involves the activation of cell surface death receptors belonging to the tumor necrosis factor (TNF) receptor family of proteins, including Fas/APO-1, tumor necrosis factor receptor 1, and TNF-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and TRAIL-R2 (1). These receptors are activated following the binding of their cognate ligands, namely, Fas ligand (FasL), TNF, and TRAIL. Death receptors contain a cytoplasmic death domain (DD) that serves as a docking site for homotypic DD interactions with DD-containing adaptor proteins (25, 54). Fas-associating protein with a DD (FADD) is the adaptor protein for Fas and is recruited to the activated receptor along with procaspase 8 to form a death-induced signaling complex. Caspase 8 is activated at the death-induced signaling complex and can then activate downstream effector caspases, resulting in apoptosis. The crucial role of FADD and caspase 8 in Fas signaling is shown in FADD- or caspase 8-deficient mice that are resistant to Fas-induced apoptosis (58, 60, 63).
The intrinsic apoptotic pathway involves the release of proapoptotic factors through pores in the mitochondrial membrane (18). Proapoptotic factors released through mitochondrial pores include cytochrome c, which triggers the activation of caspase 9, and the second mitochondrion-derived activator of caspases (SMAC), which downregulates cellular inhibitor-of-apoptosis proteins. Mitochondrial pores consist of dimers of proapoptotic members of the Bcl-2 family of proteins (Bax and Bak) and are tightly regulated by interactions with other (antiapoptotic and BH3-only) Bcl-2 family proteins (6, 19).
Cross talk can occur between the intrinsic and extrinsic apoptotic signaling pathways. Most notably, intrinsic apoptotic signaling can be initiated through the caspase 8-dependent cleavage of Bid, a BH3-only member of the Bcl-2 family of proteins. Cleaved Bid translocates to the mitochondria and facilitates the formation of pores in the mitochondrial membrane and the release of proapoptotic mitochondrial factors (35). Our previous studies have shown that in epithelial cell lines, Bid is cleaved in a caspase 8-dependent manner following reovirus infection (29). However, the contribution of Bid to the activation of intrinsic apoptotic signaling in reovirus-infected neurons in the brain remains to be determined.
Mitogen-activated protein kinases (MAPKs) constitute important mediators of signal transduction pathways that serve to coordinate the cellular response to a variety of extracellular stimuli. The MAPK superfamily includes the extracellular signal-related kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and p38 MAPKs (17). ERKs are activated in response to growth stimuli and promote growth, whereas JNK and p38 MAPKs are activated in response to a variety of environmental stresses and inflammatory signals and promote apoptosis and growth inhibition. JNK and the JNK-dependent transcription factor c-Jun are activated in reovirus-infected cells in vitro and in vivo, and JNK signaling is critical for reovirus-induced cell death (3, 9, 10). Furthermore, JNK inhibition results in the long-term survival of reovirus-infected mice and is associated with a dramatic decrease in levels of apoptosis and tissue injury in reovirus-infected brains (3).
We have previously shown that type 3 (T3) reovirus strain Dearing (T3D)-induced apoptosis of primary neuronal cultures results in the activation of caspase 8 and is blocked both by soluble Fas and by peptide inhibitors of caspase 8 (45). We now show that Fas mRNA and protein are upregulated in the brains of mice infected with encephalitic (T3), but not nonencephalitic (T1), reovirus strains. Following infection with T3D, Fas is upregulated in regions of the brain that are injured by virus and colocalizes with virus antigen in individual neurons. The death receptor-associated caspase, caspase 8, is activated in the brains of T3 reovirus-infected mice and is associated with the cleavage of Bid and the activation of caspase 9. These results suggest that the reovirus-induced upregulation of Fas triggers extrinsic apoptotic signaling in the brains of reovirus-infected mice, resulting in the activation of caspase 8 and the caspase 8-dependent activation of mitochondrial apoptotic signaling. We also show that the reovirus-induced upregulation of Fas requires JNK signaling, providing a link between reovirus-induced death receptor signaling and MAPK pathways and a potential mechanism for the therapeutic action of JNK inhibition.
MATERIALS AND METHODS
Cells and virus.
HeLa cells (ATCC CCL2) were grown in Eagle's minimal essential medium supplemented with 2.4 mM l-glutamine, nonessential amino acids, and 60 U/ml each of penicillin and streptomycin and containing 10% fetal bovine serum (Gibco-BRL, Gaithersburg, MD). Virus was grown in L929 mouse fibroblasts (ATCC CL-1), which were maintained in 2× 199 medium supplemented with 10% heat-inactivated fetal bovine serum and 4 mM l-glutamine. Reovirus T3 strain Abney (T3A), T3D, and type 1 strain Lang (T1L) are laboratory stocks, which have been plaque purified and passaged (twice) in L929 (ATCC CCL1) cells to generate working stocks. For infections, growth medium was removed from cells and was replaced with virus in 75 μl gel saline. Cells and virus were incubated at 37°C for 1 h with rocking every 15 min. After this time, fresh growth medium was added to the cells. Adenovirus expressing dominant negative (DN) c-Jun (41) and green fluorescent protein (GFP) was provided by Kim Heidenreich (University of Colorado). Adenovirus (multiplicity of infection [MOI] of 50) infections were performed 36 h prior to reovirus infection.
Inhibitors and reagents.
d-Stereoisomer c-Jun N-terminal kinase inhibitor 1 (D-JNKI-1) (trade name XG-102) and an empty TAT control protein were provided by Christopher Bonny through Xigen SA (Lausanne, Switzerland) (3). D-JNKI-1 was designed by linking a 10-amino-acid human immunodeficiency virus Tat transporter sequence to a 20-amino-acid JNK binding motif from JNK-interacting protein 1 (JIP-1). The d-retroinverso form (D-JNKI-1) has a prolonged half-life in vivo. The level of specificity of D-JNKI-1 for the JNK binding motif is extremely high based on previous studies showing that it fails to inhibit other kinases including ERK p38, protein kinase C, p34, calcium/calmodulin-dependent protein kinase, and protein kinase A. JNK inhibitor II (JNKiII) was purchased from Calbiochem (Darmstadt, Germany). D-JNKI-1 and JNKiII were used at a concentration of 10 μM, which was previously shown to inhibit reovirus-induced apoptosis or caspase 3 activation (3, 9). Cells were pretreated with inhibitors for 1 h prior to infection, and medium containing inhibitor was added to cells following infection. Caspase 8 (catalog number BF2100; R&D Systems) and caspase 9 (catalog number BF7100; R&D Systems) activity assays were performed according to the manufacturer's instructions (R&D Systems).
In vivo studies.
Two-day-old Swiss Webster mice were intracerebrally inoculated with 103 PFU of reovirus in a 10-μl volume as described previously (3). Both D-JNKI-1 and the TAT control peptide were administered by intraperitoneal injection in 10 μl phosphate-buffered saline (PBS) at a dose of 11 mg/kg of body weight/day. Treatment was started 24 h postinfection. At least three individual mice were used for all iterations of our in vivo studies. All animal experiments were approved by the institutional animal care and use committee.
Immunocytochemistry.
Immunocytochemistry experiments were performed using an MOI of 20, which resulted in some cells remaining uninfected to act as negative staining controls. Cells were grown on eight-well chamber slides coated with rat tail collagen (Becton Dickenson, Germantown, WI). Following treatment, cells were fixed with 3.7% formaldehyde in PBS for 15 min at room temperature. Following three washes in PBS, cells were permeabilized overnight at 4°C in PBS containing 0.1% Triton X. Cells were then washed in Tris-buffered saline containing 0.1% Tween 20 (TBST) before blocking for 2 to 4 h in TBST containing 5% normal goat serum (NGS) at room temperature. Cells were then washed in TBST before being incubated overnight at 4°C with antibodies directed against Fas (catalog number 610198; BD Biosciences, San Diego, CA). Antibodies were used at a 1:100 dilution in TBST with 3% bovine serum albumin. After washing (three times) in TBST, cells were incubated with secondary anti-rabbit or anti-mouse immunoglobulin G conjugated to fluorescein isothiocyanate (FITC) (catalog number FI-1200; Vector Laboratories, Burlingame, CA) for 1 h at room temperature. Cells were washed again (three times) in TBST before being counterstained with Hoechst 33342 dye (catalog number H 3570; Invitrogen-Molecular Probes, Carlsbad, CA) for 10 min at room temperature. Cells were then washed again (three times) in TBST before being mounted with Vectashield (catalog number H1000; Vector Laboratories) and digitally imaged using a Zeiss Axioplan2 epifluorescence microscope. Statistical analysis was performed using Graphpad software (Instat, San Diego, CA).
Immunohistochemistry.
Brain tissue was fixed in 10% formalin for 20 h at room temperature. Tissue was transferred into 70% ethanol before paraffin embedding and cutting of sections. Coronal brain sections (4 μm thick) were prepared and tissue injury was assessed semiquantitatively in hematoxylin- and eosin-stained tissue. Adjacent sections were then deparaffinized in xylene and rehydrated in consecutive 100%-to-75% ethanol washes. Antigen retrieval was performed using antigen-unmasking solution (Vector Laboratories) or 10 mM citrate buffer (pH 6.0). Tissue sections were permeabilized in Neuropore (Trevigen, Gaithersberg, MD) overnight at 4°C and blocked in 10% NGS in TBST for 6 to 8 h at room temperature. Sections were then incubated overnight with mouse monoclonal antibodies directed against Fas (catalog number 610198; BD Biosciences) or rabbit polyclonal T3D antibodies. Primary antibodies were used at a dilution of 1:100. For diaminobenzidine (DAB) staining, sections were washed with TBST before being incubated with biotinylated secondary antibody (Vector Laboratories) diluted in 5% NGS-TBST for 2 h at room temperature. Following further washes in TBST, sections were incubated in 0.6% H2O2 (25 min) and ABC reagent (Vector Laboratories) (for 1 h) before incubation for up to 10 min in prewarmed DAB (Trevigen, Gaithersburg, MD). Blue counterstain (Trevigen) was applied to sections before dehydrating and mounting with Vectamount (Vector laboratories). For fluorescent colabeling studies, following binding of primary antibody, sections were washed and incubated with Cy3-labeled (red) anti-mouse or FITC-labeled (green) anti-rabbit secondary antibodies.
Western blotting.
Brains were removed and transferred into 1 ml of PBS and stored at −80°C. Vials were then thawed gently to room temperature, and the PBS was aspirated off. Brains were then homogenized in 300 μl of whole-cell lysis buffer containing 15 mM Tris (pH 7.5), 2 mM EDTA, 10 mM EGTA, 20% glycerol, 0.1% NP-40, 50 mM β-mercaptoethanol, 100 μg/ml leupeptin, 2 μg/ml aprotinin, 40 μM Z-Asp-2,6-dichlorobenzoyloxymethyl ketone (Z-D-DCB), and 1 mM phenylmethylsulfonyl fluoride using a Dounce homogenizer. Lysates were then transferred into 1.5-ml Eppendorf tubes and were stored on ice until centrifugation (20,000 × g for 3 min). The supernatant was then transferred into a fresh tube containing 300 μl of 2× Laemmli buffer (125 mM Tris [pH 6.8], 4% sodium dodecyl sulfate, 10% mM β-mercaptoethanol, 20% glycerol, and 0.004% bromophenol blue). Brain lysates were boiled for 5 min and stored at −70°C. Proteins were electrophoresed overnight by sodium dodecyl sulfate-polyacrylamide gel electrophoresis at a constant voltage of 70 V. Proteins were then electroblotted onto Hybond-C nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). Immunoblots were blocked with 5% nonfat dry milk (NFDM) in TBST for 2 h at room temperature before being probed with antibodies directed against FasL (catalog number SC-834; Santa Cruz Biotechnology, Santa Cruz, CA), Bid (catalog number 2315-pc; Trevigen, Gaithersburg, MD), or activated caspase 3 (catalog number 9661; Cell Signaling Technologies, Danvers, MA). Antibodies were diluted 1:1,000 in 3% NFDM-TBST. All lysates were standardized for protein concentration with antibodies directed against actin (catalog number CP01; Oncogene, Cambridge, MA) diluted 1:10,000 in 3% NFDM-TBST. Autoradiographs were quantitated by densitometric analysis using a Fluor-S MultiImager (Bio-Rad Laboratories, Hercules, CA). Statistical analysis was performed using Graphpad software (Instat).
RNA purification and RT-PCR.
Tissues were excised from mock- and virus-infected mice at appropriate time points, immediately placed into RNAlater, and stored (short term) at −20°C. Approximately 160 mg of brain tissue (about half of the brain after the cerebellum was removed) was homogenized in 1 ml of Qiazol (RNeasy lipid tissue minikit, catalog number 74804; Qiagen) for at least 20 strokes until completely emulsified. The emulsion was transferred into a clean 1.5-ml tube and set aside for at least 5 min before the addition of 200 μl chloroform. The mixture was shaken for 15 s and set aside for 5 min before centrifugation at 12,000 × g for 15 min at 4°C. The upper aqueous phase was then carefully removed and transferred into a new tube. One volume of 70% ethanol (prepared with diethyl pyrocarbonate-treated water) was then added, and the solution was mixed before being transferred into an RNeasy column. RNA was purified according to the manufacturer's specifications, and 0.5 to 1 μl of RNasin was added to the sample, which was then stored at −80°C. Real-time (RT) PCRs were performed using the Abgene Verso kit (catalog number AB 4104/C; Epson, United Kingdom) (distributed by Fisher Scientific, Pittsburg, PA) with an Opticon 2 RT PCR machine (Bio-Rad, Hercules, CA). Data were analyzed using GENEX software (Bio-Rad). Fas and FasL primers were purchased from Stressgen (Ann Arbor, MI).
RESULTS
Fas is upregulated in the brains of mice infected with encephalitic reovirus strains.
T3D and T3A induce lethal encephalitis in 2-day-old mice following intracerebral injection. Following infection, virus is present in discrete regions of the brain, namely, the cortex, hippocampus, and thalamus, and virus-induced encephalitis is associated with tissue injury and apoptosis in these same brain regions (Fig. 1) (36, 45). We have previously shown that T3D reovirus-induced apoptosis of primary neuronal cultures is inhibited by soluble Fas (Fc:Fas), suggesting that Fas mediates apoptotic signaling in infected neurons (45). To determine how Fas signaling is initiated and is associated with disease in the brains of T3D-infected mice, RT PCR analysis was performed on mRNA extracted from mouse brains 4, 6, and 8 days following T3D infection using primers designed to amplify Fas and FasL sequences. The level of expression of Fas mRNA was significantly increased in the brains of T3D-infected compared to mock-infected mice at 8 days postinfection but not at earlier times (Fig. 2A). Fas mRNA was also found to be significantly upregulated in T3D-infected compared to mock-infected brains (40.8×; P = 0.0002) at 8 days postinfection by microarray analysis (not shown). The expression of Fas mRNA at day 8 postinfection correlated with the presentation of marked neurological symptoms (T3D-infected mice normally die between 8 and 9 days following infection). The level of expression of Fas mRNA was also significantly increased in the brains of mice infected with a second encephalitic reovirus strain, T3A, compared to that of mock-infected controls. However, the kinetics of Fas expression were increased in T3A-infected compared to T3D-infected brains, with significant increases observed at days 6 and 8 postinfection (Fig. 2A). These results reflect the fact that T3A-induced encephalitis shows increased kinetics compared to those of T3D-induced disease, with mice typically dying 7 to 8 days postinfection. Following infection with both T3D (day 8) and T3A (days 6 and 7), levels of Fas mRNA were also significantly different from those found in the brains of mice infected with nonencephalitic reovirus strain T1L (Fig. 2A). T1L does not infect neurons, and there is no virus-induced injury in the cortex, hippocampus, and thalamus of T1L-infected mouse brains (Fig. 1) (56). Indeed, microscopic examination of these regions showed no difference between T1L-infected and mock-infected brains (not shown). Instead, T1L infects the ependymal cells lining the central canal of the spinal cord and the cerebral aqueducts and ventricles (Fig. 1) to produce ventricular enlargement (hydrocephalus). Following intracerebral inoculation, T1L viral titers generally peak between days 5 and 9 and then decline, with the majority of mice surviving the infection (56).
FIG. 1.

Following intracerebral infection with T3D, virus antigen colocalizes with tissue injury in the cortex, hippocampus, and thalamus. (A) Immunohistochemical analysis indicates that reovirus antigen (green staining) is present in the cingulate cortex (CC), hippocampus (H), and thalamus (T) 8 days following intracerebral injection of T3D (103 PFU). In contrast, virus antigen is not expressed in these regions 8 days following intracerebral injection of T1L (103 PFU). (B) Viral antigen following T1L infection is found in the ependymal cells surrounding the lateral (top) and third (middle and bottom) ventricles. Tissue sections in A and B were incubated with rabbit polyclonal antireovirus antibody followed by FITC-labeled goat anti-rabbit antibody. Magnifications, ×400 (A and B, top and middle) and ×1,000 (B, bottom). The figure is representative of slides generated from at least three individual T3D- or T1L-infected animals. (C) Staining with hematoxylin and eosin indicates that tissue injury (neuronal cell death and minor inflammation) is present in the hippocampus (H), cingulate cortex (CC), and thalamus (T) 8 days following intracerebral injection of T3D (103 PFU). In contrast, injury is not evident in these areas 8 days following the intracerebral injection of T1L (103 PFU). Magnification, ×200. The figure is representative of slides generated from at least three individual T3D- or T1L-infected animals.
FIG. 2.

The level of Fas mRNA is upregulated in the brains of T3 reovirus-infected mice. mRNA was prepared from mouse brains following intracerebral injection of 103 PFU of encephalitic reovirus strains T3D (mRNA prepared on days 4, 6, and 8 postinfection [pi]) and T3A (mRNA prepared on days 4, 6, and 7 postinfection) and nonencephalitic reovirus strain T1L (mRNA prepared on day 8 postinfection). RT PCR was performed using primers designed to amplify Fas and FasL sequences. Cyclophilin was used as a control gene. The graphs show the mean increases in the levels of expression of Fas (A) or FasL (B) in reovirus-infected mouse brains compared to those seen in mock-infected brains. Error bars represent standard errors of the means. A minimum of three mock-infected and three reovirus-infected animals were used for each condition. Conditions generating statistically significant differences (P < 0.05) between reovirus-infected and mock-infected animals are indicated (*).
Having shown that levels of Fas mRNA are increased in the brains of mice following infection with encephalitic, but not nonencephalitic, reovirus strains, we next examined Fas protein expression. Immunohistochemical analysis of brains harvested from T3D-infected mice 8 days following infection revealed that Fas was upregulated in regions of the brain targeted by virus infection, including the hippocampus, cortex (Fig. 3A), and thalamus (not shown). In contrast, Fas was not upregulated in these areas following infection with T1L or in mock-infected animals. Dual-labeling studies using antibodies directed against Fas and reovirus also demonstrated that Fas and reovirus antigen colocalized in individual cells (Fig. 3B).
FIG. 3.

Fas is upregulated in the brains of T3D-infected mice in areas targeted by virus infection and colocalizes in individual cells with virus antigen. (A) Immunochemical analysis indicates that Fas (brown staining) is expressed in the cingulate cortex (CC), frontoparietal cortex (FPC), and hippocampus (H) 8 days following intracerebral injection of T3D reovirus (103 PFU). In contrast, Fas is not expressed in these regions following infection with T1L or in mock-infected mouse brains. Magnification, ×200. (B) Immunohistochemical analysis of tissue sections from the cingulate cortex demonstrates that Fas (red staining) and reovirus antigen (reo) (green staining) colocalize in individual cells (see yellow staining in merged image). Tissue sections were incubated with rabbit polyclonal antireovirus (reo) or mouse monoclonal anti-Fas primary antibody followed by FITC-labeled goat anti-rabbit or Cy3-labeled goat anti-mouse secondary antibody. Magnifications, ×400 (Fas, antireovirus, and merge [high]) and ×100 (merge [low]). The figure is representative of tissue sections from at least three individual T3D-, T1L-, and mock-infected animals.
Having shown that Fas is upregulated in mouse brains following reovirus infection, we next examined the expression of its activating ligand, FasL. Infection with both encephalitic (T3A) and nonencephalitic (T1L) reoviruses produced small but significant increases in levels of FasL mRNA (Fig. 2B) and protein (Fig. 4). However, infection with T3D did not produce a statistically significant increase in the level of expression of FasL mRNA by RT PCR (Fig. 2B) or by microarray analysis, where the level of FasL expression was increased in T3D-infected brains by 1.8-fold compared to mock-infected brains (P = 0.08) (not shown).
FIG. 4.

The level of FasL is increased in T3A-infected mouse brains. Protein lysates were prepared from mouse brains 7 days following intracerebral injection of 103 PFU of encephalitic reovirus strain T3A. (A) Lysates were analyzed by Western blot analysis using antibody directed against FasL. Antibody directed against actin was used as a protein loading control. Each lane represents an individual mock-infected (−) or T3A-infected (+) animal. (B) Densitometric analysis was used to determine the level of FasL expression in T3A- and mock-infected mouse brains, with values normalized to actin levels. The graph shows the mean level of FasL expression in mock-infected compared to T3A-infected mouse brains. Error bars represent standard errors of the means. Conditions generating statistically significant differences (P < 0.05) between reovirus-infected and mock-infected animals are indicated (*).
Caspase 8 is activated during reovirus-induced encephalitis.
T3 reovirus infection of epithelial cell lines and primary cortical cultures is associated with an increased level of activity of caspase 8 (FLICE) (29, 45) and is blocked by a cell-permeable inhibitor of caspase 8 (7, 29, 45). In addition, reovirus-induced apoptosis of epithelial cells is inhibited by the stable overexpression of DN FADD (7). The results presented above also indicate that T3 reovirus infection of the mouse brain results in increased levels of Fas mRNA. Taken together, these results indicate that caspase 8, the initiator caspase associated with extrinsic apoptotic signaling, is involved in reovirus-induced apoptosis. We next wanted to determine whether caspase 8 was activated in regions of the brain that are injured during virus-induced encephalitis. Mice were infected with reovirus (T1L, T3D, and T3A). At day 8 postinfection, brains were removed, and the cortex and hippocampal regions were harvested for caspase 8 activity assays. These regions (and not the thalamus) were chosen since they were the easiest to isolate. The cortex and hippocampal regions of brains infected with T3D and T3A had significantly increased levels of caspase 8 activity compared to those of mock-infected controls (Fig. 5). In contrast, the cortex and hippocampal regions of the brains of mice infected with T1L did not have increased levels of caspase 8 activity compared to levels of mock-infected controls (Fig. 5). These results indicate that reovirus-induced encephalitis is associated with increased levels of caspase 8 activity.
FIG. 5.
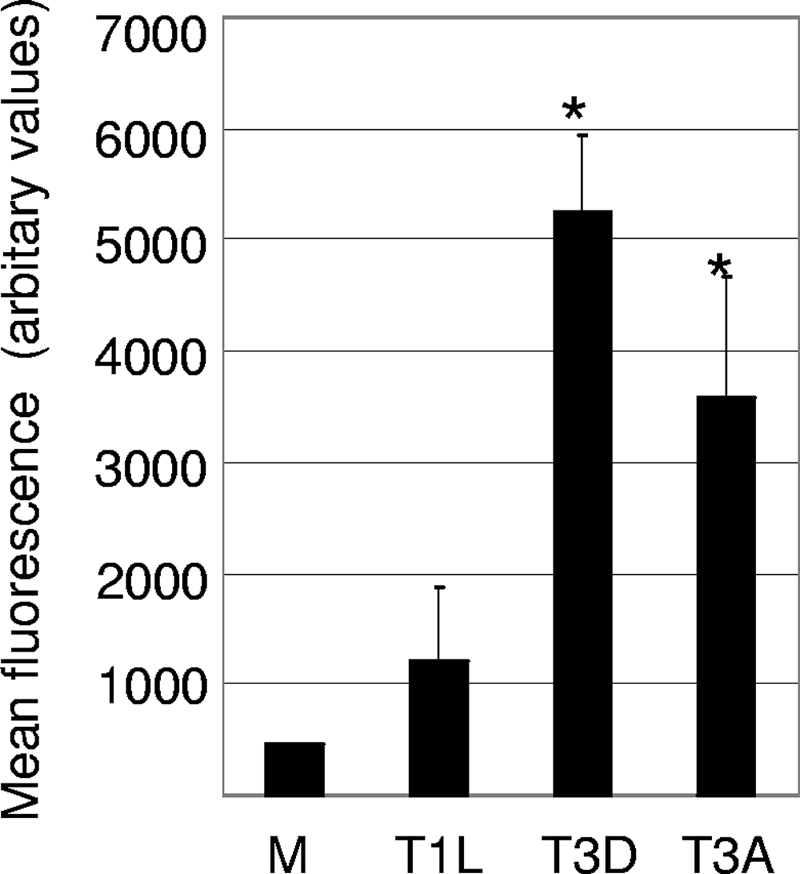
The level of caspase 8 activity is increased in the brains of T3 reovirus-infected mice. Protein extracts were prepared from reovirus-infected and mock-infected (M) mice and were analyzed for caspase 8 activity using a fluorogenic substrate assay. The graph shows the mean fluorescence (caspase 8 activity). Error bars represent standard errors of the means. A minimum of four animals were used for each condition. Conditions generating statistically significant differences (P < 0.05) between reovirus-infected and mock-infected animals are indicated (*).
Bid is cleaved in the brains of T3D-infected mice.
In some instances, caspase 8 can directly activate caspase 3, resulting in apoptosis. However, caspase 8-dependent cleavage of Bid, a BH3-only Bcl-2 family protein, is frequently involved in the activation of mitochondrial apoptotic pathways and the resulting amplification of the apoptotic response. The reovirus-induced cleavage of Bid and the activation of mitochondrial apoptotic signaling pathways are seen following reovirus infection of epithelial cell lines (29, 30). Furthermore, mitochondrial apoptotic signaling is required for reovirus-induced apoptosis in these cells (29, 30). Having shown that caspase 8 is activated in the brains of reovirus-infected mice, we next wanted to determine whether Bid is cleaved in reovirus-infected mouse brains. Brains from T3D-infected mice were harvested at 8 days postinfection and were analyzed by Western blot analysis using an antibody directed against whole (uncleaved) Bid. Decreased levels of whole Bid were seen in T3D-infected compared to mock-infected brains (Fig. 6). Levels of Bid mRNA expression were not significantly different between these animals (not shown), suggesting that the decrease in levels of whole Bid is due to cleavage rather than decreased levels of expression. Antibody directed against activated caspase 3 confirmed that apoptosis was occurring in the T3D-infected brains.
FIG. 6.

Reovirus infection of mouse brains results in the cleavage of Bid and the activation of caspase 9. (A) Protein lysates were prepared from mouse brains 8 days following intracerebral injection of 103 PFU of encephalitic reovirus strain T3D. Lysates were analyzed by Western blot analysis using antibody directed against Bid. Antibody directed against activated caspase 3 was used to demonstrate the presence of apoptosis, and antibody directed against actin was used as a protein loading control. Each lane represents an individual mock-infected (−) or T3D-infected (+) animal. (B) Densitometric analysis was used to compare Bid expression levels in T3D- and mock-infected mouse brains, with values normalized to actin levels. The graph shows the mean levels of Bid in mock-infected compared to T3D-infected mouse brains. Error bars represent standard errors of the means. Conditions generating statistically significant differences (P < 0.05) between reovirus-infected and mock-infected animals are indicated (*). (C) Protein extracts from T3D reovirus-infected mouse brains were also analyzed for caspase 9 activity. The graph shows the mean increase in caspase 9 activity levels in T3D-infected mouse brains compared to that of mock-infected controls. A minimum of three mock-infected and three reovirus-infected animals were used for each condition. Error bars represent standard errors of the means. Conditions generating statistically significant differences (P < 0.05) between reovirus-infected and mock-infected animals are indicated (*).
Caspase 9 is activated in the brains of T3D-infected mice.
Having shown that Bid is cleaved in the brains of T3D-infected mice, we next wanted to show whether caspase 9, the initiator caspase associated with mitochondrial apoptotic signaling, was activated. Brains from T3D-infected mice were obtained 6, 7, and 8 days postinfection and were analyzed for the presence of activated caspase 9. Increased levels of caspase 9 activity were detected in T3D-infected compared to mock-infected brains at day 8 postinfection (Fig. 6). These results indicate that mitochondrial apoptotic signaling is also activated in the brains of reovirus-infected mice.
Increased levels of Fas expression in reovirus-infected cells require JNK signaling.
We have previously shown that JNK and the JNK-dependent transcription factor c-Jun are activated following T3 reovirus infection of epithelial cells, primary cortical neurons, and the mouse brain (3, 9, 10). Since JNK signaling can upregulate Fas in other systems (53), we next wanted to determine whether JNK signaling was the mechanism by which reovirus increased the level of expression of Fas in reovirus-infected cells. HeLa cells have frequently been used to define apoptotic signaling pathways following reovirus infection. HeLa cells were pretreated with the JNK inhibitor D-JNKI-1 or JNKiII for 1 h prior to infection with reovirus. D-JNKI-1 is a peptide inhibitor that blocks JNK from binding to its substrate, whereas JNKiII inhibits JNK kinase activity. Infected cells were then incubated for a further 30 h in the presence of D-JNKI-1or JNKiII before being fixed and incubated with antibodies directed against Fas. Reovirus infection (T3A and T3D at an MOI of 20) resulted in a significant increase in the percentage of cells expressing Fas (Fig. 7). The presence of D-JNKI-1 significantly (P < 0.001) blocked the upregulation of Fas in T3A- and T3D-infected cells (Fig. 7). In contrast, reovirus-infected cells treated with a TAT-only control protein did not show decreased levels of upregulation of Fas. This indicates that the activation of JNK is required for the reovirus-induced upregulation of Fas. JNKiII, which inhibits JNK kinase activity, also significantly blocked the T3A-induced upregulation of Fas, an effect that was not seen in T3A-infected cells treated with vehicle (dimethyl sulfoxide) alone (Fig. 7). HeLa cells were also infected (MOI of 50) with adenoviral vectors expressing DN c-Jun or GFP. Thirty-six hours later, cells were infected with T3A, fixed after a further 30 h, and incubated with antibodies directed against Fas. The presence of DN c-Jun significantly (P < 0.001) blocked increased levels of expression of Fas in T3A-infected cells (Fig. 7). In contrast, adenoviral vectors expressing GFP had no effect on the increased level of expression of Fas in reovirus-infected cells (Fig. 7). These results indicate that JNK signaling is required for the upregulation of Fas in reovirus-infected cells and suggest that that Fas upregulation occurs through increased levels of transcription brought about by the JNK-dependent transcription factor c-Jun.
FIG. 7.

Reovirus-induced upregulation of Fas requires JNK signaling. HeLa cells were infected with encephalitic reovirus strains T3A and T3D (MOI of 20) in the presence of JNK inhibitor D-JNK-1 and JNKiII or an adenoviral vector expressing DN c-Jun (AdDNcJun). The percentage of cells expressing Fas was compared to that of reovirus-infected HeLa cells treated with control vehicles for D-JNKI-1 (a vector expressing TAT protein only) and JNKiII (dimethyl sulfoxide [DMSO]) or an adenoviral vector expressing GFP (AdGFP). After 30 h, cells were fixed and incubated with antibodies directed against Fas. The graph shows the mean percentages of immunoreactive cells from three independent experiments. Error bars represent standard errors of the means. Conditions generating statistically significant differences (P < 0.05) between treated and nontreated reovirus-infected cells are indicated (*).
To determine whether Fas upregulation was regulated by JNK signaling in vivo, 2-day-old mice were infected with reovirus (T3D) (1 × 103 PFU) followed 24 h later by treatment with D-JNKI-1 (11 mg/kg/day for 10 days) or a control peptide (11 mg/kg/day for 10 days). Mice were sacrificed at 8 days postinfection, and tissue sections were analyzed by immunohistochemistry using an antibody against Fas. Positive cells were detected by DAB staining. In reovirus-infected animals treated with control peptide, Fas was upregulated in regions of the brain known to be affected during reovirus encephalitis, including the hippocampus (Fig. 8), cortex, and thalamus (not shown). However, in mice treated with D-JNKI-1, this upregulation was blocked (Fig. 8), indicating that JNK signaling also regulates the reovirus-induced expression of Fas in vivo.
FIG. 8.

JNK is required for Fas upregulation during reovirus encephalitis. Immunohistochemical analysis indicates that the treatment of mice with D-JNKI-1 (DJNK) (11 mg/kg/day for 10 days) blocks the reovirus (103 PFU)-induced expression of Fas (brown staining) seen in the hippocampus of animals treated with a control peptide (TAT) (11 mg/kg/day for 10 days). Fas is also not expressed in mock-infected mouse brains. The figure is representative of data from three individual T3D-infected mice treated with D-JNKI-1, three individual T3D-infected mice treated with a control peptide, and three individual mock-infected animals.
DISCUSSION
T3 reovirus strains induce neuronal apoptotic cell death and lethal encephalitis in 2-day-old mice. We have previously shown that T3 reovirus-induced apoptosis of epithelial cell lines and primary cortical cultures is associated with increased activity of caspase 8 (FLICE) (29, 45) and is blocked by a cell-permeable inhibitor of caspase 8 (7, 29, 45). In addition, T3 reovirus-induced apoptosis of epithelial cells is inhibited by the stable overexpression of a DN form of the cellular adaptor protein FADD, which is required for the activation of caspase 8 (7). We have also previously shown that T3D reovirus-induced apoptosis of primary neuronal cultures is inhibited by soluble Fas (Fc:Fas) (45). These results suggest that Fas signaling activates caspase 8 during reovirus-induced neuronal apoptosis in vitro. We now show that encephalitic T3 reovirus strains upregulate Fas mRNA in the brains of mice following intracerebral injection compared to mock-infected controls. In contrast, Fas mRNA is not upregulated following intracerebral injection of nonencephalitic reovirus strain T1L. Levels of Fas mRNA in the brain differ significantly between encephalitic and nonencephalitic reovirus strains, suggesting that the upregulation of Fas dictates the onset of encephalitis following reovirus infection. The upregulation of Fas mRNA also correlates with neurovirulence between T3D and T3A. Thus, Fas is significantly upregulated at day 6 following infection with the more-neurovirulent T3A but not until day 8 following infection with T3D. Fas protein expression is also upregulated following infection with T3 reovirus strains in areas targeted and injured by reovirus infection, namely, the hippocampus, cortex, and thalamus, and colocalizes with reovirus antigen in specific cells.
In contrast, levels of FasL are significantly increased compared to those in mock-infected controls following infection with encephalitic virus strain T3A and nonencephalitic virus strain T1L. Infection with T3D also appears to increase the level of expression of FasL compared to that with mock infection, although the results did not reach statistical significance. Furthermore, levels of FasL mRNA do not differ significantly in the brains of mice following intracerebral injection of T3D, T3A, or T1L. These results suggest that although FasL may contribute to reovirus-induced neuronal apoptosis, an increased level of expression of FasL does not dictate the onset of reovirus-induced encephalitis.
Reovirus-induced, Fas-mediated apoptotic signaling appears to occur without the presence of an obvious immune response. However, a virus-induced increase in levels of Fas expression has been shown to be involved in the clearance of virus-infected cells by cytotoxic T cells and the control of viral infection following infection with a number of viruses including lymphochoriomeningitis virus (46), herpes simplex virus type 2 (14), mouse hepatitis virus (42), influenza virus (55), Theiler's murine encephalomyelitis virus (40), and West Nile virus (50). Fas-mediated apoptosis is also involved in activation-induced cell death, which acts to silence the immune response following clearance of the virus (5, 13, 51, 52). The upregulation of FasL is also commonly seen following virus infection. In some cases, the expression of FasL induced by virus infection appears to arm the infected cell, allowing it to kill infiltrating uninfected bystander cells including immune effector cells (which themselves express death receptors) and act as an immune evasion tactic. This mechanism has been established for many viruses including the human herpesviruses cytomegalovirus (49) and herpes simplex virus (44), adenovirus (20, 62), rabies virus (2), hepatitis C virus (24), and human immunodeficiency virus (26, 59).
We also show that the reovirus-induced upregulation of Fas and Fas-mediated apoptotic signaling in the mouse brain is associated with the activation of the initiator caspases caspase 8 and caspase 9 and the cleavage of Bid. This suggests that similar to what has been shown for epithelial cells, both the extrinsic and intrinsic apoptotic pathways are activated in reovirus-infected mouse brains and contribute to both virus-induced neuronal apoptosis and disease progression (7, 29, 30). Our studies suggest a model of reovirus-induced apoptosis where the virus-induced upregulation of Fas results in the activation of caspase 8. Caspase 8 can directly cleave caspase 3, resulting in apoptosis. However, our results suggest that the caspase 8-dependent cleavage of Bid and the resulting activation of caspase 9 also contribute to caspase 3 cleavage and function to amplify the apoptotic response. Evidence for the activation of caspase 8 and mitochondrial apoptotic signaling pathways in the brain following virus infection is scarce. Infection of mouse brains with ectromelia virus infection is associated with increased levels of activation of caspase 8 (32). Evidence that the intrinsic pathway of apoptosis is involved in virus-induced apoptosis in the brain comes from studies with Sindbis virus (SINV), where the expression of the antiapoptotic mitochondrial proteins Bcl-2 and peripheral benzodiazepine receptor is protective (27, 34). Furthermore, the level of peripheral benzodiazepine receptor expression is increased following infection with neurovirulent SINV strain dsTE12H compared to avirulent SINV strain dsTE12Q (27). The protective effect of gamma interferon in the brain following infection with herpes simplex virus type 1 may also be mediated, at least in part, by the downregulation of Bcl-2 (15). Finally, the Bcl-2 family members BAD and BAK and cytochrome c are differentially regulated in the brain following rabies virus infection (57).
Our results indicate that an increased level of Fas expression in reovirus-infected cells is dependent on JNK signaling and is inhibited by the inhibition of both JNK and its downstream transcription factor c-Jun. This is the first demonstration that virus-induced activation of JNK signaling is involved in the regulation of Fas, although similar observations have been made for nonviral systems (53). We have previously shown that T3 reoviruses, but not T1L, activate JNK signaling in infected cells. The lack of Fas expression in T1L-infected brains thus presumably reflects the fact that infection with T1L does not lead to the activation of JNK signaling. In addition to their involvement in reovirus infection, both JNK (23, 28, 31, 33, 43, 61) and Fas (see above) are involved in the pathogenesis of a wide variety of viruses, so it is likely that more examples of virus-induced, JNK-mediated regulation of Fas exist. Increases in levels of FasL expression seen following reovirus infection of all three reovirus strains are also likely to be due to increased levels of transcription since the level of FasL mRNA is increased in the brains of infected mice. Infection with T3A and T3D, but not T1L, is associated with the activation of JNK and c-Jun, suggesting that c-Jun is not the transcription factor that regulates the virus-induced expression of FasL (10). Other transcription factors, including NF-κB, are also activated following reovirus infection (8, 12, 37, 39) and may contribute to the upregulation of FasL seen in reovirus-infected cells (4, 16, 22, 48).
Acknowledgments
This work was supported by grants 5RO1NS050138 (K.L.T.), 5RO1NS051403 (K.L.T.), and 5KO8AI076518-02 (J.D.B.) from the National Institutes of Health and VA merit funding (K.L.T.). K.L.T. is supported by the Reuler-Lewin Family Professorship.
Microscopy imaging assistance was provided by Ron Bouchard.
Footnotes
Published ahead of print on 25 March 2009.
REFERENCES
- 1.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 2811305-1308. [DOI] [PubMed] [Google Scholar]
- 2.Baloul, L., S. Camelo, and M. Lafon. 2004. Up-regulation of Fas ligand (FasL) in the central nervous system: a mechanism of immune evasion by rabies virus. J. Neurovirol. 10372-382. [DOI] [PubMed] [Google Scholar]
- 3.Beckham, J. D., R. J. Goody, P. Clarke, C. Bonny, and K. L. Tyler. 2007. Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J. Virol. 816984-6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brozovic, S., R. Sahoo, S. Barve, H. Shiba, S. Uriarte, R. S. Blumberg, and D. F. Kinane. 2006. Porphyromonas gingivalis enhances FasL expression via up-regulation of NFkappaB-mediated gene transcription and induces apoptotic cell death in human gingival epithelial cells. Microbiology 152797-806. [DOI] [PubMed] [Google Scholar]
- 5.Brunner, T., R. J. Mogil, D. LaFace, N. J. Yoo, A. Mahboubi, F. Echeverri, S. J. Martin, W. R. Force, D. H. Lynch, C. F. Ware, et al. 1995. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 373441-444. [DOI] [PubMed] [Google Scholar]
- 6.Chipuk, J. E., and D. R. Green. 2008. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 18157-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clarke, P., S. M. Meintzer, S. Gibson, C. Widmann, T. P. Garrington, G. L. Johnson, and K. L. Tyler. 2000. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 748135-8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke, P., S. M. Meintzer, L. A. Moffitt, and K. L. Tyler. 2003. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J. Biol. Chem. 27818092-18100. [DOI] [PubMed] [Google Scholar]
- 9.Clarke, P., S. M. Meintzer, Y. Wang, L. A. Moffitt, S. M. Richardson-Burns, G. L. Johnson, and K. L. Tyler. 2004. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 7813132-13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke, P., S. M. Meintzer, C. Widmann, G. L. Johnson, and K. L. Tyler. 2001. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 7511275-11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke, P., and K. L. Tyler. 2003. Reovirus-induced apoptosis: a minireview. Apoptosis 8141-150. [DOI] [PubMed] [Google Scholar]
- 12.Connolly, J. L., S. E. Rodgers, P. Clarke, D. W. Ballard, L. D. Kerr, K. L. Tyler, and T. S. Dermody. 2000. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol. 742981-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhein, J., H. Walczak, C. Baumler, K. M. Debatin, and P. H. Krammer. 1995. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 373438-441. [DOI] [PubMed] [Google Scholar]
- 14.Dobbs, M. E., J. E. Strasser, C. F. Chu, C. Chalk, and G. N. Milligan. 2005. Clearance of herpes simplex virus type 2 by CD8+ T cells requires gamma interferon and either perforin- or Fas-mediated cytolytic mechanisms. J. Virol. 7914546-14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geiger, K. D., T. C. Nash, S. Sawyer, T. Krahl, G. Patstone, J. C. Reed, S. Krajewski, D. Dalton, M. J. Buchmeier, and N. Sarvetnick. 1997. Interferon-gamma protects against herpes simplex virus type 1-mediated neuronal death. Virology 238189-197. [DOI] [PubMed] [Google Scholar]
- 16.Ghorpade, A., S. Holter, K. Borgmann, R. Persidsky, and L. Wu. 2003. HIV-1 and IL-1 beta regulate Fas ligand expression in human astrocytes through the NF-kappa B pathway. J. Neuroimmunol. 141141-149. [DOI] [PubMed] [Google Scholar]
- 17.Graves, J. D., J. S. Campbell, and E. G. Krebs. 1995. Protein serine/threonine kinases of the MAPK cascade. Ann. N. Y. Acad. Sci. 766320-343. [DOI] [PubMed] [Google Scholar]
- 18.Green, D. R., and J. C. Reed. 1998. Mitochondria and apoptosis. Science 2811309-1312. [DOI] [PubMed] [Google Scholar]
- 19.Gross, A., J. M. McDonnell, and S. J. Korsmeyer. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 131899-1911. [DOI] [PubMed] [Google Scholar]
- 20.Hall, S. J., S. E. Canfield, Y. Yan, W. Hassen, W. A. Selleck, and S. H. Chen. 2002. A novel bystander effect involving tumor cell-derived Fas and FasL interactions following Ad.HSV-tk and Ad.mIL-12 gene therapies in experimental prostate cancer. Gene Ther. 9511-517. [DOI] [PubMed] [Google Scholar]
- 21.Hansberger, M. W., J. A. Campbell, P. Danthi, P. Arrate, K. N. Pennington, K. B. Marcu, D. W. Ballard, and T. S. Dermody. 2007. IκB kinase subunits α and γ are required for activation of NF-κB and induction of apoptosis by mammalian reovirus. J. Virol. 811360-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harwood, F. G., S. Kasibhatla, I. Petak, R. Vernes, D. R. Green, and J. A. Houghton. 2000. Regulation of FasL by NF-kappaB and AP-1 in Fas-dependent thymineless death of human colon carcinoma cells. J. Biol. Chem. 27510023-10029. [DOI] [PubMed] [Google Scholar]
- 23.Huttunen, P., T. Hyypia, P. Vihinen, L. Nissinen, and J. Heino. 1998. Echovirus 1 infection induces both stress- and growth-activated mitogen-activated protein kinase pathways and regulates the transcription of cellular immediate-early genes. Virology 25085-93. [DOI] [PubMed] [Google Scholar]
- 24.Iken, K., L. Huang, H. Bekele, E. V. Schmidt, and M. J. Koziel. 2006. Apoptosis of activated CD4+ and CD8+ T cells is enhanced by co-culture with hepatocytes expressing hepatitis C virus (HCV) structural proteins through FasL induction. Virology 346363-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itoh, N., and S. Nagata. 1993. A novel protein domain required for apoptosis. Mutational analysis of human Fas antigen. J. Biol. Chem. 26810932-10937. [PubMed] [Google Scholar]
- 26.Ji, J., J. J. Chen, V. L. Braciale, and M. W. Cloyd. 2007. Apoptosis induced in HIV-1-exposed, resting CD4+ T cells subsequent to signaling through homing receptors is Fas/Fas ligand-mediated. J. Leukoc. Biol. 81297-305. [DOI] [PubMed] [Google Scholar]
- 27.Johnston, C., W. Jiang, T. Chu, and B. Levine. 2001. Identification of genes involved in the host response to neurovirulent alphavirus infection. J. Virol. 7510431-10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, S.-M., J.-H. Park, S.-K. Chung, J.-Y. Kim, H.-Y. Hwang, K.-C. Chung, I. Jo, S.-I. Park, and J.-H. Nam. 2004. Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J. Virol. 7813479-13488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kominsky, D. J., R. J. Bickel, and K. L. Tyler. 2002. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 9926-933. [DOI] [PubMed] [Google Scholar]
- 30.Kominsky, D. J., R. J. Bickel, and K. L. Tyler. 2002. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 7611414-11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopecky-Bromberg, S. A., L. Martinez-Sobrido, and P. Palese. 2006. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 80785-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krzyzowska, M., J. Cymerys, A. Winnicka, and M. Niemialtowski. 2006. Involvement of Fas and FasL in ectromelia virus-induced apoptosis in mouse brain. Virus Res. 115141-149. [DOI] [PubMed] [Google Scholar]
- 33.Kumar, A., S. K. Manna, S. Dhawan, and B. B. Aggarwal. 1998. HIV-Tat protein activates c-Jun N-terminal kinase and activator protein-1. J. Immunol. 161776-781. [PubMed] [Google Scholar]
- 34.Levine, B., J. E. Goldman, H. H. Jiang, D. E. Griffin, and J. M. Hardwick. 1996. Bcl-2 protects mice against fatal alphavirus encephalitis. Proc. Natl. Acad. Sci. USA 934810-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94481-490. [DOI] [PubMed] [Google Scholar]
- 36.Oberhaus, S. M., R. L. Smith, G. H. Clayton, T. S. Dermody, and K. L. Tyler. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis J. Virol. 712100-2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Donnell, S. M., M. W. Hansberger, J. L. Connolly, J. D. Chappell, M. J. Watson, J. M. Pierce, J. D. Wetzel, W. Han, E. S. Barton, J. C. Forrest, T. Valyi-Nagy, F. E. Yull, T. S. Blackwell, J. N. Rottman, B. Sherry, and T. S. Dermody. 2005. Organ-specific roles for transcription factor NF-kappaB in reovirus-induced apoptosis and disease. J. Clin. Investig. 1152341-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Donnell, S. M., M. W. Hansberger, and T. S. Dermody. 2003. Viral and cellular determinants of apoptosis induced by mammalian reovirus. Int. Rev. Immunol. 22477-503. [DOI] [PubMed] [Google Scholar]
- 39.O'Donnell, S. M., G. H. Holm, J. M. Pierce, B. Tian, M. J. Watson, R. S. Chari, D. W. Ballard, A. R. Brasier, and T. S. Dermody. 2006. Identification of an NF-κB-dependent gene network in cells infected by mammalian reovirus. J. Virol. 801077-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oleszak, E. L., B. E. Hoffman, J. R. Chang, E. Zaczynska, J. Gaughan, C. D. Katsetos, C. D. Platsoucas, and N. Harvey. 2003. Apoptosis of infiltrating T cells in the central nervous system of mice infected with Theiler's murine encephalomyelitis virus. Virology 315110-123. [DOI] [PubMed] [Google Scholar]
- 41.Park, J. S., L. Qiao, Z. Z. Su, D. Hinman, K. Willoughby, R. McKinstry, A. Yacoub, G. J. Duigou, C. S. Young, S. Grant, M. P. Hagan, E. Ellis, P. B. Fisher, and P. Dent. 2001. Ionizing radiation modulates vascular endothelial growth factor (VEGF) expression through multiple mitogen activated protein kinase dependent pathways. Oncogene 203266-3280. [DOI] [PubMed] [Google Scholar]
- 42.Parra, B., M. T. Lin, S. A. Stohlman, C. C. Bergmann, R. Atkinson, and D. R. Hinton. 2000. Contributions of Fas-Fas ligand interactions to the pathogenesis of mouse hepatitis virus in the central nervous system. J. Virol. 742447-2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perkins, D., K. A. Gyure, E. F. Pereira, and L. Aurelian. 2003. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J. Neurovirol. 9101-111. [DOI] [PubMed] [Google Scholar]
- 44.Raftery, M. J., C. K. Behrens, A. Muller, P. H. Krammer, H. Walczak, and G. Schonrich. 1999. Herpes simplex virus type 1 infection of activated cytotoxic T cells: induction of fratricide as a mechanism of viral immune evasion. J. Exp. Med. 1901103-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson-Burns, S. M., D. J. Kominsky, and K. L. Tyler. 2002. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J. Neurovirol. 8365-380. [DOI] [PubMed] [Google Scholar]
- 46.Rode, M., S. Balkow, V. Sobek, R. Brehm, P. Martin, A. Kersten, T. Dumrese, T. Stehle, A. Mullbacher, R. Wallich, and M. M. Simon. 2004. Perforin and Fas act together in the induction of apoptosis, and both are critical in the clearance of lymphocytic choriomeningitis virus infection. J. Virol. 7812395-12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodgers, S. E., E. S. Barton, S. M. Oberhaus, B. Pike, C. A. Gibson, K. L. Tyler, and T. S. Dermody. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J. Virol. 712540-2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ross, M. J., S. Martinka, V. D. D'Agati, and L. A. Bruggeman. 2005. NF-kappaB regulates Fas-mediated apoptosis in HIV-associated nephropathy. J. Am. Soc. Nephrol. 162403-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sedger, L. M., D. M. Shows, R. A. Blanton, J. J. Peschon, R. G. Goodwin, D. Cosman, and S. R. Wiley. 1999. IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J. Immunol. 163920-926. [PubMed] [Google Scholar]
- 50.Shrestha, B., and M. S. Diamond. 2007. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J. Virol. 8111749-11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strasser, A., A. W. Harris, D. C. Huang, P. H. Krammer, and S. Cory. 1995. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 146136-6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swain, S. L., M. Croft, C. Dubey, L. Haynes, P. Rogers, X. Zhang, and L. M. Bradley. 1996. From naive to memory T cells. Immunol. Rev. 150143-167. [DOI] [PubMed] [Google Scholar]
- 53.Takada, E., K. Shimo, K. Hata, M. Abiake, Y. Mukai, M. Moriyama, L. Heasley, and J. Mizuguchi. 2005. Interferon-beta-induced activation of c-Jun NH2-terminal kinase mediates apoptosis through up-regulation of CD95 in CH31 B lymphoma cells. Exp. Cell Res. 304518-530. [DOI] [PubMed] [Google Scholar]
- 54.Tartaglia, L. A., T. M. Ayres, G. H. Wong, and D. V. Goeddel. 1993. A novel domain within the 55 kd TNF receptor signals cell death. Cell 74845-853. [DOI] [PubMed] [Google Scholar]
- 55.Topham, D. J., R. A. Tripp, and P. C. Doherty. 1997. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 1595197-5200. [PubMed] [Google Scholar]
- 56.Tyler, K. L. 1998. Pathogenesis of reovirus infections of the central nervous system, p. 93-125. In K. L. Tyler and M. B. A. Oldstone (ed.), Reoviruses II. Springer-Verlag, Berlin, Germany.
- 57.Ubol, S., J. Kasisith, D. Pitidhammabhorn, and V. Tepsumethanol. 2005. Screening of pro-apoptotic genes upregulated in an experimental street rabies virus-infected neonatal mouse brain. Microbiol. Immunol. 49423-431. [DOI] [PubMed] [Google Scholar]
- 58.Varfolomeev, E. E., M. Schuchmann, V. Luria, N. Chiannilkulchai, J. S. Beckmann, I. L. Mett, D. Rebrikov, V. M. Brodianski, O. C. Kemper, O. Kollet, T. Lapidot, D. Soffer, T. Sobe, K. B. Avraham, T. Goncharov, H. Holtmann, P. Lonai, and D. Wallach. 1998. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9267-276. [DOI] [PubMed] [Google Scholar]
- 59.Xu, X. N., B. Laffert, G. R. Screaton, M. Kraft, D. Wolf, W. Kolanus, J. Mongkolsapay, A. J. McMichael, and A. S. Baur. 1999. Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor zeta chain. J. Exp. Med. 1891489-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeh, W. C., J. L. Pompa, M. E. McCurrach, H. B. Shu, A. J. Elia, A. Shahinian, M. Ng, A. Wakeham, W. Khoo, K. Mitchell, W. S. El Deiry, S. W. Lowe, D. V. Goeddel, and T. W. Mak. 1998. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 2791954-1958. [DOI] [PubMed] [Google Scholar]
- 61.Zachos, G., B. Clements, and J. Conner. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 2745097-5103. [DOI] [PubMed] [Google Scholar]
- 62.Zhang, H. G., J. Xie, L. Xu, P. Yang, X. Xu, S. Sun, Y. Wang, D. T. Curiel, H. C. Hsu, and J. D. Mountz. 2002. Hepatic DR5 induces apoptosis and limits adenovirus gene therapy product expression in the liver. J. Virol. 765692-5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang, J., D. Cado, A. Chen, N. H. Kabra, and A. Winoto. 1998. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392296-300. [DOI] [PubMed] [Google Scholar]