

Abstract
Adenovirus infection activates cellular DNA damage response and repair pathways. Viral proteins that are synthesized before viral DNA replication prevent recognition of viral genomes as a substrate for DNA repair by targeting members of the sensor complex composed of Mre11/Rad50/NBS1 for degradation and relocalization, as well as targeting the effector protein DNA ligase IV. Despite inactivation of these cellular sensor and effector proteins, infection results in high levels of histone 2AX phosphorylation, or γH2AX. Although phosphorylated H2AX is a characteristic marker of double-stranded DNA breaks, this modification was widely distributed throughout the nucleus of infected cells and was coincident with the bulk of cellular DNA. H2AX phosphorylation occurred after the onset of viral DNA replication and after the degradation of Mre11. Experiments with inhibitors of the serine-threonine kinases ataxia telangiectasia mutated (ATM), AT- and Rad3-related (ATR), and DNA protein kinase (DNA-PK), the kinases responsible for H2AX phosphorylation, indicate that H2AX may be phosphorylated by ATR during a wild-type adenovirus infection, with some contribution from ATM and DNA-PK. Viral DNA replication appears to be the stimulus for this phosphorylation event, since infection with a nonreplicating virus did not elicit phosphorylation of H2AX. Infected cells also responded to high levels of input viral DNA by localized phosphorylation of H2AX. These results are consistent with a model in which adenovirus-infected cells sense and respond to both incoming viral DNA and viral DNA replication.
Cellular DNA damage response pathways protect and preserve the integrity of the genome. These pathways, which are activated in response to various forms of DNA damage, involve a number of proteins that participate in both DNA repair and cell cycle progression (62). The serine-threonine kinases ataxia telangiectasia mutated (ATM), AT- and Rad3-related (ATR), and DNA protein kinase (DNA-PK) are activated in response to distinct types of damage. The ATM pathway is activated primarily by double-stranded DNA breaks (4, 30). DNA-PK acts in conjunction with the DNA ligase IV/XRCC4 complex to mediate the ligation of double-stranded breaks through nonhomologous end joining (34). The ATR pathway can be activated in response to a wide range of genotoxic stresses, such as base or nucleotide excision, double-stranded breaks, or single-stranded breaks. Activation of ATR is generally thought to occur via the recognition of single-stranded tracks of DNA (63). Each of these pathways leads to the phosphorylation and activation of a number of cellular proteins such as the variant histone H2AX, checkpoint kinases 1 and 2 (Chk1 and Chk2), and Nijmegen break syndrome protein 1 (NBS1), among others (62). Signals transmitted by a cascade of phosphorylation events result in cell cycle arrest and the accumulation of repair protein complexes at sites of DNA damage.
Upon recognition of a double-stranded DNA break by the cell, H2AX is phosphorylated on an extended C-terminal tail at serine 139 by the phosphatidylinositol 3-kinase (PI3K)-related kinases ATM, ATR, and DNA-PK (9, 41, 44, 58). Considered one of the earliest indications of a double-stranded DNA break, phosphorylated H2AX (γH2AX) acts as a scaffolding protein to which a number of DNA repair factors can dock to facilitate repair of the damaged DNA (36, 42, 53). Areas of phosphorylated H2AX, termed γH2AX foci, are enriched for proteins involved in both homologous recombination and nonhomologous end joining, such as NBS1, BRCA1 (42), and Mdc1 (24, 50).
Although adenovirus is able to activate both ATM and ATR pathways (11), adenoviral proteins limit the extent and consequences of signaling through these pathways. The E1B-55K and E4orf6 proteins form an E3 ubiquitin ligase with the cellular proteins Cullin-5, elongins B and C, and Rbx1 (28, 43). This complex targets key cellular proteins involved in cellular response to DNA damage, including p53 (28, 43), Mre11 (51), and DNA ligase IV (3). The E4orf3 gene product targets cellular proteins central to both the cellular DNA damage response and the antiviral response. The E4orf3 protein of species C adenoviruses alters the localization of Mre11/Rad50/NBS1 (MRN) complex members within the nucleus to prevent association with centers of viral DNA replication and to ensure efficient viral DNA replication (17, 18, 52). In addition, these three viral early proteins direct members of the MRN complex (2, 35) and the single-stranded DNA-binding protein 2 (20) to cytoplasmic aggresomes, where these sequestered proteins are effectively inactivated. These viral activities, along with the inactivation of DNA-PK by E4orf3 and E4orf6 gene products (7), appear to prevent recognition of viral genomes by the MRN complex and prevent ligation of these genomes through nonhomologous end joining. In cells infected with a virus with E4 deleted, Mre11 physically binds to viral DNA in an NBS1-dependent manner and may prevent efficient genome replication (37). The overlapping means by which adenovirus disables the MRN complex and prevents DNA damage repair serves to illustrate the importance of this activity for a productive adenovirus infection. However, despite having DNA damage signaling and DNA repair pathways dismantled, adenovirus-infected cells exhibit some characteristic changes associated with DNA damage signaling events, such as the phosphorylation of H2AX (6, 15). Thus, it appears that adenovirus effectively inhibits DNA repair activity but may not fully suppress the early events of DNA damage signaling.
The focus of the present study was to elucidate the activation of DNA damage signaling pathways revealed by phosphorylation of the variant histone H2AX during wild-type adenovirus infection and to determine what stage of the virus life cycle leads to this activation. We demonstrate that infected cells respond to viral genome replication with high levels of H2AX phosphorylation throughout the cell nucleus. This phosphorylation event is not localized to viral replication centers and does not appear to be concurrent with cellular double-stranded DNA breaks; rather, H2AX phosphorylation occurs coincident with the bulk of cellular chromatin. H2AX phosphorylation follows viral DNA replication and reaches peak levels after the degradation of the Mre11. In addition, we observed that infected cells can respond to both the presence of incoming viral genomes and genome replication by initiating H2AX phosphorylation.
MATERIALS AND METHODS
Cell culture.
Cell culture media, supplements, and serum were obtained from Invitrogen Life Technologies (Carlsbad, CA) through the Tissue Culture Core Laboratory of the Comprehensive Cancer Center of Wake Forest University. HeLa cells (ATCC CCL 2; American Type Culture Collection, Manassas, VA) and HeLa cells stably expressing adenovirus preterminal protein were maintained as monolayer cultures in Dulbecco Modified Eagle medium supplemented with 10% newborn calf serum, 100 U of penicillin, and 100 μg of streptomycin per ml. Cells were maintained in subconfluent adherent cultures in a 5% CO2 atmosphere at 37C by passaging twice weekly at approximately a 1:10 dilution. Wortmannin (BioSource, Camarilla, CA) was dissolved in dimethyl sulfoxide at a concentration of 1 M and caffeine (Sigma/Aldrich, St. Louis, MO) was prepared as a stock solution of 100 mM in phosphate-buffered saline (PBS). These were diluted into culture medium at the indicated concentrations for cell treatment.
Antibodies.
The primary antibodies directed against adenovirus proteins used in the present study include mouse monoclonal antibodies against E1A (M73), E1B-55K (2A6), E2A-DNA binding protein (B6-8), and E4orf6 and E4orf6/7 (Rsa#3) proteins. Antibodies specific for cellular proteins included rabbit monoclonal antibodies against H2AX (A300-082A; Bethyl Laboratories, Montgomery, TX) and γH2AX (A300-081A; Bethyl), mouse monoclonal antibodies against γH2AX (05-636 and 16-202A; Upstate/Millipore, Billerica, MA), rabbit polyclonal antibodies against Mre11 (PC388; Calbiochem, San Diego, CA), and RPA32 (GTX70258; GeneTex, Irvine, CA) and mouse monoclonal antibodies against β-actin (A5441; Sigma). Primary antibodies were used as hybridoma tissue culture supernatant fluid (diluted 1:5 or undiluted) or at a concentration of 1 μg per ml. Secondary antibodies, raised in goats and conjugated to horseradish peroxidase, were used at 0.2 μg per ml for immunoblotting (Jackson Immunoresearch, West Grove, PA). Fluorescent antibodies were also raised in goats against mouse or rabbit immunoglobulin G and coupled to Alexa Fluor 488 or Alexa Fluor 568 (Invitrogen). Fluorescent secondary antibodies were used at a concentration of 2 μg per ml. Antibodies were diluted into Tris-buffered saline (0.137 M NaCl, 0.003 M KCl, 0.025 M Tris-Cl [pH 8.0], 0.0015 M MgCl2 0.5% bovine serum albumin, 0.1% glycine, 0.05% Tween 20, 0.02% sodium azide). Antibodies used for immunofluorescence included 10% normal goat serum (Invitrogen). Sodium azide was omitted from solutions used with horseradish peroxidase-conjugated antibodies.
Indirect immunofluorescence.
Indirect immunofluorescence was performed as previously described (40) with the modification that cells were fixed in freshly prepared 2% formaldehyde and permeabilized in 0.5% Triton X-100 in PBS. Samples were mounted with polyvinyl-alcohol based mounting medium containing the DNA dye 4′,6-diamidino-2-phenylindole (DAPI) and were analyzed by epifluorescence microscopy using a Nikon TE300 inverted microscope fitted with filters appropriate for DAPI, Alexa Fluor 488, and Alexa Fluor 568 excitation. 12-Bit images were acquired by using a Retiga EX 1350 digital camera (QImaging Corp., Burnaby, British Columbia, Canada) with a ×100 magnification/1.4-numerical aperture oil immersion objective lens. Exposure settings to avoiding saturation were determined for each experimental series using the strongest signal, typically wild-type virus-infected cells. These settings were used to photograph all other samples to permit intensity comparisons within an experimental series.
Immunoblotting.
Adenovirus-infected cells were washed, harvested by trypsin treatment, washed again, and resuspended in urea sample buffer (9 M urea, 0.05 M Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 0.01 M 1,4-dithiothreitol) at 104 cells per μl. Proteins were denatured by heating to 95°C for 5 min, and the DNA was sheared by sonication. Material derived from 5 × 104 cells was separated by electrophoresis on a 10 or 15% polyacrylamide gel. For γH2AX analysis, protein was separated under the conditions described in reference 6. After separation, the proteins were electrophoretically transferred to nitrocellulose membrane, and the nitrocellulose was stained with Ponceau S to verify quantitative transfer. The nitrocellulose membrane was blocked by incubation with 5% nonfat dry milk in Tris-buffered saline before the addition of primary antibodies at the concentrations indicated above. Hybridoma tissue culture supernatants and β-actin antibody were incubated for 3 h at room temperature. Other monoclonal and polyclonal antibodies were incubated overnight at 4°C. After incubation, the antibody was removed, and the blot was washed three times with Tris-buffered saline, followed by incubation with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibody (Jackson Immunoresearch) at the above-stated dilutions for 90 min. The antibody-antigen complex was visualized by the addition of Pierce enhanced chemiluminescence reagents (Thermo Scientific, Rockford, IL) and exposed to X-ray film.
Empty capsid treatment.
Empty adenovirus capsids were purified after sequential centrifugation to equilibrium through discontinuous and continuous a density gradients of CsCl. The absence of infectious virus was confirmed, and the concentration of viral protein in the empty capsid preparation was determined. To determine an equivalent titer, the amount of viral protein per infectious unit of the wild-type virus was measured, and an equivalent amount of empty capsid protein was used as an equivalent multiplicity of infection (MOI). Cells were treated with empty capsids for 1 h in infection media at 37°C. Infectious particles or empty capsids were aspirated, and cells were placed in normal growth medium. Note that cells exposed to empty capsid above an equivalent MOI of 1,000 showed extensive cytopathic effect.
Real-time PCR.
Total DNA was harvested from 106 adenovirus-infected cells. Briefly, cells were washed with PBS and harvested with trypsin. Cells were pelleted and resuspended in 400 μl of isotonic lysis buffer (0.01 M Tris-HCl [pH 8.3], 0.015 M MgCl2, 0.075 M KCl, 0.5% NP-40, 0.5% Tween 20, 0.5 mg of proteinase K/ml), followed by overnight incubation at 65°C. proteinase K was inactivated by 15 min of incubation at 95C. A total of 2 μl of DNA was used as a template for real-time PCR. Quantitative real-time PCR was performed with an ABI 7700 instrument (Applied Biosciences, Forest City, CA) and with the PerFeCTa qPCR FastMix (Quanta Biosciences, Gaithersburg, MD) according to the manufacturer's directions. Forward and reverse primers specific for the hexon gene of adenovirus were used in conjunction with a fluorescent probe to this same region (23). The amplification profile used was as follows: 1 cycle at 95C for 10 min and 50 cycles at 95°C for 15 s and 55°C for 1 min. A standard curve was plotted for each reaction with cycle threshold (CT) values obtained from amplification of known quantities of purified wild-type adenovirus DNA. The standard curves were used to transform the CT values of experimental samples into the relative number of genome copies. The results are presented as the mean from triplicate experiments.
Flow cytometry.
HeLa cells were harvested by treatment with trypsin and fixed in 70% ethanol for a minimum of 3 h at 4°C. The ethanol was removed, and cells were resuspended at approximately 2 × 106 cells per ml in propidium iodide buffer (0.1 M NaCl, 0.036 M sodium citrate, 50 μg of propidium iodide per ml, 0.6% Nonidet P-40) supplemented with 0.04 mg of RNase (Sigma) per ml. The DNA content of individual cells was measured by flow cytometry using a BD FACSCalibur (Becton Dickinson) with an argon laser as the excitement source (488 nm). The emitted light was analyzed for forward and side scatter, pulse width (to discriminate doublets), and red fluorescence (>630 nm) of propidium iodide to determine the DNA content per cell. A total of 10,000 events were measured in each analysis.
For analysis of H2AX phosphorylation, cells were fixed with either 70% ethanol or 2% paraformaldehyde. After fixation with PFA, cells were permeabilized with 0.5% Triton X-100. Cells were directly labeled with either a fluorescein-conjugated monoclonal antibody (16-202A; Upstate, Temecula, CA) or indirectly labeled with unconjugated monoclonal antibodies against phosphorylated H2AX (Ser-139) listed above, at room temperature for 90 min. Cells were washed three times in PBS supplemented with 1% normal calf serum (Invitrogen). If indirectly labeled, the cells were followed by incubation with fluorescein- or Alexa Fluor 488-conjugated secondary antibodies at a 1 to 1,000 dilution for 60 min. Indirectly labeled cells were again washed three times in PBS solution and immediately analyzed using a BD FACSCalibur, measuring the green fluorescence (>518 nm) of fluorescein or Alexa Fluor 488 for a total of 10,000 events. The results shown are representative of multiple experiments performed in triplicate using various combinations of the above-mentioned antibodies.
RESULTS
Adenovirus infection results in H2AX phosphorylation.
Although products of the E1B and E4 genes of adenovirus suppress DNA damage signaling at late times of infection, phosphorylation of H2AX (γH2AX) has been observed in adenovirus-infected cells (6, 15). To compare the levels of phosphorylated H2AX generated by ionizing radiation with those elicited during adenovirus infection, HeLa cells were infected with adenovirus or subjected to ∼40 Gy of ionizing radiation. Phosphorylation of H2AX was observed by immunofluorescence at 24 h postinfection (hpi) or ∼15 min after irradiation (Fig. 1A). Mock-infected cells exhibited little to no H2AX phosphorylation, with the occasional appearance of one or two γH2AX foci in a cell. As previously reported, ionizing radiation resulted in the phosphorylation of H2AX at specific foci within the cell nucleus, a finding indicative of double-stranded DNA breaks (42). Fewer γH2AX foci may be represented in the images of irradiated cells (Fig. 1A) than were actually present because of the limited depth of focus and because some foci may have resolved in the elapsed time between irradiation and processing. Adenovirus-infected cells, however, exhibited strikingly high levels of H2AX phosphorylation. This phosphorylation was spread throughout the cell nucleus and did not appear to colocalize with viral replication centers, as shown by adenovirus DNA-binding protein, DBP. Rather, phosphorylated H2AX appeared to be coincident with cellular chromatin. Because this pattern of phosphorylation was observed with three different phospho-specific antibodies raised in mice and rabbits (Bethyl A300-081A; Upstate 05-636, Upstate 16-202A), it seems unlikely that this staining pattern was due to a spurious interaction between the phospho-specific antibody and the infected cell.
FIG. 1.
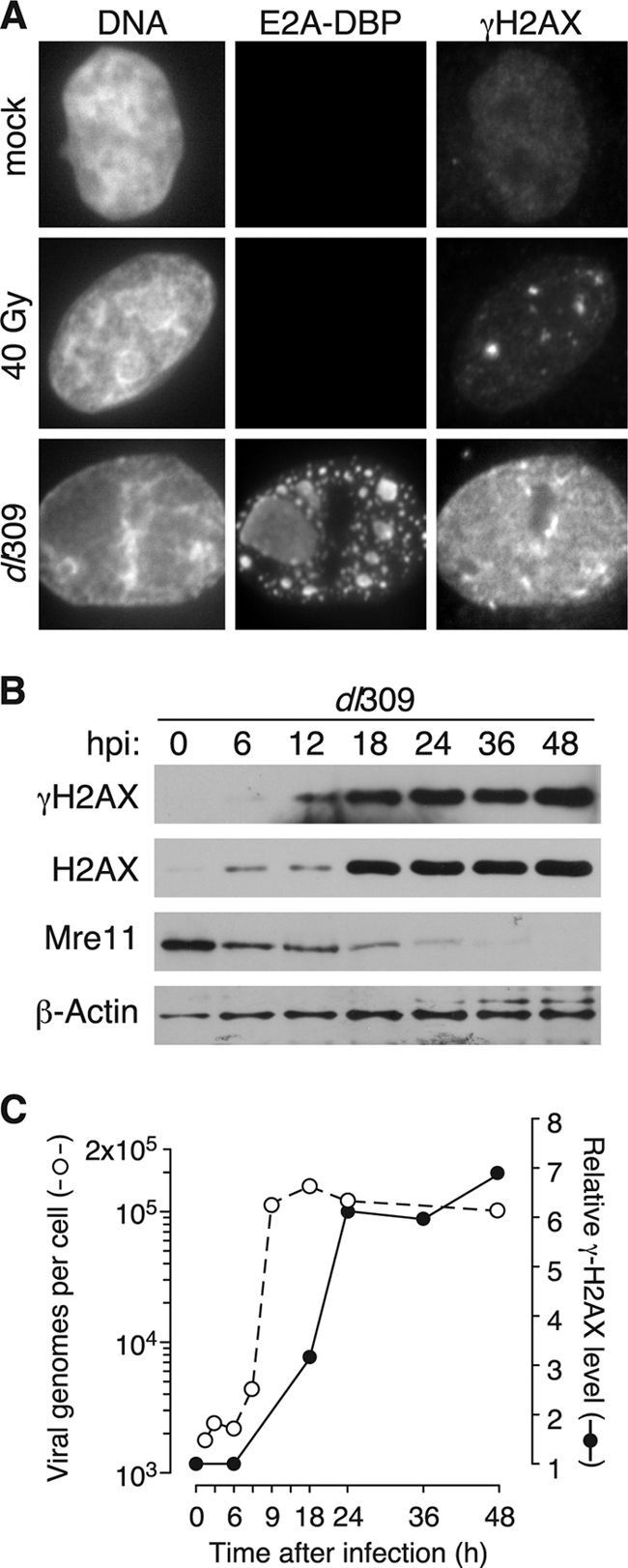
Phosphorylated H2AX accumulates at late times after infection with wild-type adenovirus. (A) HeLa cells were mock infected, exposed to 40 Gy of radiation, or infected at an MOI of 5 with the wild-type virus dl309. Cells were processed for immunofluorescence microscopy for the E2A DNA-binding protein (E2A-DBP) and phosphorylated H2AX (γH2AX) immediately after irradiation or 24 h after infection. Representative images predominantly of the nucleus are presented. Multiple foci were observed in the irradiated cells, but only those in the plane of focus are seen in this image. (B) HeLa cells were infected at an MOI of 5 and harvested at the indicate times postinfection. Material representing equivalent numbers of initially infected cells was separated by SDS-polyacrylamide gel electrophoresis and analyzed by immunoblotting with the indicated antibodies. (C) Viral DNA levels were measured by quantitative real-time PCR from HeLa cells infected at an MOI of 5 with dl309 at the indicated times after infection (open symbols). The relative level of γH2AX (closed symbols) was determined as the ratio of γH2AX signal to total H2AX signal for each time point from a blot similar to the representative blot in panel B. The ratio measured for mock-infected cells was set to 1.
Adenovirus disables cellular DNA damage responses in order to prevent ligation of cellular genomes (51). To determine whether H2AX phosphorylation occurs prior to degradation of Mre11, the levels of γH2AX and Mre11 were measured at various times during wild-type adenovirus infection. Phosphorylated H2AX first reached detectable levels at 12 hpi (Fig. 1B) and continued to increase until 24 hpi, when it appeared to peak and remain steady. Total levels of H2AX increased sharply at 18 hpi. Although this was not expected, the increase in total H2AX is consistent with a previous report noting that the level of H2AX mRNA increased during adenovirus infection (39). Histone H3 levels also increased during adenovirus infection, although to a lesser extent than H2AX (data not shown). Mre11 levels remained high at 6 hpi but began to decrease by 12 hpi. At 36 hpi, consistent with previous reports (51), Mre11 was almost completely absent from infected cells. Although low levels of phosphorylated H2AX appeared prior to complete Mre11 degradation, these results demonstrate that the high levels of H2AX phosphorylation observed during infection do not occur until after Mre11 has been significantly reduced.
To relate the kinetics of H2AX phosphorylation to the timing of viral genome replication, levels of adenovirus DNA were measured over time by real-time PCR (Fig. 1C). Viral genome replication began at ∼6 hpi. Levels of the viral genome appeared to peak at 12 hpi and remained high for the duration of the infection. Thus, phosphorylated H2AX levels peak after viral genome replication has reached its maximum and after alteration of the MRN complex (Fig. 1D), which is consistent with the idea that H2AX phosphorylation may be sustained in an MRN-independent manner.
PI3K-like kinase inhibitors decrease H2AX phosphorylation in infected cells.
The protein kinases ATM, ATR, and DNA-PK have been shown to phosphorylate H2AX (9, 41, 58). To determine the contribution of these kinases to γH2AX, cells were infected with wild-type adenovirus at high MOIs, followed at 3 hpi by treatment with the kinase inhibitors caffeine (Fig. 2A and B) and wortmannin (Fig. 2C and D) at a range of concentrations. γH2AX and total H2AX were measured by immunoblotting, and the levels of γH2AX were quantified relative to the total H2AX. The blots and quantification shown are from separate experiments, each is representative of four independent experiments.
FIG. 2.

PI3K-like kinase inhibitors decrease H2AX phosphorylation in infected cells. HeLa cells were mock infected or infected with the wild-type virus at an MOI of 5. After 3 h, either caffeine or wortmannin was added to the infected cells at the indicated concentration, and the cells were harvested for analysis after 24 h by immunoblotting for γH2AX and total H2AX. The relative levels of H2AX phosphorylation were determined as the ratio of γH2AX to total H2AX where the ratio measured for mock-infected cells was set to 1. (A to D) Representative results of four experiments for caffeine treatment are shown in panels A and B and for wortmannin treatment in panels C and D. (E and F) HeLa cells were mock infected (−) or infected with the E4 deletion virus, dl366* (+), at an MOI of 10. After 3 h, either caffeine or wortmannin was added to the infected cells at the indicated concentration, and cells were harvested for analysis at 24 h by immunoblotting for RPA32. An open arrowhead indicates nonphosphorylated RPA32, and a filled arrowhead indicates the phosphorylated form of RPA32.
Caffeine inhibits ATM at a concentration of 5 mM, ATR at 10 mM, and is only able to reduce DNA-PK activity to ca. 40% at 10 mM (45). At the lowest concentrations of caffeine, which is sufficient to inhibit only ATM, an increase in H2AX phosphorylation was observed. Such an increase has been previously reported for the use of caffeine and wortmannin in irradiated cells and is considered to be the result of increased activity of the noninhibited kinase (57). At concentrations higher than 10 mM, at which ATR activity would be almost completely inhibited, the levels of γH2AX decreased substantially to approximately the levels observed in mock-infected cells. Since caffeine is only able to reduce DNA-PK activity to 40%, it appears that the decrease in H2AX phosphorylation is the result of the inhibition of ATR activity rather than the result of the inhibition of ATM or DNA-PK.
Wortmannin inhibits ATM and DNA-PK at 30 μM and is able to reduce ATR activity to 50% at 100 μM (46). Similar to the results obtained with caffeine, inhibition of ATM and DNA-PK resulted in an initial increase in H2AX phosphorylation. As the concentration of wortmannin was increased to levels sufficient to partially inhibit ATR, H2AX phosphorylation was slightly reduced. However, wortmannin was unable to reduce H2AX phosphorylation to levels observed in mock-infected cells, potentially through continued activity of ATR. Treatment with either drug was accompanied by a reduction in amount of total H2AX, although histone levels were still higher than those of mock-infected cells.
To confirm the expected action of caffeine and wortmannin, HeLa cells were infected with dl366*, a mutant adenovirus lacking the entire E4 region (29). A similar mutant virus was shown to activate both ATM and ATR (11), resulting in the phosphorylation of both RPA32 and Chk1. Infected cells were left untreated or were treated with caffeine (Fig. 2E) or wortmannin (Fig. 2F) at the indicated concentrations. Cell lysates were harvested at 24 hpi and analyzed by immunoblotting to identify hyperphosphorylated forms of RPA32, a substrate of ATR. As expected, infection with a virus with E4 deleted resulted in RPA32 phosphorylation. This phosphorylation was ablated by treatment with all concentrations of drugs. Phosphorylation of the ATM substrate, Chk1, was similarly decreased upon drug treatment (data not shown). Accumulation of late viral proteins was diminished by ca. 40% by 30 μM wortmannin or 30 mM caffeine (data not shown).
These results indicate that the PI3K-like kinases ATM, ATR, and DNA-PK probably remain functional at some point during adenovirus infection. Since the inhibitors used here fail to specifically target individual enzymes, we cannot determine the specific contribution of each kinase to H2AX phosphorylation. However, ATR may be the kinase most responsible for H2AX phosphorylation, because inhibition of this kinase by caffeine resulted in a marked decrease in H2AX phosphorylation, whereas wortmannin, which has limited ability to inhibit ATR activity, did not significantly reduce H2AX phosphorylation. In addition, recent work of Weitzman et al. has shown that ATR is activated at early times by adenovirus infection (10).
γH2AX does not depend on early viral proteins and is independent of the cell cycle.
ATR activity is most closely linked to errors in DNA replication such as the generation of long stretches of single-stranded DNA at stalled replication forks (14, 63). Adenovirus genome replication not only generates numerous free DNA ends but also a large quantity of single-stranded DNA (21, 32). To determine whether the H2AX phosphorylation observed is a response to viral DNA replication and possibly single-stranded DNA intermediates, rather than the presence and structure of mature viral genomes, cells were infected with either the wild-type adenovirus or a mutant virus, H5wt300ΔpTP (ΔpTP). ΔpTP contains a deletion of the preterminal protein gene and thus is unable to direct the synthesis of viral DNA (47). Infection of HeLa cells with the ΔpTP mutant virus resulted in a diffuse nuclear distribution of DBP characteristic of that seen at early times of infection. The staining patterns for DBP and γH2AX confirm that the ΔpTP virus is incapable of forming replication centers and did not elicit widespread H2AX phosphorylation (Fig. 3A).
FIG. 3.
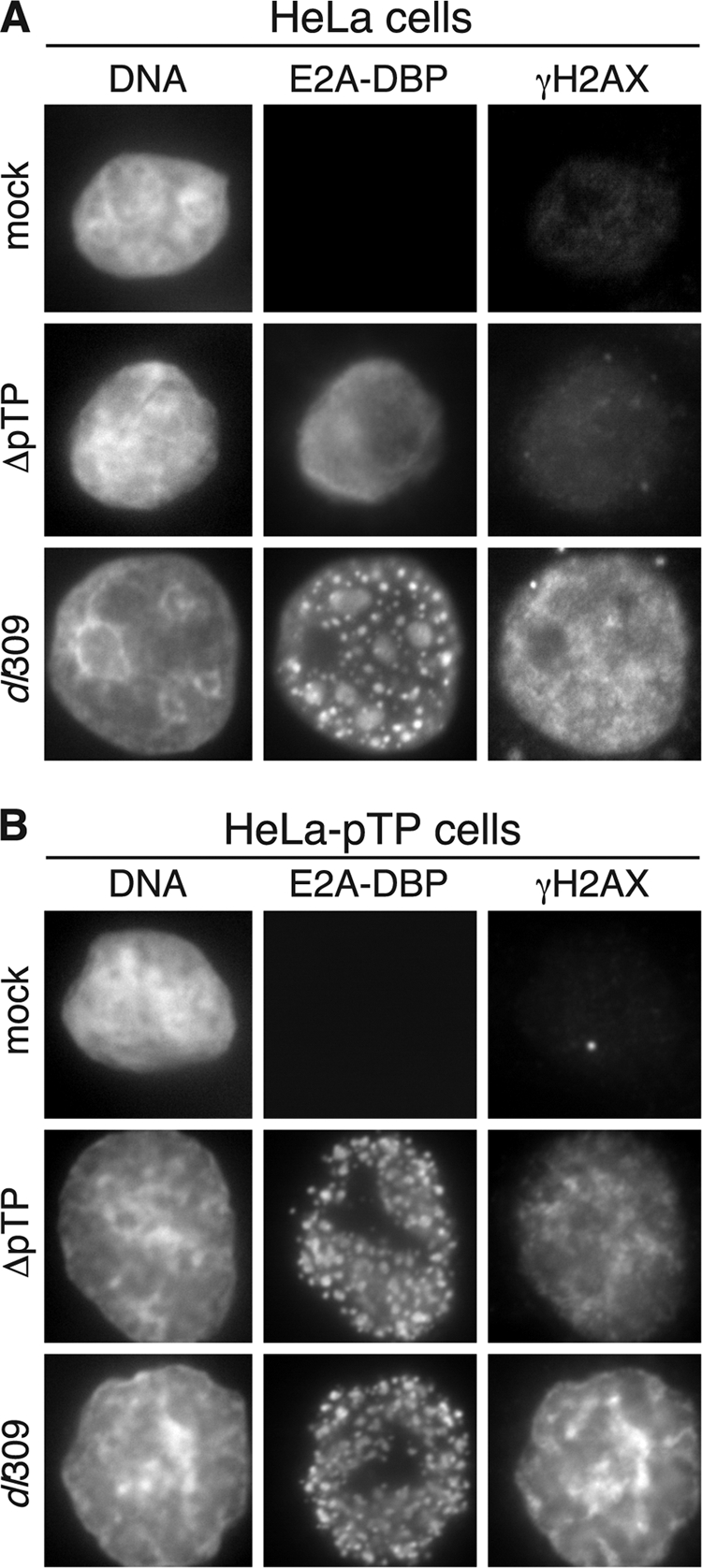
Widespread H2AX phosphorylation during adenovirus infection requires viral DNA synthesis. (A) HeLa cells were mock infected, infected with the wild-type virus dl309, or infected with the DNA replication-deficient adenovirus H5wt300-ΔpTP (ΔpTP) at an MOI of 5 and processed for immunofluorescence after 24 h for the E2A-DBP protein and γH2AX. (B) HeLa cells that stably express the E2B preterminal protein gene (HeLa-pTP) were infected and processed as in panel A. The restoration of centers of viral DNA replication is seen by the appearance of foci of E2A-DBP staining.
To exclude the possibility that unanticipated mutations in the ΔpTP viral genome contributed to the failure to elicit H2AX phosphorylation, HeLa cells that constitutively express the preterminal protein gene were infected at a high MOI with either the wild-type or ΔpTP adenovirus. Delivery of preterminal protein in trans allowed for the generation of viral replication centers in ΔpTP virus-infected cells (Fig. 3B) and resulted in nuclear wide phosphorylation of H2AX, as seen in cells infected with the wild-type virus. Since expression of the preterminal protein gene alone did not increase levels of H2AX phosphorylation, it seems likely that viral genome replication or subsequent events of the infectious cycle contributes to the extensive phosphorylation of H2AX observed in the infected cell.
Adenovirus early gene products are important for the alteration of the cellular environment and inhibition of the DNA damage response (3, 51). It has been suggested previously that H2AX phosphorylation occurs in response to adenovirus E1A gene expression (15). We have also observed that infection with adenoviruses with E1A deleted did not increase H2AX phosphorylation (data not shown). However, with the exception of the E2B preterminal protein gene, the ΔpTP virus is expected to direct the expression of all viral genes that do not depend on viral DNA synthesis. To confirm that dysregulated expression of the viral early genes did not preclude H2AX phosphorylation, the expression of early viral proteins by both ΔpTP and wild-type adenovirus was evaluated by immunoblotting. Like the wild-type virus, the ΔpTP virus expressed E1A, E1B-55K, E2A, and E4orf6 (Fig. 4A). Both viruses expressed these early gene products to largely similar levels, although ΔpTP-infected cells exhibited a slight delay in protein accumulation and expressed slightly less total protein. Unlike wild-type virus-infected cells, wherein E1A expression is high at early times and is reduced at late times of infection (48), E1A expression continued throughout the ΔpTP infection, a finding consistent with the failure of this virus to progress to late stages of infection. In addition, although the E1B-55K and E4orf6 gene products were expressed and presumably promoted the degradation of Mre11 (Fig. 4B) and p53 (data not shown), we noted that Mre11 levels remained higher in ΔpTP-infected cells than in wild-type virus-infected cells. Although the basis for this difference is not understood, the very low level of H2AX phosphorylation observed in ΔpTP-infected cells cannot be attributed to differences in early viral gene expression nor to a more rapid disappearance of Mre11. Although the ΔpTP and wild-type virus expressed early genes to slightly different levels at an equivalent multiplicity, we observed that even at high multiplicities where more viral protein would be produced, ΔpTP virus-infected cells never accumulated levels of γH2AX comparable to that observed in wild-type virus-infected cells (Fig. 5)
FIG. 4.
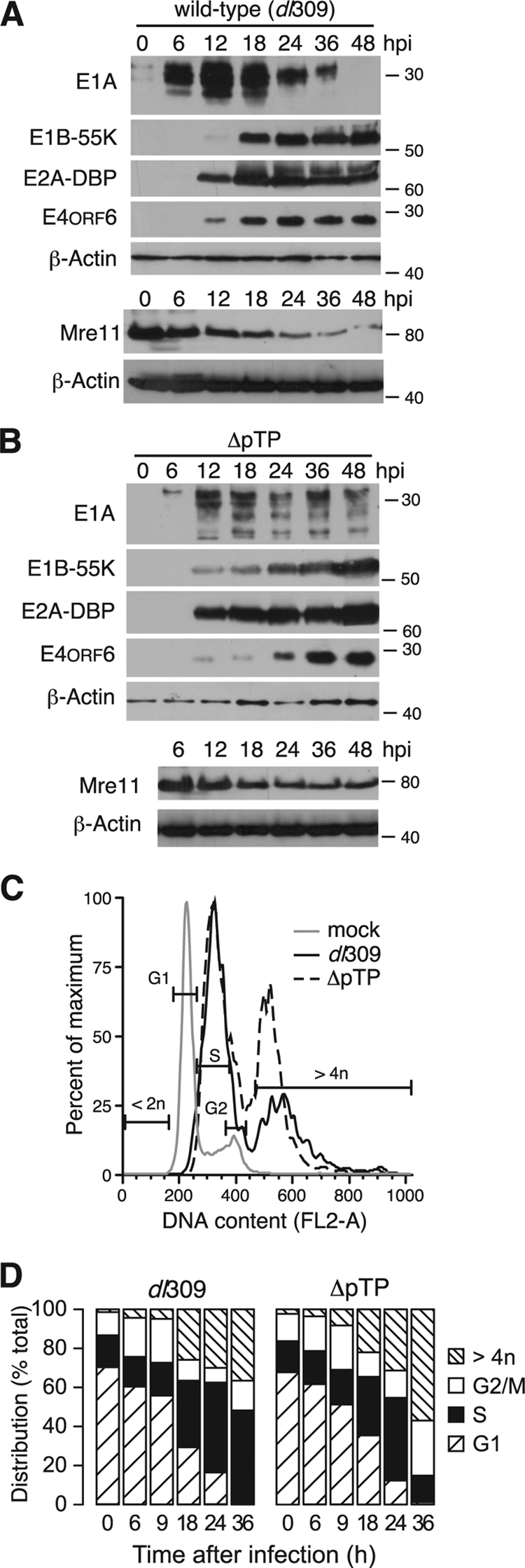
Viral DNA replication-deficient H5wt300-ΔpTP elicits similar changes to the cellular DNA profile as the wild-type virus. HeLa cells were infected with the wild-type virus dl309 (A) or H5wt300-ΔpTP (B) at an MOI of 5 and harvested at the indicated times after infection. Material representing equivalent numbers of initially infected cells was separated by SDS-polyacrylamide gel electrophoresis and analyzed by immunoblotting with antibodies for viral early proteins or cellular proteins. The approximate position of molecular mass standards is indicated on the right in kilodaltons. Mre11 levels were measured in separate infections and are shown with the corresponding β-actin loading control. (C) HeLa cells infected with indicated viruses were harvested after 24 h and the cellular DNA content analyzed by flow cytometry after staining with propidium iodide. Representative histograms are shown where the gates identify populations containing the indicated DNA content for uninfected cells. (D) HeLa cells were infected at an MOI of 5 and analyzed at the indicated times after infection by flow cytometry for DNA content as in panel C. The fraction of cells within with each gate indicated in panel C is plotted as a percentage.
FIG. 5.

Delivery of increasing amounts of nonreplicating viral genomes elicits limited and focal γH2AX. (A) HeLa cells were infected with the replication-deficient virus H5wt300-ΔpTP at the indicated MOI. Viral DNA levels were measured 24 h after infection by quantitative PCR and are presented as the number of viral genomes per infected cell. The solid line is a best-fit exponential saturation curve. The inset shows the relationship between delivered genomes and MOI at low multiplicity. The dashed line in the inset is a linear regression. (B) HeLa cells were infected with the indicated viruses and at the indicated MOI and processed 24 hpi for direct or indirect immunofluorescence for γH2AX. Levels of γH2AX were determined by flow cytometry and are normalized between experiments to the mean fluorescence intensity measured at an MOI of 20. Error bars represent the SEM from three to eight replicates performed on separate occasions. (C) HeLa cells were infected with H5wt300-ΔpTP at the indicated MOI and processed 24 hpi for immunofluorescence to visualize γH2AX and cellular DNA by DAPI staining. Representative fields are shown using equivalent exposures for each image. The pair of images in panel h is an enlargement of the inset seen in panel f. The scale bars in panels a, e, and h represent 10 μm.
E1A proteins activate cellular processes that drive the infected cell into an S-phase-like environment favorable for virus replication (48). This prolonged S-phase-like environment may resemble the cellular environment during S-phase arrest, which occurs in response to replication-associated DNA damage such as stalled replication forks (1). Arresting cells in S phase by treatment with drugs such as hydroxyurea and aphidicoline, both of which cause replication forks to stall, leads to H2AX phosphorylation (27). To establish whether the cellular S-phase-like environment during wild-type infection is responsible for H2AX phosphorylation, the DNA content of cells infected with the wild-type or ΔpTP mutant virus was measured by propidium iodide staining and flow cytometry (Fig. 4C). The apparent stage of the cell cycle was determined, and the percentage of the cell population in each stage is summarized in Fig. 4D.
At 6 hpi, cells infected with either virus exhibited distinct populations of cells containing G1, S, and G2/M content of DNA, with ca. 60% of cells containing DNA content corresponding to G1, 20% corresponding to S phase, and 20% corresponding to G2/M. As the time of infection progressed to 18 and 24 hpi, the average cellular DNA content increased such that more cells contained DNA levels corresponding to S phase (between 30 and 45%), and the amount of cells with G1 DNA content was reduced (30 to 35% at 18 hpi, 10 to 15% at 24 hpi). At late times of infection, cells accumulated DNA in excess of G2/M content (>4 n). This result has been previously described for cells infected with the E1B-55K mutant adenovirus and was referred to as endoreduplication (13). Similar DNA profiles were observed for both wild-type and ΔpTP virus infections through 24 hpi, indicating that the replication of cellular DNA by the infected cell is not responsible for H2AX phosphorylation. Note that because the ΔpTP mutant virus fails to direct viral DNA synthesis, the increase in DNA content in these cells can only be due to an increase in cellular DNA. Furthermore, examination of propidium iodide-stained wild-type virus-infected cells by fluorescence microscopy revealed that propidium iodide staining was excluded from the centers of viral DNA replication. We interpret this to mean that the viral DNA in the cell nucleus is either inaccessible or nonreactive to this fluorescent dye (data not shown). Taken together, these data are consistent with the idea that viral DNA replication acts as a stimulus for cellular DNA damage response pathways independently of any increase in cellular DNA synthesis and that viral DNA replication, or events subsequent to viral DNA replication, promote widespread phosphorylation of H2AX.
Input viral genomes generate low-level, localized H2AX phosphorylation.
Adenovirus genome replication yields thousands of copies of viral DNA and an abundance of single-stranded DNA (21, 32). Both of these DNA structures could stimulate a DNA damage response. Unlike adenoviruses with E1 deleted, which can replicate the viral genome under some circumstances (8, 25, 49), deletion of the preterminal protein gene renders the ΔpTP mutant virus completely unable to direct genome replication (47). Thus, this mutant virus provides an efficient means of delivering viral DNA at high multiplicities without the complication of viral DNA replication. To determine the efficiency of the delivery of viral genomes as a function of MOI, HeLa cell were infected with ΔpTP at a range of multiplicities. Total cell DNA was harvested at 24 hpi and used as a template for real-time PCR (Fig. 5A). At 24 hpi, when wild-type virus DNA replication has reached ∼105 copies per cell (see Fig. 1C), only input amounts of viral DNA were recovered from ΔpTP-infected cells, confirming the inability of this virus to replicate its genome. Infection and genome delivery by ΔpTP at increasing multiplicity efficiently deposited viral DNA in a dose-dependent manner, where approximately seven genomes per infectious unit were delivered at low multiplicities (Fig. 5A, inset) up to a saturating level of about 350 viral genomes per cell (Fig. 5A).
To determine the impact of input viral DNA on H2AX phosphorylation, cells were infected with various amounts of the ΔpTP mutant virus. Cells were harvested at 24 hpi and levels of γH2AX were measured by flow cytometry (Fig. 5B). At each MOI, the amount of phosphorylated H2AX was comparable to that of mock-infected cells, whereas infection with the wild-type virus at an MOI of 5 resulted in approximately twofold more H2AX phosphorylation than mock-infected cells. Similar results were obtained when phosphorylated H2AX levels were monitored by immunoblotting (data not shown).
Although no significant increase in H2AX phosphorylation was observed by flow cytometry or Western blotting at high MOI infection with the ΔpTP mutant virus, changes in H2AX phosphorylation were apparent by immunofluorescence microscopy (Fig. 5C). Delivery of up to approximately 150 copies of the viral genome (MOI ≤ 20) by infection with the ΔpTP mutant virus resulted in a very low level of H2AX phosphorylation. At these multiplicities, γH2AX was observed in a small number of distinct foci within the nucleus, somewhat similar to the pattern observed in cells exposed to ionizing radiation (see Fig. 1A) and similar to the weak staining observed in mock-infected cells (data not shown). Delivery of greater numbers of viral genomes resulted in increased numbers of γH2AX foci. Cells infected with an MOI of 50 or 100 had increased numbers of γH2AX foci compared to cells infected at lower multiplicities, and the number of foci appeared to increase with increasing multiplicities. Nonetheless, delivery of the highest amounts of viral genomes to the cell nucleus by ΔpTP infection was still insufficient to generate diffuse nuclear-wide H2AX phosphorylation that we observed during wild-type virus-infection. At these highest multiplicities, phosphorylated H2AX was still found in distinct foci within the nucleus (Fig. 5Cf to h). It remains to be determined whether these discrete foci observed at 24 hpi are associated with the input viral genomes. It also appears that the focal staining pattern observed at the higher multiplicities of infection with the ΔpTP mutant virus (Fig. 5C) was not readily detected by flow cytometry (Fig. 5B). The reason for this apparent discrepancy remains unknown. Nonetheless, the difference in H2AX phosphorylation patterns observed between wild-type and ΔpTP high MOI infection suggests that cells are able to recognize both incoming viral genomes, as presented by ΔpTP infection, and viral genome replication that occurs during wild-type virus infection.
Attachment of adenovirus to the cell surface can trigger signaling events within the cell, including PI3K activation (33). To determine whether the focal staining pattern for γH2AX observed at the highest MOIs with the ΔpTP mutant was due to the attachment of large quantities of virions to the cell surface, rather than the delivery of viral genomes, cells were exposed to the same number of viral capsids devoid of viral DNA that would be found at a comparable range of multiplicities. Empty capsid-treated cells were analyzed by immunofluorescence (Fig. 6A). In contrast to what we observed in cells infected with the ΔpTP mutant virus, exposure to empty capsids did not result in γH2AX foci at any concentration of empty particles. In addition, cell lysates were generated at 24 hpi and levels of γH2AX, H2AX, and β-actin were measured by immunoblotting. As shown previously, wild-type virus-infection resulted in a substantial increase in γH2AX levels. At all concentrations, empty virus particles did not elicit a measurable level of H2AX phosphorylation (Fig. 6B). Because infection with the ΔpTP virus at MOIs of 50 through 500 resulted in a visible change to the pattern of H2AX phosphorylation (Fig. 5C) but treatment with equivalent amounts of empty capsids did not, we conclude that the γH2AX foci observed in ΔpTP-infected cells resulted from the detection of viral genomes. Although empty capsids were unable to stimulate phosphorylation of H2AX, exposure of cells to empty capsids was not wholly innocuous, since concentrations higher than those used here were sufficient to cause cell death (data not shown).
FIG. 6.

Exposure to empty viral capsids fails to elicit γH2AX. (A) HeLa cells were exposed to empty viral capsids corresponding to the number of viral particles required for the indicated MOI of wild-type virus. After 24 h, γH2AX was visualized by immunofluorescence and photographed using the identical exposure settings in Fig. 5. (B) HeLa cells were mock infected, infected with the wild-type virus dl309, or exposed to empty capsids at the indicated equivalent MOI. After 24 h, cellular lysates were prepared and analyzed by immunoblotting for γH2AX, total H2AX, and β-actin.
Taken together, we interpret these results to indicate that adenovirus-infected cells are able to sense and respond to the entering viral DNA. Both incoming viral genomes and genome replication can be recognized. However, the cellular response to replicating viral DNA leads to activation of signaling normally associated with the DNA damage response and the concomitant phosphorylation of H2AX throughout the cell nucleus that persists through late times of infection.
DISCUSSION
Infection with wild-type adenovirus results in nuclear-wide phosphorylation of the cellular histone H2AX. This phosphorylation event is distinct from that associated with damage to cellular DNA, because the phosphorylation occurs throughout the cell nucleus rather than in specific foci. In HeLa cells, H2AX phosphorylation reaches a peak at 24 hpi. At this time Mre11 levels have been significantly reduced and viral genome replication has reached a maximum. H2AX phosphorylation appears to be mediated predominantly by ATR but with potential contributions from ATM and DNA-PK, indicating that the rate at which Mre11 is degraded may not be sufficient to fully preclude activation of these kinases in response to viral activities.
Adenovirus infection and expression of E1A gene products drives the cell into an S-phase-like state with unscheduled cellular DNA synthesis and viral DNA synthesis (48). Viral DNA does not contribute to the overall DNA content detected by propidium iodide staining and flow cytometry because the ΔpTP- and wild-type virus-infected cells exhibited similar DNA profiles after the onset of viral genome replication in wild-type virus-infected cells. Similar numbers of cells infected with either the wild-type virus or the ΔpTP virus contained a DNA content in excess of 4n while only wild-type virus-infected cells exhibited high levels of H2AX phosphorylation. From these results, we conclude that the synthesis of cellular DNA beyond a 4n content is not sufficient to elicit H2AX phosphorylation. Because only the wild-type virus was able to direct viral genome replication, the widespread H2AX phosphorylation observed during wild-type virus infection was due to viral genome replication or to subsequent events that depend on viral DNA replication.
Input viral DNA contributes little to the overall levels of H2AX phosphorylation. Infection with the ΔpTP mutant virus at various multiplicities did not result in detectable levels of γH2AX when monitored by flow cytometry or immunoblotting. However, when H2AX phosphorylation was examined in ΔpTP-infected cells by immunofluorescence microscopy, the pattern of γH2AX was distinct from that of wild-type virus-infected cells. Cells infected with the ΔpTP virus at multiplicities below 50 contained H2AX phosphorylation that was indistinguishable from uninfected cells. However, at multiplicities of 100 to 500, the ΔpTP virus elicited a moderate focal response; rather than occurring throughout the nucleus, γH2AX was found in specific, distinct foci within the nucleus. It will be interesting to determine whether these γH2AX foci, which were observed 24 h after infection, correspond to the location of the input viral DNA within the cell nucleus. Differences in the pattern of H2AX phosphorylation among uninfected cells, cells infected with the wild-type virus, and cells infected with the ΔpTP mutant virus indicate that the infected cell is capable of recognizing and responding to incoming viral DNA. However, nucleus-wide H2AX phosphorylation appears to occur only in response to viral genome replication.
Since cells appear able to respond to the incoming viral genomes independently of viral DNA replication, it seems likely that some feature of the viral DNA must be recognized promptly after delivery into the cell and trigger some facet of the DNA damage response. The cellular factor Mdc1 has been shown to localize to viral replication centers and to physically associate with viral DNA with E4 deleted (37, 38). This implies a potential role for Mdc1 in viral DNA recognition, perhaps through recruitment of the MRN complex and other DNA repair factors. However, in the absence of Mdc1, Mre11 can still localize to replication centers in cells infected with a virus with E4 deleted (38). Incoming viral genomes may activate one set of DNA damage response pathways, potentially by binding Mdc1, and viral genome replication may activate a second pathway. Our results using the ΔpTP virus support the notion that Mdc1 recognizes incoming viral DNA prior to degradation of Mre11 (38). In the case of E4 deletion viruses, this second activation due to genome replication is likely to be at least partially mediated in an MRN-dependent manner, since these proteins remain functional during infection with an E4 mutant virus. However, the results presented in this report demonstrate high levels of H2AX phosphorylation after alteration of Mre11, suggesting that adenovirus DNA replication can activate a facet of the DNA damage response independently of the MRN complex. At least two scenarios could account for the phosphorylation of H2AX observed during wild-type virus infection. Viral DNA replication could activate the responsible kinases independently of the MRN complex, with the signal for activation being a substrate other than double-stranded DNA ends. Alternatively, the MRN complex could play a role in the initial kinase activation during infection, while the propagation and persistence of H2AX phosphorylation could occur in an MRN-independent manner.
Viral genome replication yields up to 50,000 copies of linear, double-stranded viral genomes, potentially exposing the infected cell to 100,000 free DNA ends (56). In addition, a single-stranded viral DNA molecule arises for every double-stranded viral genome that is replicated (21, 32). The single-stranded viral DNA may be recognized by the infected cell as aberrant and thus contribute to the activation of cellular kinases and subsequent phosphorylation of H2AX. Previous reports demonstrated the localization of cellular replication protein A p32 subunit (RPA32), a cellular single-stranded DNA-binding protein required for multiple aspects of DNA metabolism (64), and ATR-interacting protein (ATRIP) to centers of viral DNA replication (6, 52). This localization is consistent with the recognition of single-stranded DNA within viral replication centers by cellular proteins. In addition, Blackford et al. have demonstrated a role for ATR in phosphorylation of RPA32 and H2AX during adenovirus infection (6). Recently, Carson et al. have shown that ATR is activated by adenovirus infection and that this activation is modulated by E4orf3 (10). Our results obtained with the kinase inhibitors caffeine and wortmannin are consistent with a role for ATR in H2AX phosphorylation during adenovirus infection. The requirement of viral DNA replication demonstrated in the present study and the localization of ATR and associated proteins to viral replication centers shown by others suggests a role for single-stranded DNA structures underlying the adenovirus-induced phosphorylation of H2AX. Activation of these pathways may be partially mediated by the cellular E1B-AP5 protein, an interacting partner of the viral E1B-55K protein (22). E1B-AP5 localizes to viral replication centers; binds to ATR/ATRIP, RPA, and adenovirus DBP in vivo; and is required for phosphorylation of RPA32 during adenovirus infection (6). This phosphorylation is at least partially mediated by ATR during infection with the species C type 5 virus, and is ATR dependent during infection with species A type 12 virus. Thus, E1B-AP5 may act to promote the activation and localization of ATR during adenovirus infection, resulting in the phosphorylation of various DNA damage response components.
A number of viruses activate cellular DNA damage pathways and either use these pathways for their own replication or subvert the response to ensure efficient replication (60). Murine gammaherpesvirus 68, Epstein-Barr virus, and human immunodeficiency virus type 1 (HIV-1) all activate pathways that result in the phosphorylation of H2AX (16, 55). For gammaherpesvirus 68 and HIV-1, activation is an important step in replication; gammaherpesvirus 68 uses cellular factors for circularization of the genome prior to replication (55), and HIV-1 uses cellular factors to complete integration of the provirus into cellular DNA (16). In contrast, Epstein-Barr virus replication activates DNA damage response pathways, but activation does not appear to be required for lytic viral replication (31). Although it is well understood that DNA double-stranded break repair is inhibitory to adenovirus replication, it is not yet clear whether activation of ATM and ATR pathways and the subsequent phosphorylation of H2AX has any effect on viral replication.
Since adenovirus effectively prevents genome concatenation, it is likely that H2AX phosphorylation has little effect on the outcome of virus infection in most cells. However, such widespread H2AX phosphorylation could lead to the inappropriate recruitment of DNA repair factors to cellular DNA. Phosphorylated H2AX colocalizes with a number of cellular proteins involved in repair of double-stranded DNA breaks such as Mdc1, MRN (reviewed in reference 54), and 53BP1 (59). Recruitment of repair proteins to damaged DNA is delayed in H2AX−/− cells (5, 59). These cells are also hypersensitive to ionizing radiation and exhibit mild checkpoint defects, demonstrating the importance of the specific phosphorylation of this histone to DNA repair processes (5, 12, 19). The role of H2AX in the control of homologous recombination is not as clear as in double-stranded break repair. However, sister chromatid exchange was demonstrated to be decreased ∼4-fold in H2AX−/− cell lines using a recombination reporter assay (61). Under replication stresses leading to stalled replication forks, H2AX phosphorylation occurs at sites of replication arrest and colocalizes with Rad51 and BRCA1, both of which are involved in both double-stranded break and recombination repair (42, 58).
Since γH2AX foci recruit recombination and repair proteins to areas of DNA damage, it is possible that the widespread H2AX phosphorylation seen during adenovirus infection could result in improper recruitment of DNA damage responders. This could be manifested as either the recruitment of repair proteins to undamaged cellular DNA or an impaired ability to recruit repair proteins to bona fide sites of DNA damage. Either scenario could be detrimental. The first circumstance could lead to spurious repair on undamaged DNA potentially through the homologous recombination pathway, while the second scenario could lead to the perpetuation and accumulation of double-stranded DNA breaks within the cellular chromatin as nonhomologous end joining is blocked by adenovirus infection. Because epithelial cells typically die as a result of infection, inappropriate cellular DNA repair in cells that permit a fully lytic infection would have little bearing on the fate of these cells. However, human lymphocytes can successfully harbor a productive adenovirus infection while continuing to divide and survive (D. Ornelles and L. R. Gooding, unpublished observations). We have observed nuclear-wide γH2AX in these cell types as well (data not shown). Perhaps in precursor lymphoid cells, adenovirus infection could favor the accumulation of lesions associated with inappropriate DNA repair or the persistence of double-strand DNA breaks and by this means may provide a molecular basis underlying the potential association between prenatal adenovirus infection and the development of acute lymphoblastic leukemia (26).
Acknowledgments
This study was supported by Public Health Service grant CA77342 from the National Cancer Institute to D.A.O. G.J.N. was supported during part of this study by Public Health Service grant T32AI007401 from the National Institute of Allergy and Infectious Diseases to the Department of Microbiology and Immunology of the Wake Forest University School of Medicine. Cell culture reagents were provided by the Cell and Virus Vector Core Laboratory of the Comprehensive Cancer Center of Wake Forest University, which is supported in part by the National Cancer Institute grant CA12197.
Footnotes
Published ahead of print on 25 March 2009.
REFERENCES
- 1.Andreassen, P. R., G. P. Ho, and A. D. D'Andrea. 2006. DNA damage responses and their many interactions with the replication fork. Carcinogenesis 27883-892. [DOI] [PubMed] [Google Scholar]
- 2.Araujo, F. D., T. H. Stracker, C. T. Carson, D. V. Lee, and M. D. Weitzman. 2005. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 7911382-11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker, A., K. J. Rohleder, L. A. Hanakahi, and G. Ketner. 2007. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 817034-7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421499-506. [DOI] [PubMed] [Google Scholar]
- 5.Bassing, C. H., K. F. Chua, J. Sekiguchi, H. Suh, S. R. Whitlow, J. C. Fleming, B. C. Monroe, D. N. Ciccone, C. Yan, K. Vlasakova, D. M. Livingston, D. O. Ferguson, R. Scully, and F. W. Alt. 2002. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA 998173-8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackford, A. N., R. K. Bruton, O. Dirlik, G. S. Stewart, A. M. Taylor, T. Dobner, R. J. Grand, and A. S. Turnell. 2008. A role for E1B-AP5 in ATR signaling pathways during adenovirus infection. J. Virol. 827640-7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyer, J., K. Rohleder, and G. Ketner. 1999. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263307-312. [DOI] [PubMed] [Google Scholar]
- 8.Brand, K., R. Klocke, A. Possling, D. Paul, and M. Strauss. 1999. Induction of apoptosis and G2/M arrest by infection with replication-deficient adenovirus at high multiplicity of infection. Gene Ther. 61054-1063. [DOI] [PubMed] [Google Scholar]
- 9.Burma, S., B. P. Chen, M. Murphy, A. Kurimasa, and D. J. Chen. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 27642462-42467. [DOI] [PubMed] [Google Scholar]
- 10.Carson, C. T., N. I. Orazio, D. V. Lee, J. Suh, S. Bekker-Jenson, F. D. Araujo, S. S. Lakdawala, C. E. Lilley, J. Bartek, J. Lukas, and M. D. Weitzman. 2009. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 28652-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carson, C. T., R. A. Schwartz, T. H. Stracker, C. E. Lilley, D. V. Lee, and M. D. Weitzman. 2003. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 226610-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Celeste, A., S. Petersen, P. J. Romanienko, O. Fernandez-Capetillo, H. T. Chen, O. A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M. J. Difilippantonio, C. Redon, D. R. Pilch, A. Olaru, M. Eckhaus, R. D. Camerini-Otero, L. Tessarollo, F. Livak, K. Manova, W. M. Bonner, M. C. Nussenzweig, and A. Nussenzweig. 2002. Genomic instability in mice lacking histone H2AX. Science 296922-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherubini, G., T. Petouchoff, M. Grossi, S. Piersanti, E. Cundari, and I. Saggio. 2006. E1B55K-deleted adenovirus (ONYX-015) overrides G1/S and G2/M checkpoints and causes mitotic catastrophe and endoreduplication in p53-proficient normal cells. Cell Cycle 52244-2252. [DOI] [PubMed] [Google Scholar]
- 14.Costanzo, V., and J. Gautier. 2003. Single-strand DNA gaps trigger an ATR- and Cdc7-dependent checkpoint. Cell Cycle 217. [DOI] [PubMed] [Google Scholar]
- 15.Cuconati, A., C. Mukherjee, D. Perez, and E. White. 2003. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 172922-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniel, R., J. Ramcharan, E. Rogakou, K. D. Taganov, J. G. Greger, W. Bonner, A. Nussenzweig, R. A. Katz, and A. M. Skalka. 2004. Histone H2AX is phosphorylated at sites of retroviral DNA integration but is dispensable for postintegration repair. J. Biol. Chem. 27945810-45814. [DOI] [PubMed] [Google Scholar]
- 17.Evans, J. D., and P. Hearing. 2003. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 775295-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans, J. D., and P. Hearing. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 796207-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Capetillo, O., H. T. Chen, A. Celeste, I. Ward, P. J. Romanienko, J. C. Morales, K. Naka, Z. Xia, R. D. Camerini-Otero, N. Motoyama, P. B. Carpenter, W. M. Bonner, J. Chen, and A. Nussenzweig. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4993-997. [DOI] [PubMed] [Google Scholar]
- 20.Fleisig, H. B., N. I. Orazio, H. Liang, A. F. Tyler, H. P. Adams, M. D. Weitzman, and L. Nagarajan. 2007. Adenoviral E1B55K oncoprotein sequesters candidate leukemia suppressor sequence-specific single-stranded DNA-binding protein 2 into aggresomes. Oncogene 264797-4805. [DOI] [PubMed] [Google Scholar]
- 21.Flint, S. J., S. M. Berget, and P. A. Sharp. 1976. Characterization of single-stranded viral DNA sequences present during replication of adenovirus types 2 and 5. Cell 9559-571. [DOI] [PubMed] [Google Scholar]
- 22.Gabler, S., H. Schutt, P. Groitl, H. Wolf, T. Shenk, and T. Dobner. 1998. E1B 55-kilodalton-associated protein: a cellular protein with RNA-binding activity implicated in nucleocytoplasmic transport of adenovirus and cellular mRNAs. J. Virol. 727960-7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garnett, C. T., C. I. Pao, and L. R. Gooding. 2007. Detection and quantitation of subgroup C adenovirus DNA in human tissue samples by real-time PCR. Methods Mol. Med. 130193-204. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg, M., M. Stucki, J. Falck, D. D'Amours, D. Rahman, D. Pappin, J. Bartek, and S. P. Jackson. 2003. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421952-956. [DOI] [PubMed] [Google Scholar]
- 25.Goldsmith, K. T., L. D. Dion, D. T. Curiel, and R. I. Garver, Jr. 1998. trans E1 component requirements for maximal replication of E1-defective recombinant adenovirus. Virology 248406-419. [DOI] [PubMed] [Google Scholar]
- 26.Gustafsson, B., W. Huang, G. Bogdanovic, F. Gauffin, A. Nordgren, G. Talekar, D. A. Ornelles, and L. R. Gooding. 2007. Adenovirus DNA is detected at increased frequency in Guthrie cards from children who develop acute lymphoblastic leukaemia. Br. J. Cancer 97992-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammond, E. M., S. L. Green, and A. J. Giaccia. 2003. Comparison of hypoxia-induced replication arrest with hydroxyurea and aphidicolin-induced arrest. Mutat. Res. 532205-213. [DOI] [PubMed] [Google Scholar]
- 28.Harada, J. N., A. Shevchenko, D. C. Pallas, and A. J. Berk. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 769194-9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang, M. M., and P. Hearing. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol. 632605-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kastan, M. B., and D. S. Lim. 2000. The many substrates and functions of ATM. Nat. Rev. Mol. Cell. Biol. 1179-186. [DOI] [PubMed] [Google Scholar]
- 31.Kudoh, A., M. Fujita, L. Zhang, N. Shirata, T. Daikoku, Y. Sugaya, H. Isomura, Y. Nishiyama, and T. Tsurumi. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 2808156-8163. [DOI] [PubMed] [Google Scholar]
- 32.Lavelle, G., C. Patch, G. Khoury, and J. Rose. 1975. Isolation and partial characterization of single-stranded adenoviral DNA produced during synthesis of adenovirus type 2 DNA. J. Virol. 16775-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, E., D. Stupack, R. Klemke, D. A. Cheresh, and G. R. Nemerow. 1998. Adenovirus endocytosis via alpha (v) integrins requires phosphoinositide-3-OH kinase. J. Virol. 722055-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lieber, M. R., Y. Ma, U. Pannicke, and K. Schwarz. 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell. Biol. 4712-720. [DOI] [PubMed] [Google Scholar]
- 35.Liu, Y., A. Shevchenko, and A. J. Berk. 2005. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 7914004-14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukas, C., F. Melander, M. Stucki, J. Falck, S. Bekker-Jensen, M. Goldberg, Y. Lerenthal, S. P. Jackson, J. Bartek, and J. Lukas. 2004. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 232674-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathew, S. S., and E. Bridge. 2008. Nbs1-dependent binding of Mre11 to adenovirus E4 mutant viral DNA is important for inhibiting DNA replication. Virology 37411-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathew, S. S., and E. Bridge. 2007. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365346-355. [DOI] [PubMed] [Google Scholar]
- 39.Miller, D. L., C. L. Myers, B. Rickards, H. A. Coller, and S. J. Flint. 2007. Adenovirus type 5 exerts genome-wide control over cellular programs governing proliferation, quiescence, and survival. Genome Biol. 8R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ornelles, D. A., and T. Shenk. 1991. Localization of the adenovirus early region 1B 55-kilodalton protein during lytic infection: association with nuclear viral inclusions requires the early region 4 34-kilodalton protein. J. Virol. 65424-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park, E. J., D. W. Chan, J. H. Park, M. A. Oettinger, and J. Kwon. 2003. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 316819-6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paull, T. T., E. P. Rogakou, V. Yamazaki, C. U. Kirchgessner, M. Gellert, and W. M. Bonner. 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10886-895. [DOI] [PubMed] [Google Scholar]
- 43.Querido, E., P. Blanchette, Q. Yan, T. Kamura, M. Morrison, D. Boivin, W. G. Kaelin, R. C. Conaway, J. W. Conaway, and P. E. Branton. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 153104-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogakou, E. P., D. R. Pilch, A. H. Orr, V. S. Ivanova, and W. M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 2735858-5868. [DOI] [PubMed] [Google Scholar]
- 45.Sarkaria, J. N., E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz, and R. T. Abraham. 1999. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 594375-4382. [PubMed] [Google Scholar]
- 46.Sarkaria, J. N., R. S. Tibbetts, E. C. Busby, A. P. Kennedy, D. E. Hill, and R. T. Abraham. 1998. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 584375-4382. [PubMed] [Google Scholar]
- 47.Schaack, J. 2005. Adenovirus vectors deleted for genes essential for viral DNA replication. Front. Biosci. 101146-1155. [DOI] [PubMed] [Google Scholar]
- 48.Shenk, T. 1996. Adenoviridae: the viruses and their replication, p. 2111-2148. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, PA.
- 49.Steinwaerder, D. S., C. A. Carlson, and A. Lieber. 2000. DNA replication of first-generation adenovirus vectors in tumor cells. Hum. Gene Ther. 111933-1948. [DOI] [PubMed] [Google Scholar]
- 50.Stewart, G. S., B. Wang, C. R. Bignell, A. M. Taylor, and S. J. Elledge. 2003. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421961-966. [DOI] [PubMed] [Google Scholar]
- 51.Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418348-352. [DOI] [PubMed] [Google Scholar]
- 52.Stracker, T. H., D. V. Lee, C. T. Carson, F. D. Araujo, D. A. Ornelles, and M. D. Weitzman. 2005. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J. Virol. 796664-6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stucki, M., J. A. Clapperton, D. Mohammad, M. B. Yaffe, S. J. Smerdon, and S. P. Jackson. 2005. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 1231213-1226. [DOI] [PubMed] [Google Scholar]
- 54.Stucki, M., and S. P. Jackson. 2006. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 5534-543. [DOI] [PubMed] [Google Scholar]
- 55.Tarakanova, V. L., V. Leung-Pineda, S. Hwang, C. W. Yang, K. Matatall, M. Basson, R. Sun, H. Piwnica-Worms, B. P. Sleckman, and H. W. T. Virgin. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1275-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van der Vliet, P. C. 1995. Adenovirus DNA replication. Curr. Top. Microbiol. Immunol. 199(Pt. 2)1-30. [DOI] [PubMed] [Google Scholar]
- 57.Wang, H., M. Wang, W. Bocker, and G. Iliakis. 2005. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell Physiol. 202492-502. [DOI] [PubMed] [Google Scholar]
- 58.Ward, I. M., and J. Chen. 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 27647759-47762. [DOI] [PubMed] [Google Scholar]
- 59.Ward, I. M., K. Minn, K. G. Jorda, and J. Chen. 2003. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 27819579-19582. [DOI] [PubMed] [Google Scholar]
- 60.Weitzman, M. D., C. T. Carson, R. A. Schwartz, and C. E. Lilley. 2004. Interactions of viruses with the cellular DNA repair machinery. DNA Repair 31165-1173. [DOI] [PubMed] [Google Scholar]
- 61.Xie, A., N. Puget, I. Shim, S. Odate, I. Jarzyna, C. H. Bassing, F. W. Alt, and R. Scully. 2004. Control of sister chromatid recombination by histone H2AX. Mol. Cell 161017-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang, J., Y. Yu, H. E. Hamrick, and P. J. Duerksen-Hughes. 2003. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis 241571-1580. [DOI] [PubMed] [Google Scholar]
- 63.Zou, L., and S. J. Elledge. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 3001542-1548. [DOI] [PubMed] [Google Scholar]
- 64.Zou, Y., Y. Liu, X. Wu, and S. M. Shell. 2006. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J. Cell Physiol. 208267-273. [DOI] [PMC free article] [PubMed] [Google Scholar]