

Abstract
During acute infection, West Nile virus (WNV) has been reported to infect a variety of cell types in various tissues of both experimentally and naturally infected hosts. Virus infects epithelial cells in the skin, kidney, intestine, and testes, although the importance of these findings is unclear. In the current study, we have observed that WNV infection of kidney tubules in mice coincides with the loss of expression of several members of the claudin family. Proteins of this family are often involved in epithelial barrier formation and function. WNV infection of epithelial cells in culture resulted in a decrease in the transepithelial electrical resistance, an increase in the efflux of mannitol across the monolayer, and a loss of intracellular levels of claudin-1 to -4. WNV capsid alone was sufficient for the degradation event, which was mediated through lysosomal proteases. Since epithelial cells are frequent sites of WNV infection, these observations imply a potential mechanism for virus dissemination and extraneural pathogenesis.
West Nile Virus (WNV) is a mosquito-borne flavivirus that first appeared in the United States in 1999. Since that time, the virus has spread across the continental United States, causing significant morbidity and mortality. Roughly 20% of WNV infections are symptomatic, and approximately 1 out of every 150 infections progresses to encephalitis and/or meningitis (39, 42). Following the bite of a carrier mosquito, WNV infection of the host is thought to initiate in the Langerhans cells of the skin (13). Viral replication continues in the regional tissue and lymph nodes, which results in the dissemination of the virus into the bloodstream. Replication then proceeds at several sites throughout the host, including the kidneys, heart, connective tissue, smooth muscle, spleen, and ultimately the brain (46). Infection of the nervous system is characteristic of the most severe cases of WNV disease, often resulting in death or long-term neurologic sequelae (26). Pathologies associated with extraneural sites of infection have also been reported, including acute renal failure (11, 24).
Epithelial cells are major targets of WNV infection in vivo in both humans and experimentally infected rodents. In a hamster model of WNV infection, virus can be detected in the epithelial cells of the kidney up to 60 days postinfection, suggesting that this tissue may be a site of viral persistence (53). Infection of kidney epithelium has also been found in WNV-infected mice (20) and dogs (12). In WNV-infected humans, epithelial cell infection has also been demonstrated in several organs, including the kidney, lung, pancreas, thyroid, intestine, and testes (4). These studies suggest that the epithelial cells may play an important role in WNV pathogenesis. However, the growth characteristics of WNV in epithelial cells and the effect of WNV on the polarity and permeability functions of polarized epithelial cells have not been investigated. One important feature of epithelial cells is the presence of tight junctions (TJ). TJ are intercellular contacts between endothelial or epithelial cells which allow the formation of polarized cells with discrete apical and basolateral plasma membrane domains and separate physiologically distinct apical and basolateral fluid compartments (22). Macromolecules larger than 30 Å in diameter are generally excluded from the TJ, but permeability to small ions varies depending on tissue-specific requirements and local physiological stimuli (2, 23). TJ are thus not static seals but dynamic structures subject to transient changes in permeability (30). The TJ is composed of the transmembrane proteins occludin, claudin (a family of more than 20 members), and junctional adhesion molecule (a family of four known members). These proteins mediate cell-cell interactions and regulate junctional permeability. The transmembrane proteins interact on their cytoplasmic side with several other components of the TJ. These include zonula occludens 1, 2, and 3 (ZO-1 to -3, which belong to a family of membrane-associated guanylate kinase homologues). ZO-1 is thought to nucleate the initial formation of the TJ and to provide a scaffold for TJ assembly. This process is thought to require the interaction of ZO-1 with the adherens junction (1, 7).
In this study, we show that WNV infection of epithelial cells in vivo and in vitro results in a loss of claudin protein expression. The loss of claudin expression coincides with perturbation of the permeability functions of cultured epithelial cells as measured by a reduction in transepithelial electrical resistance (TER) and an increase in the efflux of [14C]mannitol across the cell monolayer. Expression of WNV capsid alone was sufficient to mediate these events. Epithelial cells are likely to be targets for WNV infection from the earliest times of exposure (e.g., the keratinocytes at the site of the mosquito bite) and also may be sites of long-term persistence. Therefore, the effect of the virus on the TJ, a crucial component of epithelial function, suggests a possible mechanism of virus spread, as well as potentially contributing to pathogenesis.
MATERIALS AND METHODS
Cell culture and virus stocks.
Caco-2 cells were grown in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate (Invitrogen). C6/36 mosquito cells were grown in the above medium at 28°C. The origin of WNV strain 385-99 used in this study has been described (35, 62). Infectious virus was propagated twice on Vero cells and once in C6/36 cells. Virus stock was concentrated and purified by centrifugation through a 20% sorbitol cushion at 28,000 rpm for 1.5 h in an SW28 rotor (Beckman). Following centrifugation, virus was resuspended in 1/100 of the original volume in DMEM supplemented with 0.1% bovine serum albumin, and aliquots were stored at −80°C. The virus titer was determined by plaque assay. Generation of WNV virus-like particles (WNV-VLPs) has been described previously (17, 36). WNV-VLP titers (infectious units/ml) were determined on Vero cells. For UV inactivation, virus was exposed to 254 nm UV light for 10 min in a Stratalinker 1800 UV cross-linker (Stratagene). The absence of WNV gene expression in cells infected with UV-irradiated virus was verified by immunofluorescent staining (data not shown).
Infection of transwell cultures and measurement of TER.
Caco-2 cells (4 × 105) were seeded onto 12-mm diameter, 3-μm-pore-size polycarbonate filters (Costar) and fed at 3-day intervals. TER of the monolayers was measured daily using an Evom epithelial voltohmeter with an Endohm-12 chamber (World Precision Instruments). Monolayers were allowed to attain maximum TER values before infection with WNV (typically 250 to 300 Ω·cm2). Cells were infected at a multiplicity of 10 PFU/cell. Virus-containing medium was added to the upper chamber of the transwell inserts (100 μl) and medium without virus was added to the lower chamber (1 ml), and cells were incubated at 37°C on the rocker for 2 h. Virus-containing medium was removed, and cells were treated with citrate acid buffer (40 mM Na citrate, 135 mM NaCl, 10 mM KCl, pH 3.2) for 1 min washed twice in phosphate-buffered saline (PBS), and refed with normal growth medium (0.5 ml in the upper chamber, 1.5 ml in the lower chamber).
Indirect immunofluorescence.
Forty-eight h postinfection (p.i.), transwell inserts were washed twice with PBS and incubated with methanol at −20°C for 5 min, followed by washing with PBS. Cells were further incubated with IF wash solution (PBS with Ca2+ and Mg2+ containing 0.1% Triton X-100 and 0.2% bovine serum albumin) for 5 min at room temperature (RT), and all further washes were performed with IF wash solution. Nonspecific antibody binding sites were blocked by incubating with IF wash solution containing 5% normal goat serum for 10 min at RT. Cells were washed three times, followed by incubation with 1:100 dilutions of anti-E (purified supernatant from hybridoma D1-4G2-4-15; ATCC), rabbit anti-claudin-1 (Invitrogen), mouse anti-ZO-1 (BD Biosciences), and rabbit antioccludin (Invitrogen) for 1 h at RT, followed by Alexafluor-conjugated secondary antibodies for 1 h at RT. Membranes were separated from the transwell supports using a razor blade, mounted on glass slides in Prolong antifade reagent with 4′,6′-diamidino-2-phenylindole (Invitrogen) to stain the nuclei, and covered with coverslips. Samples were left to dry for 24 h at RT. Images were acquired as described previously (35). Projections of appropriate Z stacks are shown.
Measurement of mannitol flux.
Mannitol flux was measured largely as described previously (34). Cells were infected as described above with WNV. At 48 h p.i., medium in the bottom compartment was replaced with 1.5 ml of DMEM supplemented with 2% FBS and 1 mM cold mannitol. The top compartment was replaced with 0.5 ml of the same medium containing 0.2 μCi [14C]mannitol (51 mCi/mmol; Perkin Elmer Life Sciences). At 0, 60, 120, and 180 min, 50-μl duplicate aliquots were removed from the bottom compartment and the medium was replenished with 100 μl fresh DMEM-2% FBS-1 mM mannitol. Samples were mixed with 3 ml EcoLite scintillation fluid (ICN Biomedicals) and counted in a Beckman LS5000 TD liquid scintillation counter. The rate of mannitol flux was determined by linear regression analysis using Microsoft's Excel software program.
VLP experiments.
Cells were infected with 10 IU/cell of VLPs and TER monitored as described above. At 48 h p.i., cells were fixed and processed for immunofluorescence as described above except that the cells were stained with monoclonal antibody specific for NS1 (22NS1, a kind gift from Michael Diamond) instead of the WNV E-specific antibody.
Removal of virus from culture supernatants.
Cells were infected as described above, and culture supernatant was collected at 48 h p.i. To remove infectious virus, supernatants were centrifuged at 28,000 rpm for 2.5 h in a Beckman SW28 rotor. In general, 99.9% of virus was removed by centrifugation. In order to neutralize remaining virus, a monoclonal antibody specific for E (E16, a kind gift from Michael Diamond [43]) was added to a final concentration of 10 μM, which has been shown to be sufficient for virus neutralization. Removal of virus from culture supernatants was verified by immunofluorescent staining and plaque assay.
Pulse-chase and Western blotting.
For pulse-chase assays, cells were grown in 35-mm dishes to confluence for 2 days and infected with 10 PFU/cell of WNV as described above. Forty-two h p.i., cells were washed twice with PBS and incubated in DMEM without methionine and cystine containing 5% dialyzed FBS for one hour, followed by incubation with the same medium containing 250 μCi/ml 35S-EasyTag Express protein labeling mix (Perkin-Elmer) for 1 h. Cells were washed twice with PBS and incubated with normal growth medium containing excess of methionine and cysteine (100 μg/ml). At 0, 2.5, 5 and 10 h postlabeling, cells were washed twice with cold PBS on ice and frozen at −80°C. Cell lysis was performed by scraping cells into 500 μl of RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS] with aprotinin, leupeptin, pepstatin, and 1 mM phenylmethylsulfonylfluoride). Lysates were incubated on ice for 10 min and centrifuged at 12,000 × g for 15 min at 4°C. Supernatants were incubated with 2 μg of rabbit immunoglobulin G and 20 μl of protein A Sepharose beads for 2 h at 4°C on a rotator. Samples were centrifuged at 500 × g for 5 min to pellet the beads. Precleared lysates were incubated overnight with 1 μg of rabbit claudin-1 antibody (Invitrogen), followed by incubation with 20 μl of protein A Sepharose beads for 2 h at 4°C. Beads were pelleted as described above, and supernatant was used for immunoprecipitation with rabbit antioccludin antibody (Invitrogen) as described above. Beads were washed twice with lysis buffer and once with Tris-buffered saline, boiled in 1× Laemmli buffer, and resolved on a 12% SDS-polyacrylamide gel electrophoresis (PAGE) (for claudin-1) or 10% SDS-PAGE (for occludin). Gels were transferred to Immobilon-polyvinylidene difluoride (PVDF) (Millipore) membranes for 2 h. Membranes were exposed on films for autoradiography and analyzed by Western blotting by probing with claudin-1 and occludin antibody to determine the efficiency of immunoprecipitation. Signal intensities of the autoradiograph and Western blots were quantified using the IPLab Gel H software program (Analytics Corp.).
For direct analysis of protein expression, cells were lysed in RIPA buffer as described above and proteins resolved by SDS-PAGE followed by transfer to Immobilon-PVDF membranes and Western blotting by standard protocols. Where indicated, bafilomycin A1 (Sigma) was added to cultures at 24 h p.i. to a final concentration of 200 nM. Antibodies to claudin-1 to -4 and occludin were obtained from Invitrogen, anti-β actin was from Sigma, and anti-NS3 and -NS5 polyclonal antisera were provided by Ian Lipkin, Columbia University.
Quantitation of claudin-1 mRNA.
Caco-2 cells were infected at a multiplicity of 10, and RNA was harvested at 48 h p.i. using the TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Reverse transcription and quantitative PCR were performed with One-Step RT-PCR master mix (Applied Biosystems) in an ABI Prism 7700 sequence detector. Primer and probe sets specific for claudin-1 and β-actin were obtained from Applied Biosystems.
Capsid-expressing cell lines.
The capsid coding sequence without the C- terminal transmembrane domain was amplified from a full-length WNV cDNA clone (48) using forward (5′-GGAAGGATCCATGTCTAAGAAACCAGGAGGG-3′) and reverse (5′-GCCCTCTAGACTTCTTTTCTTTTGTTTTGAGCTCCGCCG-3′) primers using PCR and Expand HiFi polymerase (Roche). The amplified product was cloned into the BamHI and XbaI sites of the expression vector pEF1myc-his-B (Invitrogen). MDCK cells were transfected by electroporation using a Bio-Rad gene pulser with a 0.4-cm cuvette at 260 V, 960 μF. Cells were selected in 600 μg/ml G418 starting at 48 h posttransfection. Individual colonies were ring cloned and further purified with two additional rounds of subcloning.
Infection and analysis of mice.
B6.129S7-Rag1tm1Mom/J mice (38) were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred at the VGTI vivarium (Oregon Health & Science University); they were used at 12 weeks of age. All animals were housed and bred under specific-pathogen-free conditions at the Oregon Health & Science University. All WNV experiments were completed within a U.S. Department of Agriculture-approved biosafety level 3 facility and were approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee in accordance with the applicable federal, state, and local regulations. Mice were infected with 250 PFU WNV subcutaneously. At 10 to 16 days p.i., when mice appeared moribund, they were sacrificed and individual organs harvested and fixed in Streck tissue fixative (Streck Laboratories). WNV-infected cells were detected by immunofluorescent staining with rabbit anti-WNV antiserum and claudin-2 and occludin antibodies as described previously (4).
Statistical analysis.
The GraphPad Prism and Microsoft Excel software programs were used for graphical representations and statistical analyses. P values were obtained from a two-tailed, unpaired Student t test.
RESULTS
WNV infection of kidney tubule epithelial cells results in loss of claudin in infected mice.
Analyses of tissues obtained from WNV-infected humans, hamsters, or mice have revealed that epithelial cells are important targets of viral infection (4, 20, 51) in such organs as the kidney, intestine, testes, and bile duct. In these tissues, epithelial cells form TJ, resulting in barrier formation. In order to determine if TJ protein expression is disrupted during WNV infection in vivo, we examined staining of claudins and occludin in infected kidney tubules in mice. We have observed staining of the kidney tubule epithelium in both rag1−/− knockout and immunocompetent (C57BL/6) mice (Fig. 1 and data not shown). The rag1−/− knock out mice are homozygous null for recombination activation gene 1 (rag1) and cannot effect V(D)J recombination of immunoglobulin or T-cell receptor genes (38). In order to separate the direct effect of the virus on TJ protein expression from exogenous effects exerted by T cells or other components of the adaptive immune response, we examined TJ protein and WNV antigen localization in infected rag1−/− mice. In this model, animals develop terminal disease at 9 to 12 days. At that time, animals show a fairly widespread infection, including infection of most neurons except for cerebellar granular neurons (data not shown). There is a broad distribution of other infected organs, similar to that reported in autopsies of infected immunosuppressed humans (4). Immunofluorescent staining of kidney sections using antibodies to WNV demonstrate occasional infected renal tubules (Fig. 1). Various claudins compose portions of TJ in different portions of the nephron. In particular, claudin-2 is expressed predominantly in the proximal tubules (25). Double-label immunofluorescent staining for claudin-2 and WNV antigens demonstrated that in most cases, WNV-infected tubules demonstrated an absence of claudin-2 staining (Fig. 1A). Quantitation of claudin-2 and WNV staining in regions of infection showed that 75% of uninfected tubules stained positive for claudin-2 while 0% of WNV-infected tubules are claudin-2 positive (Table 1). We also examined the localization of other claudin family members in infected kidney sections. Claudin-1 is found primarily in Bowman's capsule, in which we have not observed any evidence of WNV epithelial infection. Claudin-3 is found predominantly in distal segments of the nephron. Double-label immunofluorescent staining for claudin-3 and WNV antigens demonstrated that in most cases, similar to the case with claudin-2, WNV-infected tubules demonstrated an absence of claudin-3 staining (data not shown); however, rarely both WNV and claudin-3 antigens colocalized in epithelia. Although we have not stained for all 24 members of the claudin family in WNV-infected tissues due to technical limitations, our data suggest that WNV-infected tubules showed a loss of (at least some members of) claudin family proteins. In contrast, the TJ protein occludin was present at the luminal surfaces of both WNV-infected and uninfected kidney tubules (Fig. 1 and Table 1), demonstrating that not every TJ-associated protein has been disrupted. In some cases, a qualitative difference in occludin staining was observed, suggesting that less occludin is present in infected tubules. This may suggest some effect of WNV infection on occludin expression, although the limits of this assay prevent us from drawing quantitative conclusions beyond the presence or absence of specific proteins.
FIG. 1.

Claudin-2 expression is decreased in infected kidney tubule epithelial cell in vivo. Kidney sections derived from rag1−/− mice were doubly labeled with anti-WNV and anti-claudin-2 (A) or anti-occludin (B). Single arrows indicate WNV-positive tubules showing an absence of claudin-2 staining. Double arrows indicate claudin-2 stain at the apical surface of uninfected tubules. In panel B, single arrows point to infected tubules that retain occludin staining. Details of an infected tubule (white box, bottom left) and an uninfected tubule (yellow box, bottom right) are shown, with luminal occludin staining indicated by arrows.
TABLE 1.
Claudin-2 staining in WNV-positive and -negative kidney tubulesa
| Protein assayed | WNV antigen staining result | No. of tubules counted | No. (%) claudin-2 positive | No. (%) occludin positive | P value |
|---|---|---|---|---|---|
| Claudin-2 | Positive | 17 | 0 (0) | <0.001 | |
| Negative | 102 | 77 (75) | |||
| Occludin | Positive | 41 | 31 (76) | 0.8755 | |
| Negative | 88 | 70 (79.5) |
Sections from kidneys of WNV-infected rag1−/− mice (n = 3) were stained for WNV antigen and claudin-2 or occludin as described in Methods. Individual tubules were scored for the presence of WNV and claudin-2 or occludin staining. Statistical significance was determined by the Cochran-Mantel-Haenzel test of independence.
WNV infection of polarized Caco-2 epithelial cells results in loss of barrier function.
We next assessed the effect of WNV on the permeability barrier functions of Caco-2 epithelial cells. Caco-2 cells grown on transwell filters support high levels of WNV growth and demonstrate preferential virus entry on the apical surface (data not shown). In order to characterize the effect of WNV infection on the epithelial permeability barrier, cells were infected with WNV and TER was measured at the indicated time points p.i. WNV infection was found to compromise the barrier functions, as observed by a drop in the TER at 48 h p.i. (Fig. 2A). Exposure of cells to UV-inactivated virus did not result in significant changes in TER, demonstrating that viral protein expression or replication is required for TJ disruption (Fig. 2A). We also determined the functional effect of TER disruption in WNV-infected cells by measuring the efflux of [14C]mannitol across the monolayer. Compared to mock-infected control cells, WNV-infected monolayers showed a 12-fold increase in the rate of mannitol efflux across the epithelial barrier (Fig. 2B). Cell viability assays showed no difference between the control and infected cells at 48 h p.i., suggesting that the observed effect on TER is not due to a cytopathic effect of the virus (Fig. 2C).
FIG. 2.

WNV affects epithelial barrier functions. (A) Caco-2 cells were infected with WNV or UV-inactivated WNV (MOI of 10 PFU/cell), and TER was monitored at the indicated times p.i. Error bars represent means ± SD. Data are representative of three or more experiments performed with triplicate samples. (B) Caco-2 cells were infected at a multiplicity of 10 PFU/cell, and permeability to mannitol was assayed at 48 h p.i. as described in Methods. The results shown represent the average for three independent infections ± SD. *, P = 5 × 10−6. (C) Caco-2 cells grown and infected as described above were trypsinized at 48 h p.i., and cell viability was determined by trypan blue exclusion. A total of 500 cells were counted in duplicate samples of each. Error bars represent means ± SEM. (D) Supernatants (sup) from mock-, WNV- or WNV-VLP-infected cells were collected at 48 h p.i., and virus was removed as described in Materials and Methods. Cleared supernatants were added to naive cells. TER was monitored at the indicated times postaddition. Error bars represent means ± SD. Data are representative of two experiments performed with triplicate samples.
Secreted factors such as cytokines have been implicated in the disruption of epithelial TJ (32, 55). Additionally, dengue virus, a flavivirus related to WNV, has been reported to affect endothelial TJ permeability in vitro via the production of interleukin 8 (50) and monocyte chemoattractant protein 1 (28). To determine if the observed effect of WNV on epithelial barrier functions is a paracrine effect due to the secreted factors in the media of infected cells, we collected culture supernatants from mock- or WNV-infected cells at 48 h p.i., and virus was removed through centrifugation and addition of neutralizing antibody. Virus-free culture supernatant was transferred onto naive cells grown on transwells. If cytokines and other factors in the media were responsible for TJ disruption, we would expect to see a reduction in the TER values. However, we did not observe any effect on the TER values when cells were incubated with the WNV-infected media for up to 48 h (Fig. 2D). Additionally, we infected cells with WNV-VLPs that contain a genome deleted for the prM and E coding regions packaged by WNV structural proteins provided by a helper virus (17). These particles are therefore able to infect cells but unable to make infectious progeny. As shown, supernatants from cells infected with these VLP also have no effect on TER. Therefore, in contrast to dengue virus infection of endothelial cells, WNV-mediated disruption of epithelial barrier functions seems to be a direct effect of virus infection and not due to the action of secreted inflammatory cytokines or other factors.
WNV infection of epithelial cells induces relocalization and degradation of claudin proteins.
The barrier functions of polarized epithelial cells are mainly attributed to the presence of functional TJ at the apical-basolateral interface of these cells. Therefore, we examined the integrity of TJs by analyzing the localization of TJ proteins in mock-infected and WNV- infected cells by indirect immunofluorescence. In agreement with our in vivo observations, WNV infection disrupted the localization of the transmembrane TJ-associated protein claudin-1 (Fig. 3A). In contrast, localization of the TJ proteins ZO-1, occludin, and JAM-A was unchanged following WNV infection (Fig. 3B and C and data not shown). Additionally, localization of β-catenin, a protein associated with the cytoplasmic face of the adherens junctions, was also unaffected (data not shown). We next prepared total cell lysates from mock- and WNV-infected cells and determined the levels of claudin-1 and ZO-1 by Western blot analysis. Claudin-1 levels were reduced by 90% in infected cells compared to those for controls. ZO-1 protein levels were not altered further, demonstrating that not all TJ proteins are degraded in infected cells (Fig. 4A). Claudin-1 degradation was also observed in the WNV-infected MDCK (Fig. 4B) and HaCat (data not shown) cell lines, which are derived from epithelia of the kidney and skin, respectively. Analysis of claudin mRNA levels in infected Caco-2 cells showed equivalent amounts of claudin-1 mRNA in infected and uninfected cells at 48 h p.i., demonstrating that reduction of claudin protein levels is not due to effects on claudin mRNA transcription or stability (data not shown).
FIG. 3.

WNV disrupts claudin-1 localization. Caco-2 cells were infected at a multiplicity of 10 PFU/cell, and localization of claudin-1 (A), ZO-1 (B), or occludin (C) was analyzed by indirect immunofluorescence at 48 h p.i. using WNV E (green), claudin-1, ZO-1, or occludin (red) antibody. Nuclei were visualized by Hoescht staining (blue). Scale bar: 10 μm.
FIG. 4.

WNV causes degradation of claudin family proteins. Control and WNV-infected Caco-2 (A) or MDCK (B) cell lysates were analyzed by Western blotting to determine the levels of claudin-1, ZO-1, and NS5 (Caco-2) or claudin-1, β-actin, and NS3 (MDCK). (C) Control and WNV-infected Caco-2 cell lysates were analyzed by Western blotting to determine the levels of claudin-2, -3, and -4, occludin, NS3, and β-actin. (D) Control and WNV-infected cells were metabolically labeled with [35S]methionine and chased for the indicated times. Claudin-1 was immunoprecipitated from the lysates, and supernatants of claudin-1 immunoprecipitates were used for occludin immunoprecipitation. Protein complexes were resolved by SDS-PAGE and transferred onto PVDF membranes. Claudin-1 and occludin half-lives were quantified by autoradiography. The positions of the molecular weight marker bands are shown for both gels; an arrow points to the occludin band. Western blot analysis of the membranes with respective antibodies confirmed equal amounts of protein immunoprecipitation in all samples.
The claudin family consists of at least 24 known members (54). In order to determine if the WNV-mediated effect on claudin stability extends to other members of the family, particularly those that were absent from WNV-infected epithelial cells in vivo, we examined changes in protein levels of claudin-2, -3, and -4 (all of which are expressed in Caco-2 cells) following WNV infection. As shown in Fig. 4C, levels of each of these claudins are reduced in cells infected with WNV relative to the levels in uninfected cells. These data demonstrate that several claudin family members are degraded during WNV infection. To determine if WNV infection was affecting the stability of claudin-1, we performed metabolic labeling and pulse-chase analysis of claudin-1 from cell lysates prepared from 35S-labeled control and WNV-infected cells. We observed a marked increase in the rate of claudin-1 degradation in WNV-infected cells (Fig. 4D). While there was a 15% reduction in the amount of claudin-1 at the 10-h chase period in control cells compared to that at time zero hours, claudin-1 levels were decreased by about 60% in WNV- infected lysates, indicating that WNV infection increased the turnover rates of claudin-1. The half-life of occludin was not significantly affected in the observed periods of chase in our experiments (Fig. 4D). Western blot analysis of the membranes showed that equal amounts of claudin-1 and occludin were immunoprecipitated at all time points in control and WNV-infected lysates; however, consistent with the pulse-chase results, claudin-1 levels were about 60% less in WNV-infected cells than in uninfected cells. Claudin family members have been demonstrated to affect TJ-mediated barrier function by regulation of paracellular permeability to small ions (3), and claudin-1 has been shown to be essential for establishment of proper barrier function in mammalian epidermis (18). Therefore, our results suggest that the effect of WNV on epithelial barrier functions could be due in part to the relocalization and degradation of claudin family members in infected cells. However, because we have not examined the effect of WNV on every TJ component protein, we cannot rule out the possibility that other TJ proteins are disrupted during WNV infection and contribute to barrier dysfunction.
WNV capsid is sufficient to disrupt barrier functions.
In order to identify the viral protein(s) responsible for the disruption of barrier functions and claudin degradation, cells were infected with WNV or WNV-VLPs. The WNV-VLPs contain a genome that lacks the region coding for the prM and E proteins [VLP(−prME)]) or the C, prM, and E proteins [WLP(−CprME)]. The genomes are packaged in cell lines producing high levels of WNV structural proteins (17). We initially infected cells with both types of VLPs and measured TER 48 h p.i., followed by fixation and staining for NS1 and TJ proteins. WNV-VLPs were capable of disrupting the barrier functions, as observed by a drop in the TER values (Fig. 5A) compared to those for mock-infected cells, but TER reduction was significantly impaired (i.e., TER was higher) in cells infected with VLP(−CprME) than in those infected with VLP(−prME) or intact WNV. Similarly to WNV infection, claudin-1 localization was altered in cells infected with both types of VLP (Fig. 5B and data not shown) and ZO-1 staining remained unchanged (data not shown). However, significantly more claudin remained in VLP(−CprME)-infected cells than in WNV- or VLP(−prME)-infected cells (Fig. 5C), suggesting that capsid may play a role in the degradation, but possibly not in the mislocalization, of claudins. We also observed that C expression is reduced in VLP(−prME) relative to that in WNV (data not shown), which may contribute to the inability of VLP(−prME) to degrade claudin to the same extent as intact WNV.
FIG. 5.

WNV-VLPs are capable of disrupting TJ functions. (A) Caco-2 cells were infected with WNV or WNV-VLPs deleted for the prME or CprME regions of the genome at a multiplicity of 10 PFU/cell. TER was monitored at 48 h p.i. Error bars represent means ± SD. Data are representative of two experiments performed with triplicate samples. (B) Caco-2 cells were infected with WNV-VLPs (−prME) as described above, and localization of claudin-1 was analyzed by indirect immunofluorescence at 48 h p.i. using WNV-NS1 (green) or claudin-1 (red) antibody. Nuclei were visualized by Hoescht staining (blue). Scale bar, 10 μm. (C) Left panels: protein lysates were harvested from control, WNV-infected, or VLP(−prME) at 48 h p.i. Right panels: protein lysates were harvested from control, WNV-infected, or VLP(−CprME) at 48 h p.i. Lysates were analyzed for claudin-1, NS3, C, and β-actin by Western blotting. The band intensity for claudin-1 was quantified, and relative claudin-1 expression is shown.
We next examined claudin-1 levels in MDCK cells stably transfected with a plasmid vector expressing the soluble form of the WNV C protein. As shown in Fig. 6A, claudin-1 levels in two independently derived C-expressing lines were <90% of the levels in a cell line stably transfected with the empty expression vector, while β-actin and occludin levels were similar in the C-expressing and vector cell lines. Additionally, we examined the ability of these cell lines to establish an intact barrier. TER was measured from cells plated on transwells at 24 and 48 h postseeding. TER established by the C-expressing clone was approximately 10% of that in control transwells (Fig. 6B).
FIG. 6.
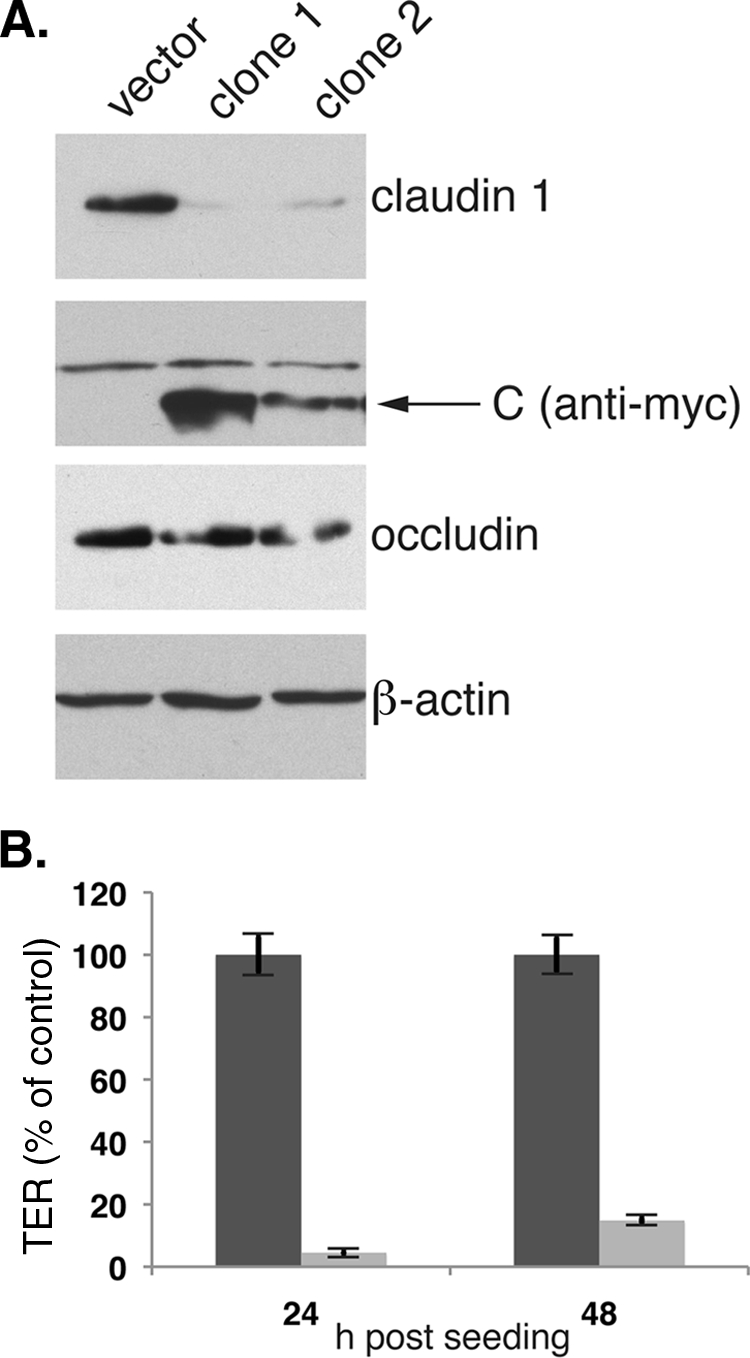
WNV C is sufficient to induce claudin degradation. (A) Total cell lysates from cell lines expressing Myc-tagged WNV C (clones 1 and 2) or the empty expression vector were analyzed by Western blotting for claudin-1, C, occludin, and β-actin expression. The arrow indicates the specific band for Cmyc. (B) cells (4 × 105) were plated on transwell filters and TER measured at 24 and 48 h postseeding. Values are the averages for three transwells for each cell line ± SD.
WNV causes lysosomal degradation of claudin.
There are several pathways that may be involved in the degradation of claudins in WNV-infected cells (63). One possibility is the targeted ubiquitination of the claudins, followed by proteosome-dependent degradation. In order to test this possibility, we treated WNV-infected Caco-2 cells with the proteosome inhibitor MG132 at 24 h p.i. and harvested cell lysates at 48 h p.i. Analysis of claudin-1 levels in the infected or control lysates showed that MG132 treatment did not reduce claudin degradation in infected cells (data not shown). We next tested the possibility that claudins are targeted to the lysosome and degraded by lysosomal proteases. Examination of claudin remaining in infected cells showed colocalization with LAMP-2, a marker of late endosomes and lysosomes, suggesting that this pathway may be involved in claudin degradation (Fig. 7A). In order to further confirm the role of lysosomes in this process, we used bafilomycin A1, an inhibitor of the vacuolar ATPase proton pump, which prevents endosome/lysosome acidification and subsequent action by resident acid proteases (10). Because increasing the endosomal pH is expected to interfere with viral entry (14), bafilomycin A1 was not added until 24 h p.i. Cells were lysed and claudin-1 levels were examined at 48 h p.i. As shown in Fig. 7B, infected cells treated with bafilomycin A1 show levels of claudin-1 similar to the levels in uninfected cells, suggesting that claudin-1 is degraded via the lysosomal pathway. We next tested if claudin expression in C-expressing cells can be rescued by treatment with bafilomycin A1. As expected, bafilomycin-treated cells show levels of claudin-1 comparable to those of the empty-vector control (Fig. 7C). Therefore, the lysosomal pathway mediates the degradation of claudin in C-expressing cell lines as in WNV-infected cells.
FIG. 7.

Lysosomes are involved in WNV-mediated claudin degradation. (A) Caco-2 cells were infected at a multiplicity of 10 PFU/cell and localization of LAMP-2 (green), claudin-1 (red), and E (red in far right panels) determined. Nuclei were visualized by Hoescht staining (blue). Scale, 15 μm. The highlighted area was rotated 90° using the Softworx imaging software program and is shown below to highlight colocalization of LAMP-2 and claudin-1 along the Y plane. (B) Caco-2 cells were infected with WNV at a multiplicity of 10. At 24 h p.i., cells were treated with bafilomycin A1 (BAF) at a final concentration of 200 nM or dimethyl sulfoxide. At 48 h p.i., cell lysates were prepared and analyzed by Western blotting. cldn-1, claudin-1. (C) C-expressing cell line (clone 1) or a cell line made with the empty expression vector was grown to confluence and treated with 200 nM bafilomycin A1 for 24 h. Cell lysates were prepared and analyzed by Western blotting.
DISCUSSION
We have shown that WNV infection of epithelial cells resulted in degradation of several claudin family proteins in vivo and in vitro, with corresponding disruption of the permeability barrier. Degradation of the TJ proteins occludin and ZO-1 and the adherens junction protein β-catenin was not observed following WNV infection, demonstrating that this effect is selective for a subset of TJ proteins, including members of the claudin family, although it remains to be determined if other TJ resident proteins are also degraded. Perturbation of lysosome acidification by bafilomycin A1 inhibited the degradation of claudin-1, implicating lysosomal proteases in this process. WNV-VLPs that lack the C protein are impaired in their ability to degrade (but not mislocalize) claudin compared to that of WNV- or C-containing VLPs. Additionally, the C protein alone was capable of mediating claudin degradation, suggesting that C is involved in targeting claudin proteins to the lysosome. We have noted that following infection of cells with WNV-VLP(−CprME), some residual claudin degradation is still observed. This may indicate that the C protein present in the packaged VLP may contribute to claudin degradation as well. Alternatively, C may be partially redundant with one or more nonstructural proteins with regard to claudin degradation. In vitro, TJ disruption was observed at 48 h p.i. The delay in the loss of TER may be due to the possibility that while the half-life of claudin-1 in infected cells is significantly shortened, sufficient claudin-1 may remain in the cells to maintain barrier function until 48 h p.i.
The TJ is a highly regulated structure that can alter permeability based on inter- and intracellular signals (27, 33). Such stimuli as cytokines, calcium depletion, and bacterial toxins have been shown to induce internalization of subsets of TJ proteins through multiple pathways (reviewed in reference 63). A number of viruses infecting polarized epithelial cells have been shown to use proteins of the TJ as receptors for entry (6, 9), and among flaviviruses, hepatitis C virus has recently been shown to use claudin-1 at late stages in entry (16, 37). Additionally, claudin-6 and -9 have also been shown to function as coreceptors for entry into endothelial cells (37). Recently the hepatitis C virus envelope protein E2 was shown to interact with occludin and cause its retention in the endoplasmic reticulum, leading to altered TJ organization (8). Disruption of TJs at later stages of viral infection has been hypothesized to facilitate viral egress, as has been observed in the cases of adenovirus (57) and rotavirus (49). Interestingly, several enveloped viruses that infect polarized epithelial cells exit from these cells in a manner that does not compromise TJ integrity (for example, Crimean-Congo hemorrhagic fever virus [15], respiratory syncytial virus [45], and vaccinia virus [56]). Therefore, we speculate that TJ disruption by WNV may not be a requirement for viral egress but rather may have a role in viral replication and dissemination.
Several viruses have been demonstrated to disrupt with TJ, either directly through contact of viral proteins with the TJ or indirectly through paracrine effectors. Proteins of TJ complex have been shown to be the targets of various viral proteins. Simian virus 40 small-T antigen specifically affects TJs, but not adherens junctions, by causing altered distribution and reduction in the amounts of the TJ proteins ZO-1, occludin, and claudin-1 (41) in MDCK cells, possibly playing a role in the transformation of epithelial cells by simian virus 40. Human immunodeficiency virus type 1 (HIV-1) gp120 has been shown to cause proteasomal degradation of the TJ proteins ZO-1 and ZO-2, leading to disruption of barrier functions in human brain microvascular endothelial cells (40). In similarity to our observations, expression of HIV-1 Tat in cultured human retinal pigment epithelial cells increased the permeability and decreased the TER of the epithelial monolayer. HIV-1 Tat also disrupted and downregulated the TJ proteins claudin-1, claudin-3, and claudin-4 in these cells, whereas the expression of occludin was unaffected (5).
In our study, we show that the WNV C protein is able to mediate disruption of TJ in epithelial cells. The mechanism of TJ disruption by C and the role it plays in WNV pathogenesis remain to be characterized. Previous observations have implicated exogenous cytokines in increasing the permeability of endothelial TJ during flavivirus infection (31, 50, 58). Disruption of TJs by WNV capsid is therefore likely to act in conjunction with cytokine-mediated effects to contribute to WNV pathogenesis in vivo, suggesting that there is a complex interplay of multiple factors that contribute to WNV pathogenesis.
Previously published data from naturally and experimentally infected mammals demonstrate that epithelial cells are major targets of WNV infection in vivo. It is likely that epithelial cells will be infected soon after the bite of a carrier mosquito (i.e., keratinocytes [29]). In infected mice, replication of the virus in kidney tubules has been demonstrated before the virus can be detected in the brain (20), and in humans and hamsters, virus can be detected in the epithelial cells of the kidney and other tissues for weeks p.i. (4, 53). Replication in epithelial cells, therefore, is likely to be important for virus dissemination throughout the organism. Recent observations have demonstrated that WNV can be shed in the urine of infected mammals (44, 51-53, 60). Replication of the virus in the kidney and the perturbation of barrier function may contribute to this phenomenon, suggesting a potential mechanism for viral persistence and spread.
In addition, epithelial-cell involvement has been documented for infected mosquitoes and birds, the natural hosts for WNV (21, 47, 59, 61, 64). It is possible that TJ disruption by WNV has evolved to allow spread of the virus through the epithelium of the salivary glands and midgut of the mosquito host. The septate junctions of insect epithelium are functionally homologous to mammalian TJ and contain several proteins homologous to mammalian TJ proteins (19). Therefore, disruption of mammalian TJ may be a by-product of the normal WNV life cycle which nonetheless has important implications for viral replication in the mammalian host.
Acknowledgments
We thank W. Ian Lipkin for generously providing us with the polyclonal NS3 and NS5 antibodies, Aurelie Snyder for her assistance with deconvolution microscopy, Guoji Wang for help with immunofluorescent staining of tissue sections, and Byung Park for statistical analysis. We thank members of the Nelson lab for useful discussions.
This project has been funded in whole or in part by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN266200500027C.
Footnotes
Published ahead of print on 15 April 2009.
REFERENCES
- 1.Aijaz, S., M. S. Balda, and K. Matter. 2006. Tight junctions: molecular architecture and function. Int. Rev. Cytol. 248261-298. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, J. M., and C. M. Van Itallie. 1995. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 269G467-G475. [DOI] [PubMed] [Google Scholar]
- 3.Angelow, S., R. Ahlstrom, and A. S. Yu. 2008. Biology of claudins. Am. J. Physiol. Renal Physiol. 295F867-F876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armah, H. B., G. Wang, B. I. Omalu, R. B. Tesh, K. A. Gyure, D. J. Chute, R. D. Smith, P. Dulai, H. V. Vinters, B. K. Kleinschmidt-DeMasters, and C. A. Wiley. 2007. Systemic distribution of West Nile virus infection: postmortem immunohistochemical study of six cases. Brain Pathol. 17354-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai, L., Z. Zhang, H. Zhang, X. Li, Q. Yu, H. Lin, and W. Yang. 2008. HIV-1 Tat protein alter the tight junction integrity and function of retinal pigment epithelium: an in vitro study. BMC Infect. Dis. 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barton, E. S., J. C. Forrest, J. L. Connolly, J. D. Chappell, Y. Liu, F. J. Schnell, A. Nusrat, C. A. Parkos, and T. S. Dermody. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104441-451. [DOI] [PubMed] [Google Scholar]
- 7.Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84869-901. [DOI] [PubMed] [Google Scholar]
- 8.Benedicto, I., F. Molina-Jimenez, O. Barreiro, A. Maldonado-Rodriguez, J. Prieto, R. Moreno-Otero, R. Aldabe, M. Lopez-Cabrera, and P. L. Majano. 2008. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 481044-1053. [DOI] [PubMed] [Google Scholar]
- 9.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 2751320-1323. [DOI] [PubMed] [Google Scholar]
- 10.Bowman, E. J., A. Siebers, and K. Altendorf. 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 857972-7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brener, Z. Z., N. B. Harbord, I. Zhuravenko, A. D. Nicastri, M. Bergman, A. Dubrow, D. Feinfeld, and J. Winchester. 2007. Acute renal failure in a patient with West Nile viral encephalitis. Nephrol. Dial. Transplant. 22662-663. [DOI] [PubMed] [Google Scholar]
- 12.Buckweitz, S., S. Kleiboeker, K. Marioni, J. Ramos-Vara, A. Rottinghaus, B. Schwabenton, and G. Johnson. 2003. Serological, reverse transcriptase-polymerase chain reaction, and immunohistochemical detection of West Nile virus in a clinically affected dog. J. Vet. Diagn. Investig. 15324-329. [DOI] [PubMed] [Google Scholar]
- 13.Byrne, S. N., G. M. Halliday, L. J. Johnston, and N. J. King. 2001. Interleukin-1β but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Investig. Dermatol. 117702-709. [DOI] [PubMed] [Google Scholar]
- 14.Chu, J. J., and M. L. Ng. 2004. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J. Virol. 7810543-10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connolly-Andersen, A. M., K. E. Magnusson, and A. Mirazimi. 2007. Basolateral entry and release of Crimean-Congo hemorrhagic fever virus in polarized MDCK-1 cells. J. Virol. 812158-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans, M. J., T. von Hahn, D. M. Tscherne, A. J. Syder, M. Panis, B. Wolk, T. Hatziioannou, J. A. McKeating, P. D. Bieniasz, and C. M. Rice. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446801-805. [DOI] [PubMed] [Google Scholar]
- 17.Fayzulin, R., F. Scholle, O. Petrakova, I. Frolov, and P. W. Mason. 2006. Evaluation of replicative capacity and genetic stability of West Nile virus replicons using highly efficient packaging cell lines. Virology 351196-209. [DOI] [PubMed] [Google Scholar]
- 18.Furuse, M., M. Hata, K. Furuse, Y. Yoshida, A. Haratake, Y. Sugitani, T. Noda, A. Kubo, and S. Tsukita. 2002. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J. Cell Biol. 1561099-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuse, M., and S. Tsukita. 2006. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 16181-188. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Tapia, D., D. E. Hassett, W. J. Mitchell, Jr., G. C. Johnson, and S. B. Kleiboeker. 2007. West Nile virus encephalitis: sequential histopathological and immunological events in a murine model of infection. J. Neurovirol. 13130-138. [DOI] [PubMed] [Google Scholar]
- 21.Girard, Y. A., V. Popov, J. Wen, V. Han, and S. Higgs. 2005. Ultrastructural study of West Nile virus pathogenesis in Culex pipiens quinquefasciatus (Diptera: Culicidae). J. Med. Entomol. 42429-444. [DOI] [PubMed] [Google Scholar]
- 22.Gumbiner, B. 1987. Structure, biochemistry, and assembly of epithelial tight junctions. Am. J. Physiol. 253C749-v758. [DOI] [PubMed] [Google Scholar]
- 23.Gumbiner, B. M. 1993. Breaking through the tight junction barrier. J. Cell Biol. 1231631-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang, C., B. Slater, R. Rudd, N. Parchuri, R. Hull, M. Dupuis, and A. Hindenburg. 2002. First isolation of West Nile virus from a patient with encephalitis in the United States. Emerg. Infect. Dis. 81367-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiuchi-Saishin, Y., S. Gotoh, M. Furuse, A. Takasuga, Y. Tano, and S. Tsukita. 2002. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J. Am. Soc. Nephrol. 13875-886. [DOI] [PubMed] [Google Scholar]
- 26.Klee, A. L., B. Maidin, B. Edwin, I. Poshni, F. Mostashari, A. Fine, M. Layton, and D. Nash. 2004. Long-term prognosis for clinical West Nile virus infection. Emerg. Infect. Dis. 101405-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kniesel, U., and H. Wolburg. 2000. Tight junctions of the blood-brain barrier. Cell Mol. Neurobiol. 2057-76. [DOI] [PubMed] [Google Scholar]
- 28.Lee, Y. R., M. T. Liu, H. Y. Lei, C. C. Liu, J. M. Wu, Y. C. Tung, Y. S. Lin, T. M. Yeh, S. H. Chen, and H. S. Liu. 2006. MCP-1, a highly expressed chemokine in dengue haemorrhagic fever/dengue shock syndrome patients, may cause permeability change, possibly through reduced tight junctions of vascular endothelium cells. J. Gen. Virol. 873623-3630. [DOI] [PubMed] [Google Scholar]
- 29.Limon-Flores, A. Y., M. Perez-Tapia, I. Estrada-Garcia, G. Vaughan, A. Escobar-Gutierrez, J. Calderon-Amador, S. E. Herrera-Rodriguez, A. Brizuela-Garcia, M. Heras-Chavarria, A. Flores-Langarica, L. Cedillo-Barron, and L. Flores-Romo. 2005. Dengue virus inoculation to human skin explants: an effective approach to assess in situ the early infection and the effects on cutaneous dendritic cells. Int. J. Exp. Pathol. 86323-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lum, H., and A. B. Malik. 1994. Regulation of vascular endothelial barrier function. Am. J. Physiol. 267L223-l241. [DOI] [PubMed] [Google Scholar]
- 31.Luplertlop, N., D. Misse, D. Bray, V. Deleuze, J. P. Gonzalez, V. Leardkamolkarn, H. Yssel, and F. Veas. 2006. Dengue-virus-infected dendritic cells trigger vascular leakage through metalloproteinase overproduction. EMBO Rep. 71176-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma, T. Y., G. K. Iwamoto, N. T. Hoa, V. Akotia, A. Pedram, M. A. Boivin, and H. M. Said. 2004. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 286G367-G376. [DOI] [PubMed] [Google Scholar]
- 33.Matter, K., and M. S. Balda. 2003. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4225-236. [DOI] [PubMed] [Google Scholar]
- 34.McCarthy, K. M., S. A. Francis, J. M. McCormack, J. Lai, R. A. Rogers, I. B. Skare, R. D. Lynch, and E. E. Schneeberger. 2000. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J. Cell Sci. 1133387-3398. [DOI] [PubMed] [Google Scholar]
- 35.Medigeshi, G. R., A. J. Hirsch, D. N. Streblow, J. Nikolich-Zugich, and J. A. Nelson. 2008. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of αvβ3 integrin. J. Virol. 825212-5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medigeshi, G. R., A. M. Lancaster, A. J. Hirsch, T. Briese, W. I. Lipkin, V. Defilippis, K. Fruh, P. W. Mason, J. Nikolich-Zugich, and J. A. Nelson. 2007. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 8110849-10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meertens, L., C. Bertaux, L. Cukierman, E. Cormier, D. Lavillette, F. L. Cosset, and T. Dragic. 2008. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 823555-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mombaerts, P., J. Iacomini, R. S. Johnson, K. Herrup, S. Tonegawa, and V. E. Papaioannou. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68869-877. [DOI] [PubMed] [Google Scholar]
- 39.Mostashari, F., M. L. Bunning, P. T. Kitsutani, D. A. Singer, D. Nash, M. J. Cooper, N. Katz, K. A. Liljebjelke, B. J. Biggerstaff, A. D. Fine, M. C. Layton, S. M. Mullin, A. J. Johnson, D. A. Martin, E. B. Hayes, and G. L. Campbell. 2001. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet 358261-264. [DOI] [PubMed] [Google Scholar]
- 40.Nakamuta, S., H. Endo, Y. Higashi, A. Kousaka, H. Yamada, M. Yano, and H. Kido. 2008. Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J. Neurovirol. 14186-195. [DOI] [PubMed] [Google Scholar]
- 41.Nunbhakdi-Craig, V., L. Craig, T. Machleidt, and E. Sontag. 2003. Simian virus 40 small tumor antigen induces deregulation of the actin cytoskeleton and tight junctions in kidney epithelial cells. J. Virol. 772807-2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petersen, L. R., and A. A. Marfin. 2002. West Nile virus: a primer for the clinician. Ann. Intern. Med. 137173-179. [DOI] [PubMed] [Google Scholar]
- 43.Pierson, T. C., Q. Xu, S. Nelson, T. Oliphant, G. E. Nybakken, D. H. Fremont, and M. S. Diamond. 2007. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1135-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platt, K. B., B. J. Tucker, P. G. Halbur, B. J. Blitvich, F. G. Fabiosa, K. Mullin, G. R. Parikh, P. Kitikoon, L. C. Bartholomay, and W. A. Rowley. 2008. Fox squirrels (Sciurus niger) develop West Nile virus viremias sufficient for infecting select mosquito species. Vector Borne Zoonotic Dis. 8225-233. [DOI] [PubMed] [Google Scholar]
- 45.Roberts, S. R., R. W. Compans, and G. W. Wertz. 1995. Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J. Virol. 692667-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samuel, M. A., and M. S. Diamond. 2006. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 809349-9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scholle, F., Y. A. Girard, Q. Zhao, S. Higgs, and P. W. Mason. 2004. trans-packaged West Nile virus-like particles: infectious properties in vitro and in infected mosquito vectors. J. Virol. 7811605-11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi, P. Y., M. Tilgner, M. K. Lo, K. A. Kent, and K. A. Bernard. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 765847-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Svensson, L., B. B. Finlay, D. Bass, C. H. von Bonsdorff, and H. B. Greenberg. 1991. Symmetric infection of rotavirus on polarized human intestinal epithelial (Caco-2) cells. J. Virol. 654190-4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talavera, D., A. M. Castillo, M. C. Dominguez, A. E. Gutierrez, and I. Meza. 2004. IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. J. Gen. Virol. 851801-1813. [DOI] [PubMed] [Google Scholar]
- 51.Tesh, R. B., M. Siirin, H. Guzman, A. P. Travassos da Rosa, X. Wu, T. Duan, H. Lei, M. R. Nunes, and S. Y. Xiao. 2005. Persistent West Nile virus infection in the golden hamster: studies on its mechanism and possible implications for other flavivirus infections. J. Infect. Dis. 192287-295. [DOI] [PubMed] [Google Scholar]
- 52.Tonry, J. H., C. B. Brown, C. B. Cropp, J. K. Co, S. N. Bennett, V. R. Nerurkar, T. Kuberski, and D. J. Gubler. 2005. West Nile virus detection in urine. Emerg. Infect. Dis. 111294-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tonry, J. H., S. Y. Xiao, M. Siirin, H. Chen, A. P. da Rosa, and R. B. Tesh. 2005. Persistent shedding of West Nile virus in urine of experimentally infected hamsters. Am. J. Trop. Med. Hyg. 72320-324. [PubMed] [Google Scholar]
- 54.Turksen, K., and T. C. Troy. 2004. Barriers built on claudins. J. Cell Sci. 1172435-2447. [DOI] [PubMed] [Google Scholar]
- 55.Utech, M., M. Bruwer, and A. Nusrat. 2006. Tight junctions and cell-cell interactions. Methods Mol. Biol. 341185-195. [DOI] [PubMed] [Google Scholar]
- 56.Vermeer, P. D., J. McHugh, T. Rokhlina, D. W. Vermeer, J. Zabner, and M. J. Welsh. 2007. Vaccinia virus entry, exit, and interaction with differentiated human airway epithelia. J. Virol. 819891-9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walters, R. W., P. Freimuth, T. O. Moninger, I. Ganske, J. Zabner, and M. J. Welsh. 2002. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 110789-799. [DOI] [PubMed] [Google Scholar]
- 58.Wang, Y., M. Lobigs, E. Lee, and A. Mullbacher. 2004. Exocytosis and Fas mediated cytolytic mechanisms exert protection from West Nile virus induced encephalitis in mice. Immunol. Cell Biol. 82170-173. [DOI] [PubMed] [Google Scholar]
- 59.Weingartl, H. M., J. L. Neufeld, J. Copps, and P. Marszal. 2004. Experimental West Nile virus infection in blue jays (Cyanocitta cristata) and crows (Corvus brachyrhynchos). Vet. Pathol. 41362-370. [DOI] [PubMed] [Google Scholar]
- 60.Wu, X., L. Lu, H. Guzman, R. B. Tesh, and S. Y. Xiao. 2008. Persistent infection and associated nucleotide changes of West Nile virus serially passaged in hamsters. J. Gen. Virol. 893073-3079. [DOI] [PubMed] [Google Scholar]
- 61.Wunschmann, A., J. Shivers, L. Carroll, and J. Bender. 2004. Pathological and immunohistochemical findings in American crows (Corvus brachyrhynchos) naturally infected with West Nile virus. J. Vet. Diagn. Investig. 16329-333. [DOI] [PubMed] [Google Scholar]
- 62.Xiao, S. Y., H. Guzman, H. Zhang, A. P. Travassos da Rosa, and R. B. Tesh. 2001. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg. Infect. Dis. 7714-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu, D., and J. R. Turner. 2008. Stimulus-induced reorganization of tight junction structure: the role of membrane traffic. Biochim. Biophys. Acta 1778709-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang, Z., F. Wilson, R. Read, L. Pace, and S. Zhang. 2006. Detection and characterization of naturally acquired West Nile virus infection in a female wild turkey. J. Vet. Diagn. Investig. 18204-208. [DOI] [PubMed] [Google Scholar]