

Abstract
Nonstructural protein 4B (NS4B) plays an essential role in the formation of the hepatitis C virus (HCV) replication complex. It is a relatively poorly characterized integral membrane protein predicted to comprise four transmembrane segments in its central portion. Here, we describe a novel determinant for membrane association represented by amino acids (aa) 40 to 69 in the N-terminal portion of NS4B. This segment was sufficient to target and tightly anchor the green fluorescent protein to cellular membranes, as assessed by fluorescence microscopy as well as membrane extraction and flotation analyses. Circular dichroism and nuclear magnetic resonance structural analyses showed that this segment comprises an amphipathic α-helix extending from aa 42 to 66. Attenuated total reflection infrared spectroscopy and glycosylation acceptor site tagging revealed that this amphipathic α-helix has the potential to traverse the phospholipid bilayer as a transmembrane segment, likely upon oligomerization. Alanine substitution of the fully conserved aromatic residues on the hydrophobic helix side abrogated membrane association of the segment comprising aa 40 to 69 and disrupted the formation of a functional replication complex. These results provide the first atomic resolution structure of an essential membrane-associated determinant of HCV NS4B.
With 120 to 180 million chronically infected individuals worldwide, hepatitis C virus (HCV) infection represents a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (38). HCV contains a 9.6-kb positive-strand RNA genome that encodes a polyprotein of about 3,000 amino acids (reviewed in references 36 and 51). The polyprotein precursor is co- and posttranslationally processed by cellular and viral proteases to yield the mature structural and nonstructural proteins. The structural proteins include the core and the envelope glycoproteins E1 and E2. The nonstructural proteins include the p7 ion channel polypeptide, the NS2-3 and NS3-4A proteases, an RNA helicase located in the C-terminal two-thirds of NS3, the NS4B and NS5A proteins, and the NS5B RNA-dependent RNA polymerase. HCV replication takes place in a membrane-associated complex, composed of viral proteins, replicating RNA, altered cellular membranes, and other host factors (7, 18, 31, 43). Determinants for membrane association of the HCV nonstructural proteins have been mapped and a likely endoplasmic reticulum (ER)-derived membrane alteration, designated the membranous web, was found to harbor the HCV replication complex (7, 18; reviewed in reference 36).
NS4B is a 27-kDa integral ER membrane protein (21). The expression of NS4B alone induces the formation of the membranous web (7). Thus, an essential function of NS4B is the induction of the specific membrane alteration that serves as a scaffold for the HCV replication complex. In addition, a nucleotide-binding motif has been proposed to reside in the middle of NS4B (8), and RNA binding properties have recently been reported for NS4B (9).
Both the N and the C termini of NS4B are believed to be oriented toward the cytosol, and prediction algorithms indicate the presence of four putative transmembrane segments in the central portion of the protein (21, 27, 28, 42). The cytosolic orientation of the bulk of the protein was confirmed experimentally (21), but a more refined membrane topology is so far elusive. The introduction of glycosylation acceptor sites at various positions in NS4B validated the prediction of ER luminal loops around amino acid positions 112 and 161 (27, 28). Intriguingly, the N terminus of NS4B was reported to be translocated into the ER lumen at least partially, presumably by a posttranslational mechanism (28). Interestingly, the coexpression of the other HCV proteins appears to limit this translocation (27). A recent report indicates that NS4B is palmitoylated at C-terminal residues Cys 257 and Cys 261 and forms oligomers (59).
In order to define the membrane topology of NS4B we have analyzed a comprehensive panel of green fluorescent protein (GFP) fusion constructs comprising different segments of NS4B (N. Arora, V. Castet, and D. Moradpour, unpublished data). In the course of these studies, we unexpectedly found that a fusion construct comprising the N-terminal 74 amino acids (aa) of NS4B was associated with membranes, while most prediction methods located the beginning of the first transmembrane segment around aa 74 (21, 28, 42). Here, we demonstrate that an amphipathic α-helix extending from aa 42 to 66 (α-helix 42-66) in the N-terminal portion of NS4B mediates this membrane association and plays an essential role in the formation of the HCV replication complex.
MATERIALS AND METHODS
Sequence analyses and structure predictions.
Sequence analyses were performed using tools available at the Institut de Biologie et Chimie des Protéines (IBCP) Network Protein Sequence Analysis (NPSA) website (http://npsa-pbil.ibcp.fr) (5). HCV NS4B sequences were retrieved from the European HCV Database (http://euhcvdb.ibcp.fr/) (6). Multiple-sequence alignments were performed with ClustalW (52) using default parameters. The repertoire of residues at each amino acid position and their frequencies observed in natural sequence variants were computed by the use of a program developed at the IBCP (F. Dorkeld, C. Combet, F. Penin, and G. Deléage, unpublished data). Protein secondary structures were deduced from a large set of prediction methods available at the NPSA website, including DSC, HNNC, SIMPA96, MLRC, SOPM, PHD, and Predator (see http://npsa-pbil.ibcp.fr/NPSA and references therein). Interfacial hydrophobicity plots were generated with MPEx (http://blanco.biomol.uci.edu/mpex/) by using the scale developed by Wimley and White (55). The sequence of the HCV H77 consensus clone (24) (GenBank accession number AF009606) was used to predict the secondary structure and membrane binding properties of the N-terminal portion of NS4B.
Plasmids.
All NS4B fragments were derived from the HCV H77 consensus clone present in pBRTM/HCV1-3011con (24) (kindly provided by Charles M. Rice, the Rockefeller University, New York, NY). Primers NS4B-1-fwd and NS4B-40-fwd as well as NS4B-40-rev, NS4B-69-rev, NS4B-74-rev, and NS4B-261-rev (Table 1) were used to amplify full-length NS4B or fragments corresponding to aa 1 to 74, 1 to 40, and 40 to 69. The amplification products were digested with EcoRI and BamHI and cloned into pCMVKEB-GFP (2), yielding GFP fusion constructs pCMVNS4B-GFP, pCMVNS4B1-74-GFP, pCMVNS4B1-40-GFP, and pCMVNS4B40-69-GFP, respectively.
TABLE 1.
Oligonucleotide sequences
| Oligonucleotide | Restriction site | Sequencea |
|---|---|---|
| NS4B-1-fwd | EcoRI | 5′ ATTCGAATTCACCATGTCTCAGCACTTACCGTAC 3′ |
| NS4B-40-fwd | EcoRI | 5′ ATTCGAATTCACCATGCAGACCAAC 3′ |
| NS4B-40-rev | BamHI | 5′ ATTCGGATCCCTGGACAGCAGGGGTGATAAC 3′ |
| NS4B-69-rev | BamHI | 5′ ATTCGGATCCCGTTGACAGGCCCGC 3′ |
| NS4B-74-rev | BamHI | 5′ TTCGGATCCGGGGTTACCAGGCAGCGT 3′ |
| NS4B-261-rev | BamHI | 5′ TGAACCGGATCCGCATGGAGTGGTACA 3′ |
| AH2mut-rev | NdeI | 5′ ATTCCACATATGCTTCGCCGCGGCGACCTCGAGTTTCTGCGCGTT 3′ |
| AH2mut-fwd | NdeI | 5′ GCGAAGCATATGGCGAATGCCATCAGTGGGATACAAGCCTTG 3′ |
| HCV5275-fwd | 5′ AGATGTGGAAGTGTT 3′ | |
| HCV6819-rev | 5′ ATTCGGGCGATGGGATC 3′ | |
| repAH2mut-fwd | 5′ CGAAGCCGCCGCGGCGAAGCATATGGCGAATGCCATCAGCGGGATACAAGCTTTAGCAGGC 3′ | |
| repAH2mut-rev | 5′ CGCTGATGGCATTCGCCATATGCTTCGCCGCGGCGGCTTCGAGGGTCCGCGCCTTGGATTCC 3′ | |
| Rep1984-fwd | 5′ TCAAAGACCCTTGCCGGC 3′ | |
| Rep5099-rev | 5′ ACTTCTGTGAAGAATTCG 3′ | |
| gt-fwd | HindIII | 5′ ATGATGAAGCTTACCATGGCTAACAGTACCAGTCAAGGATCACAGGCTCC 3′ |
| gtmut-fwd | HindIII | 5′ ATGATGAAGCTTACCATGGCTCAGAGTACCAGTCAAGGATCACAGGCTCC 3′ |
| gt-rev | KpnI | 5′ GGTACCTTGTGATCCAGCAACTGGAGCCTGTGATCCTTGACTGGTAC 3′ |
| NS4Bgt-40-fwd | KpnI | 5′ CTCCAGTTGCTGGATCACAAGGTACCCAGACCAACTGGCAGAAACTCGAG 3′ |
| NS4BAH2mut-40-fwd | KpnI | 5′ CTCCAGTTGCTGGATCACAAGGTACCCAGACCAACGCGCAGAAACTCGAG 3′ |
Restriction enzyme recognition sites are underlined, and mutated nucleotides are shown in bold.
Alanine substitutions were introduced by PCR-directed mutagenesis. A 5′ fragment was amplified from pBRTM/HCV1-3011con with forward primer HCV5275-fwd and reverse primer AH2mut-rev (Table 1). A 3′ fragment was amplified with forward primer AH2mut-fwd and reverse primer HCV6819-rev (Table 1). The central primers included a silent C to T change at HCV nucleotide position 5292, creating an NdeI restriction site. The amplification products were cloned into pCR2.1TOPO (Invitrogen, La Jolla, CA), followed by digestion with NsiI and NdeI or NdeI and SnaBI, respectively, and ligation into the NsiI and SnaBI sites of pUHDHCV(H)con (46), yielding the tetracycline-controlled transactivator (tTA)-dependent polyprotein expression construct pUHDHCV(H)conAH2mut (see Fig. 6A). This plasmid served as the template for the PCR amplification of fragments representing aa 40 to 69 of NS4B or full-length NS4B. Primers NS4B-40-fwd and NS4B-69-rev or NS4B-1-fwd and NS4B-261-rev, respectively, were employed for this purpose (Table 1). The amplification products were digested with EcoRI and BamHI and cloned into pCMVKEB-GFP, yielding GFP fusion constructs pCMVNS4B40-69-AH2mut-GFP and pCMVNS4B-AH2mut-GFP, respectively.
FIG. 6.
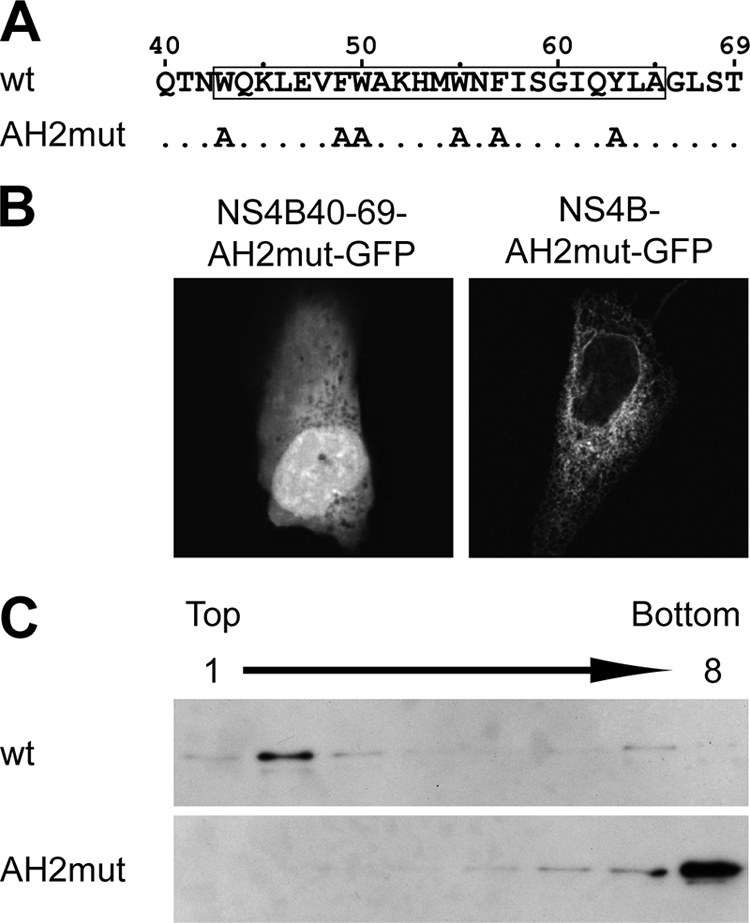
Aromatic residues are required for membrane association of the NS4B aa 40-69 segment. (A) Sequence of mutant AH2mut. (B) U-2 OS cells were transiently transfected with pCMVNS4B40-69-AH2mut-GFP or pCMVNS4B-AH2mut-GFP as indicated, followed by confocal laser scanning microscopy as described in Materials and Methods. See Fig. 2 for the subcellular distribution of the corresponding wild-type constructs. (C) U-2 OS cells were transiently transfected with pCMVNS4B40-69-GFP or pCMVNS4B40-69-AH2mut-GFP, followed by membrane flotation analysis, 12% SDS-polyacrylamide gel electrophoresis, and immunoblotting using MAb JL-8 against GFP. wt, wild type.
An NST consensus motif for N-linked glycosylation, followed by a 14-aa spacer sequence (2), was added to the N terminus of a fragment comprising NS4B aa 40 to 261 by overlap extension PCR. A construct harboring glutamine instead of asparagine within the glycosylation acceptor site (gtmut) served as the control. A 5′ fragment was amplified from pBRTM/HCV1-3011con with forward primer gt-fwd or gtmut-fwd and reverse primer gt-rev (Table 1). A 3′ fragment was amplified with forward primer NS4Bgt-40-fwd or NS4BAH2mut-40-fwd and reverse primer NS4B-261-rev (Table 1). The overlap extension products obtained by PCR with forward primer gt-fwd or gtmut-fwd and reverse primer NS4B-261-rev (Table 1) were digested with HindIII and BamHI, followed by ligation into the HindIII/BamHI sites of pCMVKEB-GFP, yielding constructs pCMVgt-NS4B40-261-GFP, pCMVgtmut-NS4B40-261-GFP, and pCMVgt-NS4B40-261-AH2mut-GFP. Plasmid pCMVNS5Bcon-gt was described previously (23).
A subgenomic HCV replicon construct harboring the alanine substitutions described above was derived from pCon1/SG-Neo(I)/AflII (34) (kindly provided by Charles M. Rice) by overlap extension PCR. A 5′ fragment was amplified with forward primer Rep1984-fwd and reverse primer repAH2mut-rev, and a 3′ fragment was amplified with forward primer repAH2mut-fwd and reverse primer Rep5099-rev (Table 1). The fragments were joined by PCR with primers Rep1984-fwd and Rep5099-rev, followed by digestion with BsrGI and MluI and ligation into the corresponding sites of pCon1/SG-Neo(I)/AflII, yielding construct pCon1/SG-Neo(I)/AflII-AH2mut.
Antibodies.
Monoclonal antibody (MAb) JL-8 against GFP was from Clontech (Palo Alto, CA), and MAb JG1 against mitochondrial heat shock protein 70 (mtHsp70) was from Affinity Bioreagents (Golden, CO). Polyclonal antibodies against protein disulfide isomerase (PDI) and mitofilin were from StressGen (Victoria, British Columbia, Canada) and Abcam (Cambridge, United Kingdom), respectively. MAb 5B-3B1 against HCV NS5B was described previously (33). MAb G1/296 against p63 (47) was a gift of Hans-Peter Hauri (University of Basel, Switzerland). MAb 11H against HCV NS5A (3) was a gift of Jan Albert Hellings (bioMérieux, Boxtel, The Netherlands). Polyclonal antiserum 86 against HCV NS4B was a gift of Ralf Bartenschlager (University of Heidelberg, Germany).
Cell lines.
Tetracycline-regulated cell lines were generated as described previously (35). In brief, the constitutively tTA-expressing, U-2 OS human osteosarcoma-derived founder cell line UTA-6 (11) was cotransfected with tTA-dependent expression construct pUHDHCV(H)con-AH2mut and pBabepuro (37), followed by selection with G418 and puromycin. Antibiotic double-resistant clones were pooled and analyzed by immunofluorescence and confocal laser scanning microscopy 48 h following tetracycline withdrawal.
Immunofluorescence and confocal laser scanning microscopy.
U-2 OS human osteosarcoma cells grown on glass coverslips were transfected with GFP fusion constructs, fixed 24 to 48 h posttransfection with 2% paraformaldehyde, and mounted in SlowFade (Molecular Probes, Eugene, OR). Immunofluorescence staining was performed as described previously (35). Bound primary antibody was revealed with Alexa-488-conjugated goat anti-mouse or Alexa-594-conjugated goat anti-rabbit antibody (both from Molecular Probes, Eugene, OR) or Cy3-conjugated goat anti-rabbit antibody (Jackson Laboratories, West Grove, PA). The coverslips were mounted in SlowFade and examined using a Leica SP5 AOBS confocal laser scanning microscope.
Membrane flotation assays.
Membrane flotation assays were performed as described previously (30, 40). In brief, transiently transfected U-2 OS cells were subjected to Dounce homogenization in hypotonic buffer (10 mM Tris-HCl [pH 7.4], 2 mM MgCl2) containing a protease inhibitor cocktail (Roche Diagnostics, Penzberg, Germany), followed by equilibrium centrifugation of postnuclear supernatants in 5 to 37.5% (wt/vol) Nycodenz (Axis Shield, Oslo, Norway) gradients. Subsequently, the fractions were collected from the top, and equal volumes were subjected to immunoblotting, as described previously (35). For the membrane extraction experiments, postnuclear supernatants were adjusted to 0.25 M sucrose and centrifuged at 100,000 × g for 45 min at 4°C. The pellet containing mitochondrial and microsomal membranes was resuspended in NTE buffer (100 mM NaCl, 10 mM Tris-HCl [pH 8.0], 1 mM EDTA), 100 mM sodium carbonate (pH 11.5), or 1% Triton X-100 and incubated for 20 min at 4°C. Finally, membrane flotation analyses were performed as described above.
Peptide synthesis and purification.
Peptide NS4B[40-69] (QTNWQKLEVFWAKHMWNFISGIQYLAGLST) and mutant peptide NS4B[40-69]-AH2mut (QTNAQKLEVAAAKHMANAISGIQALAGLST) (where AH2mut harbors the alanine substitutions described above) were synthesized by the stepwise solid phase method of Merrifield, employing Fmoc (N-[9-fluorenyl]methoxycarbonyl) chemistry. The peptides were highly purified by reversed-phase high-performance liquid chromatography on a Vydac C8 column (300 Å, 10 μm, 10 by 250 mm) using a water-acetonitrile gradient containing 0.1% trifluoroacetic acid. Purified peptides exhibited the expected masses, as determined by mass spectroscopy. However, the spontaneous cyclization of the N-terminal glutamine with loss of its NH3 group (45) occurred rapidly upon peptide solubilization. This N-terminal residue remained flexible, and its cyclization had no influence on the peptide structure.
CD.
Circular dichroism (CD) spectra were recorded on a Jobin Yvon CD6 spectrometer calibrated with 1S-(+)-10-camphorsulfonic acid. Measurements were carried out at room temperature in a 0.1-cm path length quartz cuvette, with peptide concentrations ranging from 30 to 60 μM. Spectra were recorded in the 190- to 250-nm wavelength range with a 0.2-nm increment and a 2-s integration time. Spectra were processed with CD6 software, baseline corrected, and smoothed using a third-order least-square polynomial fit. Spectral units were expressed as the molar ellipticity per residue by using peptide concentrations determined by measuring the UV light absorbance of tyrosine and tryptophan at 280 nm. The secondary structure content was estimated with various deconvolution methods by using DICHROWEB tools (http://www.cryst.bbk.ac.uk/cdweb/) (54).
NMR spectroscopy.
Purified NS4B[40-69] was dissolved either in 100 mM sodium dodecyl sulfate (SDS)-d25 (>98% isotopic enrichment) or 100 mM dodecylphosphocholine (DPC)-d38 (>98%) or in a mixture of 50% 2,2,2-trifluoroethanol (TFE)-d2 (>99%) in H2O (vol/vol), and 2,2-dimethyl-2-silapentane-5-sulfonate was added to the nuclear magnetic resonance (NMR) samples as an internal 1H chemical shift reference. NMR spectra were acquired at 15, 25, and 35°C. Multidimensional experiments were performed on a Varian UNITYplus 500-MHz spectrometer using standard homonuclear pulse sequences, including nuclear Overhauser enhancement spectroscopy (NOESY) (mixing times between 100 and 250 ms) and clean total correlation spectroscopy (isotropic mixing time of 80 ms), as detailed previously (12, 41). Water suppression was achieved by presaturation. Varian VNMR software was used to process all data, and Sparky was used for spectral analysis (http://www.cgl.ucsf.edu/home/sparky/). Intraresidue backbone resonances and aliphatic side chains were identified from homonuclear 1H clean total correlation spectroscopy experiments and confirmed with 1H-13C heteronuclear single quantum correlation experiments in 13C natural abundance. Sequential assignments were determined by correlating intraresidue assignments with interresidue cross peaks observed in bidimensional 1H NOESY. NMR-derived 1Hα and 13Cα chemical shifts are reported relative to the random coil chemical shifts in TFE (29).
NMR-derived constraints and structure calculation.
NOE intensities used as input for structure calculations were obtained from the NOESY spectrum recorded with a 150-ms mixing time and checked for spin diffusion on spectra recorded at shorter mixing times (50 ms). NOEs were partitioned into three categories of intensities that were converted into distances ranging from a common lower limit of 1.8 Å to upper limits of 2.8, 3.9, and 5.0 Å. Protons without stereospecific assignments were treated as pseudoatoms, and the correction factors were added to the upper distance constraints (57). Neither additional dihedral angle nor hydrogen bond restraints were introduced. Three-dimensional structures were generated from the NOE distances by the dynamical simulated annealing protocol with the XPLOR-NIH 2.9.7 program (49) using the standard force fields and default parameter sets. A set of 50 structures was initially calculated to widely sample the conformational space, and the structures of low energy with no distance restraint violations (>0.5 Å) were retained. The selected structures were compared by pairwise root mean square deviation (RMSD) over the backbone atom coordinates (N, Cα, and C′). Local analogies were analyzed by calculating the local RMSD of a tripeptide window slid along the sequence. Statistical analyses, the superimposition of structures, and structural analyses were performed with MOLMOL version 2.6 (25), AQUA version 3.2, and PROCHECK-NMR version 3.5.4 (26).
Preparation of proteoliposomes.
Peptide NS4B[40-69] was dissolved in 50% TFE at a concentration of 10 mg per ml, and 50 μl of this solution (i.e., 0.5 mg of peptide) was mixed with 5 mg 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Avanti Polar Lipids, Alabaster, AL) in 50 μl chloroform in a glass tube to obtain a final peptide/lipid ratio of 1:10 (wt/wt). The mixture was dried under nitrogen and lyophilized to remove any traces of solvent. The dry film was hydrated and shaken with 0.5 ml of 20 mM HEPES buffer at pH 7.4, followed by sonication for 15 min and extrusion through a LiposoFast-Basic extruder (Avestin, Ottawa, Canada) equipped with a polycarbonate membrane with a 100-nm pore diameter.
Attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR).
Spectra were recorded on a Bruker IFS 55 infrared spectrophotometer equipped with a reflectance accessory and a polarizer mount assembly with a gold wire grid element. The internal reflection element was a germanium ATR plate (50 by 20 by 2 mm) with an aperture angle of 45° yielding 25 internal reflections. To improve the signal-to-noise ratio, 128 accumulations were performed. Spectra were recorded at a nominal resolution of 2 cm−1. Measurements were performed at room temperature. Oriented multilayers were formed by the slow evaporation of 10 μl of the sample (1 mg peptide per ml) on one side of the ATR plate.
Secondary structure quantification was performed on samples subjected to 1 h of deuteration, as described previously (16). Briefly, Fourier self-deconvolution was applied to increase the resolution of the spectra in the amide I region. Least square iterative curve fitting was performed to fit components of the amide I band revealed by self-deconvolution to the nondeconvolved spectrum between 1,700 and 1,600 cm−1.
For the amount of material used in the present studies, film thickness remains small compared with the infrared wavelength. This allows the “thin film” approximation to be used for the establishment of equations describing the dichroic ratio as a function of the orientational order parameter (17). In an α-helix, the main transition dipole moment lies approximately parallel to the helical axis. It is therefore possible to derive the orientation of an α-helix from the orientation of the peptide bond C=O group (17). This information was obtained, and the mean angle of the helix axes was then calculated as previously described (14, 17).
In vitro transcription and RNA electroporation.
Replicon constructs were linearized with ScaI, followed by in vitro transcription using the T7 RiboMAX express large-scale RNA production system from Promega (Madison, WI). The transcripts were purified using the RNeasy minikit (Qiagen, Hilden, Germany). The replicon RNA was electroporated into Huh-7.5 cells (1) (kindly provided by Charles M. Rice), followed by plating into 100-mm-diameter dishes at 6 × 105, 6 × 104, and 6 × 103 cells per dish and crystal violet staining after 3 weeks of G418 selection, as described previously (34).
Nucleotide sequence accession numbers.
The atomic coordinates for the NMR structure of NS4B[40-69] and the NMR restraints in 50% TFE are available in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank under accession number 2JXF (RCSB100411). The proton chemical shifts of all residues have been deposited in the BioMagResBank (BMRB) under the accession number 15559. For consistency with standard protein chemistry nomenclature, the NS4B peptide is referred to as NS4B[40-69], and residues within this peptide are numbered from the start of the NS4B protein (HCV polyprotein residues 1751 to 1780, strain H77, genotype 1a, GenBank accession number AF009606).
RESULTS
Sequence analyses and structure predictions.
Fluorescence microscopy as well as membrane extraction and flotation analyses of a comprehensive panel of GFP fusion constructs comprising different segments of NS4B revealed the presence of an unexpected determinant for membrane association within the N-terminal 74 aa of NS4B (data demonstrated below). Based on this observation, we performed sequence analyses and structure predictions to assess the degree of conservation of the N-terminal portion of NS4B and to identify motifs potentially involved in membrane association. The amino acid repertoire deduced from the analysis of 448 HCV isolates of various subtypes of genotype 1 revealed that amino acids are strictly conserved in 76% of the sequence positions (denoted by asterisks in Fig. 1A). The apparent variability is limited at most positions since the observed residues exhibit similar physicochemical properties, as indicated both by the similarity pattern (colons and dots) and the hydropathic pattern where o, i, and n denote hydrophobic, hydrophilic, and neutral residues, respectively (see the legend to Fig. 1 for details). The degree of conservation among different genotypes was investigated by ClustalW alignment of 27 reference sequences representative of the major HCV genotypes and subtypes (see the legend to Fig. 1A). The amino acid repertoire derived from this alignment revealed that the segment comprising aa 50 to 80 (aa 50-80 segment) is well conserved, whereas the aa 1-50 segment appears highly variable, except for some well-conserved positions (Fig. 1A). However, despite this apparent variability, conservation of the hydropathic character at many positions indicates that the overall structure is conserved among the different HCV genotypes. This is supported by secondary-structure analyses that always predicted the presence of two α-helices for the segments from approximately aa 5 to 34 and 40 to 65, as shown in Fig. 1B for HCV strain H77. In addition, ideal α-helix projections indicated that both predicted helices are mainly amphipathic (data not shown). None of these predicted helices carried an obvious signal peptide or transmembrane character.
FIG. 1.

Sequence analyses as well as predictions of secondary-structure and membrane binding properties. (A) Amino acid repertoires. The sequence comprising NS4B aa 1 to 80 from the HCV H77 consensus clone (GenBank accession number AF009606), which was used to prepare the constructs described in this study, is indicated. Amino acids are numbered with respect to NS4B and the HCV polyprotein (top row). The amino acid repertoire deduced from the ClustalW multiple alignments of 448 NS4B sequences of genotype 1 is reported at the top. Amino acids observed at a given position with a frequency <1% were not included. Below is the amino acid repertoire of the 27 representative NS4B sequences from confirmed HCV genotypes and subtypes (listed with accession numbers in Table 1 in reference 50), including the recently described genotype 7a (accession number EF108306; see the European HCV Database [http://euhcvdb.ibcp.fr/] for details). The degree of amino acid and physicochemical conservation at each position can be inferred from the extent of variability (with the observed amino acids listed in decreasing order of frequency from top to bottom) together with the similarity index according to ClustalW convention (asterisk, invariant; colon, highly similar; dot, similar) (52) and the consensus hydropathic pattern deduced from the consensus amino acid repertoire. o, hydrophobic position (F, I, W, Y, L, V, M, P, and C); n, neutral position (G, A, T, and S); i, hydrophilic position (K, Q, N, H, E, D, and R); v, variable position (i.e., when both hydrophobic and hydrophilic residues are observed at a given position). (B) Secondary-structure predictions of NS4B are indicated as helical (h, blue), extended (e, red), turn (t, green), or undetermined (coil [c], yellow). Predictions were made by using the Web-based algorithms DSC, HNNC, MLRC, PHD, Predator, SOPM, and SIMPA96 available at the NPSA website (http://npsa-pbil.ibcp.fr; reference 5 and references therein). The segment highlighted in gray corresponds to the peptide NS4B[40-69]. (C) Interfacial hydrophobicity plot generated with MPEx (http://blanco.biomol.uci.edu/mpex/) using the Wimley and White interfacial hydrophobicity scale (55). The plot shows the mean values using a window of 19 residues. The segment exhibiting a propensity to partition into the lipid bilayer is indicated by the red bar.
To examine the propensity of the N-terminal portion of NS4B to interact with membranes, the interfacial hydrophobicity was calculated using the algorithm developed by Wimley and White (55). As shown in Fig. 1C for NS4B from HCV strain H77, the interfacial hydrophobicity plots calculated for sequences of various genotypes indicated that the aa 42-80 segment exhibits a clear propensity to partition into the interface of a phospholipid bilayer (Fig. 1C, red bar). In contrast, the N-terminal aa 1-41 segment does not show any propensity to partition into a lipid bilayer. From the above sequence analyses and predictions of secondary structure and lipotropic properties, we concluded that the NS4B aa 40-69 segment could potentially fold into a membrane-associated amphipathic α-helix.
Identification of a determinant for membrane association in the N-terminal portion of NS4B.
GFP fusion constructs were analyzed by fluorescence microscopy to validate the above predictions. As shown in Fig. 2, full-length NS4B fused to GFP showed a fluorescence pattern that included the nuclear membrane, was strongest in the perinuclear region, and extended in a reticular fashion through the cytoplasm. This pattern corresponds to that previously described for full-length NS4B (21) and is consistent with the pattern observed for similar GFP fusion constructs (10, 20, 27, 28). As described previously, NS4B-GFP fluorescence colocalized mainly with the ER marker PDI but also likely included ER-derived modified membranes that have excluded the marker (Fig. 2, left panel). NS4B aa 1 to 74 fused to GFP showed a similar pattern. In addition, this construct partially colocalized with the mitochondrial marker mitofilin (Fig. 2, right panel). By contrast, NS4B aa 1 to 40 fused to GFP showed a diffuse fluorescence pattern, suggesting a lack of membrane association of this segment. GFP alone and a construct harboring NS4B aa 1 to 29 fused to GFP showed the same diffuse fluorescence pattern (data not illustrated). Conversely, NS4B aa 40 to 69 fused to GFP (40-69-GFP) showed a pattern very similar to NS4B aa 1 to 74 fused to GFP, with colocalization with PDI and mitofilin. These results demonstrate the presence of a novel determinant for membrane association between NS4B aa 40 and 69.
FIG. 2.

Subcellular localization of NS4B-GFP fusion constructs. U-2 OS cells were transiently transfected with pCMVNS4B-GFP, pCMVNS4B1-74-GFP, pCMVNS4B1-40-GFP, or pCMVNS4B40-69-GFP, as indicated. Cells were processed for immunofluorescence staining using polyclonal antibodies against the ER marker PDI or the mitochondrial marker mitofilin. Slides were analyzed by confocal laser scanning microscopy as described in Materials and Methods. Horizontal sections taken through the center of the nuclei are shown. Insets show higher magnifications of selected areas.
Membrane flotation analyses were performed to confirm and extend these observations, as shown in Fig. 3. GFP fusion constructs were expressed in U-2 OS cells, followed by hypotonic cell lysis and equilibrium centrifugation in Nycodenz gradients. Under these conditions, membrane proteins float to the upper, low-density fractions, as indicated by p63, an integral ER membrane protein (47) (Fig. 3A, top panel). As expected, full-length NS4B fused to GFP floated to the membrane-containing low-density fractions. By contrast, the construct harboring NS4B aa 1 to 40 fused to GFP was found in the high-density fractions, confirming its lack of membrane association (Fig. 3A, middle panel). Analogous results were obtained for GFP alone and for a construct harboring aa 1 to 29 fused to GFP (data not illustrated). Finally, construct 40-69-GFP was recovered mainly in the upper, low-density fractions, confirming its membrane association. Taken together, the flotation assays confirmed our fluorescence microscopy analyses demonstrating that the NS4B aa 40-69 segment associates with cellular membranes.
FIG. 3.
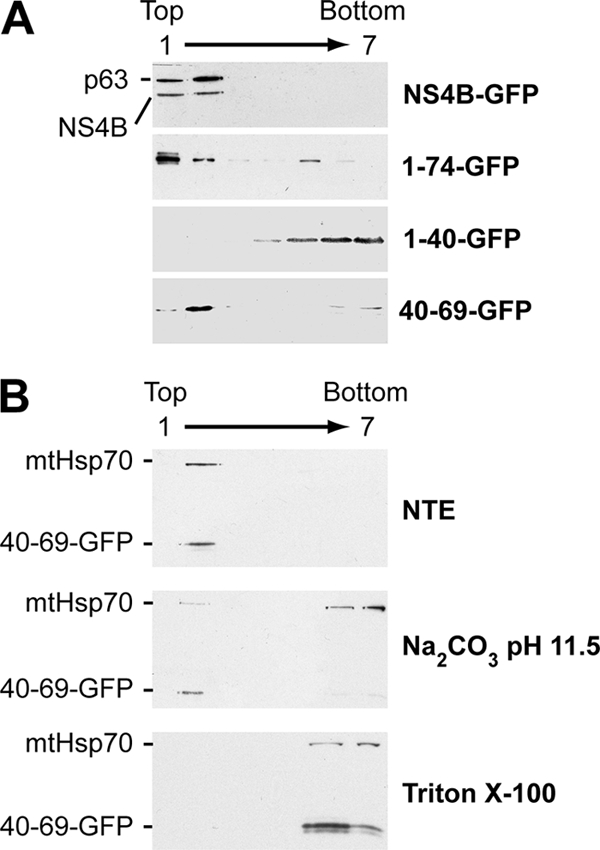
The NS4B aa 40-69 segment mediates tight membrane association. (A) The hypotonic lysates of U-2 OS cells transiently transfected with pCMVNS4B-GFP, pCMVNS4B1-74-GFP, pCMVNS4B1-40-GFP, or pCMVNS4B40-69-GFP were analyzed by centrifugation through Nycodenz gradients, as described in Materials and Methods. Seven fractions were collected from the top and analyzed by immunoblotting using MAb JL-8 against the GFP. The endogenous integral ER membrane protein p63 was detected using MAb G1/296. (B) U-2 OS cells transiently transfected with pCMVNS4B40-69-GFP were lysed in a hypotonic buffer, and the membrane fraction was obtained by ultracentrifugation as described in Materials and Methods. Membrane pellets were resuspended in physiological NTE buffer (top panel), 100 mM sodium carbonate (pH 11.5) (middle panel), or 1% Triton X-100 (bottom panel) and incubated for 20 min at 4°C. Subsequently, membrane flotation analyses were performed as described in Materials and Methods. Seven fractions were collected from the top and analyzed by immunoblotting using MAb JL-8 against GFP. mtHsp70 was detected with MAb JG1 and served as a control for a peripherally membrane-associated protein.
To extend these findings and to further characterize the membrane association of 40-69-GFP, differential membrane extractions followed by flotation assays were performed. For these, membrane fractions were isolated by ultracentrifugation of the hypotonic lysates from transfected cells, followed by differential extraction of the pellet fraction. NTE was used as a physiologic buffer, while alkaline extraction (100 mM sodium carbonate [pH 11.5]) was used to release peripheral proteins from the membranes. As shown in Fig. 3B (top panel), both the 40-69-GFP construct and endogenous mtHsp70 as a control for a peripherally membrane-associated protein (48) were recovered in the membrane fraction under physiological extraction conditions. Under alkaline extraction conditions, the 40-69-GFP construct remained membrane associated, while mtHsp70 was solubilized (Fig. 3B, middle panel). Finally, both proteins were recovered in the high-density fractions, following extraction with 1% Triton X-100 which disrupts cellular membranes (Fig. 3B, bottom panel). Taken together, these observations indicate that the NS4B aa 40-69 segment mediates a tight membrane association.
Conformation analyses of NS4B[40-69] by CD.
To gain insight into the structure and lipotropic properties of the NS4B aa 40-69 segment, the corresponding peptide, designated NS4B[40-69], was chemically synthesized, purified, and analyzed by CD. The solubility of this peptide in water was limited, and it displayed a complex spectrum with two large negative bands around 208 and 222 nm, indicating the presence of an α-helix with some less-defined secondary structures (Fig. 4A). An α-helix content of approximately 45% together with 10 to 15% β-structure was estimated with the various CD deconvolution methods used. Such a secondary structure content for a short peptide suggests that it likely exists as soluble, micelle-like aggregates in aqueous solution due to its amphipathic nature. The secondary structure of NS4B[40-69] was also examined in the presence of either lysophosphatidyl choline (LPC) or various detergents (SDS, n-dodecyl β-d-maltoside [DM], DPC) or cosolvents (TFE-water mixture) that mimic the membrane environment (Fig. 4A and B). TFE is known to solubilize hydrophobic peptides as monomers and to stabilize the folding of peptidic sequences with an intrinsic propensity to adopt an α-helical structure (4, 32). Dissolving the peptide in 50% TFE generated a spectrum that was consistent with an α-helical folding, with two minima at 208 and 222 nm and a maximum at 192 nm (Fig. 4A and B). However, a shoulder is observed at about 230 to 235 nm that is likely due to tryptophan residues that have been shown to give rise to CD bands in this region (53). Indeed, the CD spectra of peptide NS4B[40-69]-AH2mut, in which the six aromatic residues were changed to alanine, displayed a typical α-helix folding in the presence of 50% TFE or SDS, without such a shoulder at 230 to 235 nm (Fig. 4C). The shapes of the spectra of peptide NS4B[40-69] recorded in LPC, SDS, DM, and DPC are similar and also consistent with an α-helical structure. Because of the significant contribution of aromatic residues in the far UV region, predictions of the amounts of various types of secondary structures are inaccurate for peptide NS4B[40-69]. It should be noted, however, that the various CD deconvolution methods used indicate a predominant α-helix content ranging from 52 to 60% in LPC as well as in detergents and from 64 to 73% in 50% TFE. In summary, CD spectral analyses indicated the high propensity of NS4B[40-69] to interact with lipids and to adopt an α-helical structure upon lipid binding. As expected, mutation of the six aromatic residues into alanine in peptide NS4B[40-69]-AH2mut did not change the propensity of this sequence to fold into an α-helix when probed in 50% TFE or SDS (Fig. 4C).
FIG. 4.

Far-UV CD analyses of synthetic peptides NS4B[40-69] (A, B) and NS4B[40-69]-AH2mut (C) in membrane mimetic environments. CD spectra were recorded in 5 mM sodium phosphate buffer (pH 7.5) (H2O, dashed line), complemented with either 50% TFE (alternate dashed line) or 1% LPC (solid line) or the following detergents: 100 mM SDS (small dashed line), 100 mM DM (large dashed line), or 100 mM DPC (dotted line). To facilitate data comparisons for NS4B[40-69], the spectra recorded for 50% TFE were used as references for the data shown in panels A and B. See Materials and Methods as well as Fig. 6 for the amino acid sequence of peptide NS4B[40-69]-AH2mut.
The NS4B aa 40-69 segment comprises an amphipathic α-helix.
Deuterated micellar SDS and DPC are popular membrane mimetic media for structure analyses of membrane peptides by liquid NMR (39). Unfortunately, samples of NS4B[40-69] prepared in SDS and DPC displayed broad, poorly resolved NMR spectra that may be due to some oligomerization of the peptide. Nevertheless, a complete amino acid sequential attribution and 1Hα chemical shift variation analysis were possible from spectra recorded in DPC. Both spectra recorded in SDS and DPC did not permit the modeling of the NS4B[40-69] peptide structure. We thus studied the three-dimensional structure of NS4B[40-69] dissolved in 50% TFE-d2, which yielded well-resolved NMR spectra (data not shown). Sequential attribution of all spin systems was complete and an overview of the sequential and medium range NOE connectivities is shown in Fig. 5A. The NOE connectivity patterns clearly demonstrate that the main body of the peptide, including residues 42 to 66, displays characteristics typical of an α-helix, including strong dNN(i, i + 1) and medium dαN(i, i + 1) sequential connectivities as well as weak dαN(i, i + 2), medium or strong dαN(i, i + 3) and dαβ(i, i + 3), and weak dαN(i, i + 4) medium-range connectivities. The NOE-based indications of α-helical conformation were supported by the deviation of the 1Hα chemical shifts from random coil values (56). The long series of negative variation of 1Hα chemical shifts (Δδ1Hα < −0.1 ppm) observed for residues Asn 42 to Gly 66, both in 50% TFE and 100 mM DPC (Fig. 5B and C, respectively), are indeed typical of an α-helical conformation. Based on the NOE-derived interproton distance constraints, a set of 50 structures was calculated with X-PLOR, and a final set of 30 low-energy structures that fully satisfied the experimental NMR data were retained. The number and types of NOE constraints used for the structure calculations as well as the statistics for this final set of 30 structures are given in Table 2. The superimposition of the 30 structures (Fig. 5D) shows that the main part of the helix (residues 44 to 65) is well defined, with an RMSD of 0.8 Å (Table 2). As illustrated in Fig. 5E to H, the asymmetric distribution of polar and hydrophobic residues on each side of α-helix 42-66, especially in the region from aa 49 to 64, clearly reveals the amphipathic character of this α-helix. Moreover, aromatic residues Trp 43, Phe 49, Trp 50, Trp 55, Phe 57, and Tyr 63 are well positioned within or on the edges of the hydrophobic side, suggesting their essential role in the interaction with the membrane interface. Indeed, tyrosine and tryptophan are frequently found at the membrane interface (19, 58). These structural features, together with the clear propensity of the NS4B[40-69] peptide to adopt an α-helical structure upon binding to lipid-like molecules, suggest that NS4B amphipathic α-helix 42-66 associates with the membrane interface, at least transiently, in an in-plane topology.
FIG. 5.

NMR analysis and structure calculation of the HCV NS4B[40-69] peptide. (A) Summary of the sequential (i, i + 1) and medium-range (i, i + 2 to i, i + 4) NOEs in 50% TFE. The intensities of the NOEs are indicated by the thickness of the bars. Asterisks indicate that the presence of a NOE cross peak was not confirmed because of overlapping resonances. (B, C) 1Hα chemical shift differences (in parts per million) in 50% TFE (B) and 100 mM DPC (C), respectively. Chemical shift differences were calculated by subtraction of the experimental values from the random coil conformation values. The dashed lines indicate the standard threshold value of ΔHα for an α-helix (−0.1 ppm). (D) Superimposition of the backbone heavy atoms (N, Cα, and C′) of the 30 final structures calculated from the NMR data recorded in 50% TFE. The 30 structures were superimposed for the best overlap of residues 44 to 65. The average positions of the Trp, Tyr, and Phe aromatic side chains are displayed. (E) Amino acid sequence of NS4B[40-69], including α-helix 42-66 (box). The residues are color coded according to their physicochemical properties. Hydrophobic residues (V, L, F, M, I, W, and Y) are dark gray, and alanine and glycine are light gray. Polar residues (S, N, Q, and T) are yellow, and positively and negatively charged groups of basic (K) and acidic (E) residues are blue and red, respectively. Histidine is cyan. (F to H) Structure of NS4B amphipathic α-helix 42-66 in 50% TFE. (F) Side view of the peptide backbone (ribbon representation) and residue side chains (stick representation). Residue side chains are colored as described for panel E, except for tryptophan, tyrosine, and phenylalanine, which are colored magenta, purple, and black, respectively. (G, H) Top (hydrophilic side) and bottom (hydrophobic side) views of the surface of amphipathic α-helix 42-66. Residue side chains are colored as described in panel E. Panels F to H were generated from structure coordinates (PDB entry 2JXF) using VMD (http://www.ks.uiuc.edu/Research/vmd/) (22) and rendered with POV-Ray (http://www.povray.org).
TABLE 2.
Statistics of final simulated annealing structures of peptide NS4B[40-69]
| Restraint or statistic | Value |
|---|---|
| No. of distance restraints | |
| Intraresidue | 0 |
| Sequential | 141 |
| Medium range | 160 |
| Total | 301 |
| Statistics for the final X-PLOR structures | |
| No. of structures in the final set | 30 |
| X-PLOR energy (kcal·mol−1) | −166 ± 7 |
| NOE violations | |
| No. >0.5 Å | None |
| RMSD (Å) | 0.056 ± 0.004 |
| Deviation from idealized covalent geometry | |
| Angles (degrees) | 1.04 ± 0.03 |
| Impropers (degrees) | 0.21 ± 0.01 |
| Bonds (Å) | 0.0043 ± 0.0001 |
| RMSD (Å) | |
| Backbone (C′, Cα, N) | |
| Helix segment 44-65 | 0.80 ± 0.27 |
| All residues | 2.35 ± 0.76 |
| Heavy atoms | |
| Helix segment 44-65 | 1.56 ± 0.29 |
| All residues | 2.99 ± 0.67 |
| Ramachandran dataa (aa 44-65) (for 630 residues) | |
| Residues in most favored regions (%) | 98.3 |
| Residues in allowed regions (%) | 1.7 |
| Residues in generously allowed regions (%) | 0 |
| Residues in disallowed regions (%) | 0 |
| Ramachandran dataa (for 780 residues) | |
| Residues in most favored regions (%) | 89.7 |
| Residues in allowed regions (%) | 6.7 |
| Residues in generously allowed regions (%) | 1.7 |
| Residues in disallowed regions (%) | 1.9 |
From PROCHECK-nmr (26), excluding glycine and proline residues.
Aromatic residues are required for membrane association of the NS4B aa 40-69 segment.
As demonstrated above, the NMR structure of NS4B[40-69] revealed an amphipathic α-helix. To validate the role of the hydrophobic side in membrane association, we replaced the fully conserved aromatic residues on the hydrophobic side of the helix with alanine (Fig. 6, AH2mut). As documented above by CD of the corresponding mutant synthetic peptide (Fig. 4C, NS4B[40-69]-AH2mut), these changes preserved the α-helical folding propensity while abrogating the hydrophobic character of the corresponding positions.
Mutant AH2mut was expressed as a fusion with GFP (NS4B40-69-AH2mut-GFP) and analyzed by fluorescence microscopy. In addition, the same alanine substitutions were introduced into a full-length NS4B construct fused to GFP (NS4B-AH2mut-GFP). As shown in Fig. 6B, the membrane association of construct NS4B40-69-AH2mut-GFP was completely abrogated, as documented by the diffuse fluorescence signal. This observation was confirmed by membrane flotation analyses. As shown in Fig. 6C, the wild-type NS4B40-69-GFP construct was found in the membrane-containing upper fraction, while mutant NS4B40-69-AH2mut-GFP was found only in the lower fractions harboring soluble proteins. These data confirm the importance of the aromatic residues on the hydrophobic side of amphipathic α-helix 42-66 in mediating the membrane association of the aa 40-69 segment. Interestingly, the same mutations did not interfere with the membrane association of full-length NS4B (Fig. 6B), indicating that one or more internal determinants in NS4B can ensure membrane association in the context of the full-length protein.
Membrane topology of the NS4B aa 40-69 segment.
In principle, amphipathic α-helices can associate with membranes in an in-plane topology or can form transmembrane segments upon oligomerization. The membrane topology of the NS4B aa 40-69 segment was determined by ATR-FTIR of synthetic peptide NS4B[40-69] reconstituted in DMPC proteoliposomes as well as by glycosylation tagging experiments.
By ATR-FTIR, the amide I band (1,700 to 1,600 cm−1) revealed a peak centered at 1,656 cm−1 characteristic of an α-helix (14, 15) (Fig. 7A). In good agreement with the data obtained by CD in membrane mimetics, the α-helix content was found to reach 52%. To determine the helix orientation, spectra were recorded with parallel and perpendicular polarized light. The dichroic spectrum was obtained by subtracting the spectrum recorded with perpendicular polarized light from the spectrum recorded with parallel polarized light (Fig. 7A, dashed line). Initially, the orientation of the DMPC acyl chains was assessed using characteristic bands associated with lipid molecules. We used the νs(CH2) peak (2,849 cm−1) (13) to quantify the lipid chain orientation (data not shown). The measured dichroic ratio for this band is 0.97 with an isotropic dichroic ratio of 1.30. On the basis of these measurements, one can assume that the maximum tilt between the acyl chains and a vector normal to the germanium surface is 15 to 20°. As any additional source of disorder would reduce this tilt (44), the value of 15 to 20° is a maximum tilt. In the case of NS4B[40-69], helix orientation was determined using the amide I band. In the dichroic spectrum (Fig. 7A, dashed line), a strong positive deviation at 1,656 cm−1 is observed, indicating a perpendicular orientation of the associated dipole to the surface of the germanium plate and, therefore, a parallel orientation with respect to a vector normal to the surface of the DMPC bilayer. From the secondary structure of NS4B[40-69] and the frequency of the positive deviation, we conclude that the dipole responsible for this deviation is associated with α-helices. This dipole is oriented in the direction of the helical axes (14). To quantitatively determine the orientation of the NS4B[40-69] helical segment, we calculated a dichroic ratio associated with the amide I peak equal to 2.42. With such a dichroic ratio, the maximum mean tilt between the helical axis and a vector normal to the germanium surface is ∼20°. This indicates that NS4B[40-69] adopts a transmembrane orientation.
FIG. 7.
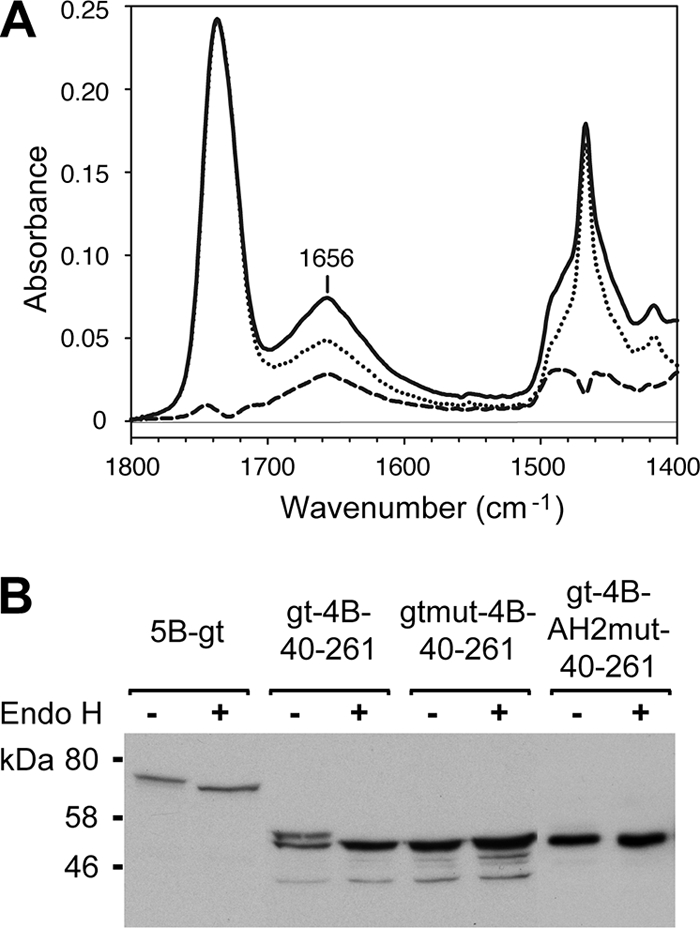
Membrane topology of the NS4B aa 40-69 segment. (A) ATR-FTIR of NS4B[40-69]-DMPC proteoliposomes. The spectrum represented by the solid line was obtained with parallel polarized light and the spectrum represented by the dotted line with perpendicular polarized light. Both spectra were rescaled and superimposed based on the C=O lipid band at 1,740 cm−1. The dashed line represents the calculated dichroic spectrum. (B) Glycosylation tagging experiments. Lysates of U-2 OS cells transfected with pCMVNS5Bcon-gt (5B-gt), pCMVgt-NS4B40-261-GFP (gt-4B-40-261), pCMVgtmut-NS4B40-261-GFP (gtmut-4B-40-261), and pCMVgt-NS4B40-261-AH2mut-GFP (gt-4B-AH2mut-40-261) were treated (+) or not (−) with endoglycosidase H (Endo H) following the manufacturer's recommendations (New England Biolabs, Beverly, MA), followed by 12% SDS-polyacrylamide gel electrophoresis and immunoblotting with MAbs 5B-3B1 against NS5B or JL-8 against GFP. Molecular mass standards are indicated on the right.
To investigate the membrane topology of the NS4B aa 40-69 segment in cellulo, an NST consensus motif for N-linked glycosylation, followed by a 14-aa spacer sequence, was added to the N terminus of a construct representing NS4B aa 40 to 261 (gt-NS4B40-261-GFP). A construct harboring a glycosylation acceptor tag at the C terminus of HCV NS5B, a well-characterized tail-anchored protein, served as the positive control (23), and a construct harboring glutamine instead of asparagine within the glycosylation acceptor site served as the negative control (gtmut-NS4B40-261-GFP). As shown in Fig. 7B, an additional 3-kDa band was observed for construct gt-NS4B40-261-GFP, indicating that the added asparagine residue was glycosylated by oligosaccharyltransferase present in the ER lumen. Digestion with endoglycosidase H abrogates this band, with the remaining band migrating at the same position as gtmut-NS4B40-261-GFP. Importantly, a construct harboring an N-terminal glycosylation acceptor tag and the alanine substitutions described above (AH2mut) was not glycosylated, indicating that membrane association via the hydrophobic side of amphipathic α-helix 42-66 is essential for the transmembrane orientation of this segment. These results indicate that the NS4B aa 40-69 segment can traverse the phospholipid bilayer as a transmembrane segment and that, given the endoglycosidase H-sensitive nature of the transferred carbohydrate moiety, it is retained in the ER. The amphipathic character of this segment suggests that transmembrane orientation is achieved in the form of an oligomer.
Alanine substitution of the aromatic residues in NS4B amphipathic α-helix 42-66 disrupts the formation of a functional HCV replication complex.
We previously characterized cell lines inducibly expressing the entire HCV polyprotein derived from the HCV H77 consensus clone (46). In these cells, HCV nonstructural proteins are properly processed and accumulate in dotlike structures which, at the ultrastructural level, correspond to membranous webs (7) (Fig. 8A, white arrowheads). The membranous webs formed in these cells are very similar to the ones observed in Huh-7 cells harboring HCV replicons and represent viral replication complexes (18). To investigate the impact of the alanine substitutions in NS4B amphipathic α-helix 42-66 on HCV replication complex formation, cell lines were generated that inducibly express NS4B harboring these substitutions in the context of the entire HCV polyprotein. Immunoblot analyses demonstrated correct polyprotein processing by these cell lines (data not illustrated). The subcellular localization of NS4B and NS5A was investigated by double-label immunofluorescence microscopy. As shown in Fig. 8A, wild-type and mutant NS4B colocalized with NS5A in all cell lines. However, the formation of dotlike structures as a correlate of membranous webs was completely abrogated in the mutant. In addition, the impact of these alanine substitutions on HCV RNA replication was assessed by introduction into a subgenomic HCV replicon. As shown in Fig. 8B, the RNA replication of this construct was completely abrogated. Taken together, these data indicate that membrane association and/or protein-protein interactions mediated by the aromatic residues in NS4B amphipathic α-helix 42-66 are required for the formation of a functional HCV replication complex.
FIG. 8.

Alanine substitution of aromatic residues in NS4B amphipathic α-helix 42-66 disrupts the formation of a functional HCV replication complex. (A) Stable cell lines inducibly expressing the entire polyprotein derived from the HCV H77 consensus clone, wild-type (wt) or harboring NS4B mutations (AH2mut) as indicated, were cultured for 36 h in the absence of tetracycline, followed by double-label immunofluorescence and confocal laser scanning microscopy, as described in Materials and Methods. HCV NS4B and NS5A were detected using polyclonal antiserum 86 and MAb 11H, respectively. Note the complete absence of cytoplasmic dotlike structures in mutant AH2mut. (B) RNA was in vitro transcribed from a subgenomic replicon construct harboring replacements of the conserved aromatic residues by alanine within α-helix 42-66 (AH2mut), followed by plating into 100-mm-diameter dishes at 6 × 105, 6 × 104, and 6 × 103 cells per dish and crystal violet staining after 3 weeks of G418 selection, as described in Materials and Methods. wt denotes the parental wild-type replicon. A negative control replicon with an inactivating mutation in the NS5B RNA-dependent RNA polymerase did not yield any viable clones (not shown).
DISCUSSION
NS4B is an integral membrane protein that plays a central role in the formation of the HCV replication complex. By fluorescence microscopy as well as membrane extraction and flotation analyses, we identified a novel determinant for membrane association in the N-terminal portion of NS4B. This determinant was mapped to aa 40 to 69 and was found to comprise an amphipathic α-helix (aa 42 to 66) that displays lipotropic properties and has the potential to traverse the phospholipid bilayer as a transmembrane segment, likely upon oligomerization. Site-directed mutagenesis revealed a key role of conserved aromatic residues in mediating membrane association and transmembrane topology as well as an essential role of this amphipathic α-helix in HCV replication complex assembly.
Elazar et al. previously reported that an amphipathic α-helix predicted within NS4B aa 1 to 26 mediates membrane association (10). At variance with this report, we found that GFP fusion constructs harboring NS4B aa 1 to 29 (data not illustrated) or 1 to 40 were not membrane associated. Consistent with this finding, the interfacial hydrophobicity plot shown in Fig. 1C does not predict any membrane binding propensity in this region. A closer inspection of the expression construct of Elazar et al. revealed that their 42-aa sequence (SQHLPYIEQGMMLAEQFKQKALGLLQLRRLHQWISSECTTPC) corresponds to the N-terminal 26 aa fused to the C-terminal 16 aa of NS4B (indicated in italics in the above sequence). According to all secondary-structure prediction methods tested, this sequence has the potential to fold into a long α-helix (underlined in the sequence above) with amphipathic properties (data not shown). Moreover, the interfacial hydrophobicity plot (data not shown) clearly predicts lipotropic properties in the C-terminal half of this sequence. In addition, this 42-aa sequence includes two C-terminal cysteine residues that have recently been reported to be palmitoylated (59). Taken together, these predictions are in keeping with the membrane binding properties of the 42-aa construct reported by Elazar et al., but we conclude that the very N-terminal region of NS4B, including aa 1 to 40, does not harbor a bona fide membrane binding determinant. One cannot exclude, however, the possibility that the predicted first amphipathic helix may participate in the dynamic transmembrane reorganization of amphipathic α-helix 42-66 and/or its oligomerization.
Our data, based on prediction algorithms, CD, NMR, ATR-FTIR, glycosylation acceptor site tagging, and site-directed mutagenesis, are compatible with a transient in-plane topology of NS4B amphipathic α-helix 42-66 that can evolve to a transmembrane orientation. These findings are in agreement with the observation by Lundin et al. of a partial translocation of the N terminus of NS4B through the membrane, as documented by the glycosylation of an artificial acceptor site inserted at aa position 33 (28).
The transmembrane orientation of NS4B amphipathic α-helix 42-66 is likely associated with oligomerization. Such oligomer formation is expected based on physicochemical considerations which would invoke the formation of a hydrophilic internal core by the hydrophilic helix sides and of an outer hydrophobic face formed by the hydrophobic sides. However, one cannot exclude the formation of heterooligomers with other viral or cellular membrane elements. In both scenarios, it is likely that specific protein-protein interactions are involved, suggesting a potential for regulated multifunctionality of this segment. As suggested by Lundin et al., translocation may be regulated by the coexpression and processing of other HCV nonstructural proteins (27). An exciting consequence of the putative oligomerization of α-helix 42-66 would be the formation of membrane pores which could play a role in the as yet poorly characterized functions of HCV NS4B. Structural data on full-length NS4B are expected to yield insight into these intriguing issues.
Of note, the membrane association of the aa 40-69 segment is dispensable for the membrane targeting of full-length NS4B, indicating that one or more internal determinants can ensure membrane association. However, HCV polyprotein expression constructs harboring mutations that abolish membrane association of the aa 40-69 segment showed a striking defect in the dotlike structure formation as a correlate of membranous web formation. In addition, the introduction of these mutations into a subgenomic replicon completely abrogated HCV RNA replication. These observations point toward an important role of NS4B amphipathic α-helix 42-66 in HCV replication complex assembly. Clearly, further studies are required to elucidate the mechanistic role of NS4B amphipathic α-helix 42-66 in HCV replication complex assembly and to resolve the complete membrane topology of NS4B.
Acknowledgments
We gratefully acknowledge Ralf Bartenschlager, Hans-Peter Hauri, Jan Albert Hellings, and Charles M. Rice for reagents; Christophe Combet for bioinformatics support; and Eve Pécheur for expert advice on liposome preparation. CD experiments were performed on the platform Production et Analyse des Protéines of the IFR 128 BioSciences Gerland-Lyon Sud.
This work was supported by the Swiss National Science Foundation (3100A0-107831 and 3100A0-122447), the Swiss Cancer League/Oncosuisse (OCS-01762-08-2005), the Leenaards Foundation, the Désirée and Niels Yde Foundation, the European Commission (LSHM-CT-2004-503359, VIRGIL Network of Excellence on Antiviral Drug Resistance), the French Centre National de la Recherche Scientifique, the French National Agency for Research on AIDS and Viral Hepatitis, and the program Biotherapeutics of Lyon Biopole. N.A. was supported by the Alexander von Humboldt Foundation. V.R. is a senior research associate of the National Fund for Scientific Research (Belgium).
Footnotes
Published ahead of print on 8 April 2009.
REFERENCES
- 1.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 7613001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brass, V., J. M. Berke, R. Montserret, H. E. Blum, F. Penin, and D. Moradpour. 2008. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3-4A complex. Proc. Natl. Acad. Sci. USA 10514545-14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brass, V., E. Bieck, R. Montserret, B. Wölk, J. A. Hellings, H. E. Blum, F. Penin, and D. Moradpour. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2778130-8139. [DOI] [PubMed] [Google Scholar]
- 4.Buck, M. 1998. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 31297-355. [DOI] [PubMed] [Google Scholar]
- 5.Combet, C., C. Blanchet, C. Geourjon, and G. Deléage. 2000. NPS@: network protein sequence analysis. Trends Biochem. Sci. 25147-150. [DOI] [PubMed] [Google Scholar]
- 6.Combet, C., N. Garnier, C. Charavay, D. Grando, D. Crisan, J. Lopez, A. Dehne-Garcia, C. Geourjon, E. Bettler, C. Hulo, P. Le Mercier, R. Bartenschlager, H. Diepolder, D. Moradpour, J.-M. Pawlotsky, C. Rice, C. Trépo, F. Penin, and G. Deléage. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res. 35D363-D366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egger, D., B. Wölk, R. Gosert, L. Bianchi, H. E. Blum, D. Moradpour, and K. Bienz. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 765974-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Einav, S., M. Elazar, T. Danieli, and J. S. Glenn. 2004. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J. Virol. 7811288-11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Einav, S., D. Gerber, P. D. Bryson, E. H. Sklan, M. Elazar, S. J. Maerkl, J. S. Glenn, and S. R. Quake. 2008. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat. Biotechnol. 261019-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elazar, M., P. Liu, C. M. Rice, and J. S. Glenn. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J. Virol. 7811393-11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Englert, C., X. Hou, S. Maheswaran, P. Bennett, C. Ngwu, G. G. Re, A. J. Garvin, M. R. Rosner, and D. A. Haber. 1995. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. EMBO J. 144662-4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Favier, A., B. Brutscher, M. Blackledge, A. Galinier, J. Deutscher, F. Penin, and D. Marion. 2002. Solution structure and dynamics of Crh, the Bacillus subtilis catabolite repression HPr. J. Mol. Biol. 317131-144. [DOI] [PubMed] [Google Scholar]
- 13.Fringeli, U., and H. H. Gunthard. 1981. Infrared membrane spectroscopy. In E. Grell, ed., Membrane spectroscopy. Springer Verlag, Berlin, Germany.
- 14.Goormaghtigh, E., V. Cabiaux, and J. M. Ruysschaert. 1994. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. I. Assignments and model compounds. Subcell. Biochem. 23329-362. [DOI] [PubMed] [Google Scholar]
- 15.Goormaghtigh, E., V. Cabiaux, and J. M. Ruysschaert. 1994. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Subcell. Biochem. 23405-450. [DOI] [PubMed] [Google Scholar]
- 16.Goormaghtigh, E., V. Cabiaux, and J. M. Ruysschaert. 1990. Secondary structure and dosage of soluble and membrane proteins by attenuated total reflection Fourier-transform infrared spectroscopy on hydrated films. Eur. J. Biochem. 193409-420. [DOI] [PubMed] [Google Scholar]
- 17.Goormaghtigh, E., V. Raussens, and J. M. Ruysschaert. 1999. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1422105-185. [DOI] [PubMed] [Google Scholar]
- 18.Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 775487-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granseth, E., G. von Heijne, and A. Elofsson. 2005. A study of the membrane-water interface region of membrane proteins. J. Mol. Biol. 346377-385. [DOI] [PubMed] [Google Scholar]
- 20.Gretton, S. N., A. I. Taylor, and J. McLauchlan. 2005. Mobility of the hepatitis C virus NS4B protein on the endoplasmic reticulum membrane and membrane-associated foci. J. Gen. Virol. 861415-1421. [DOI] [PubMed] [Google Scholar]
- 21.Hügle, T., F. Fehrmann, E. Bieck, M. Kohara, H.-G. Kräusslich, C. M. Rice, H. E. Blum, and D. Moradpour. 2001. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 28470-81. [DOI] [PubMed] [Google Scholar]
- 22.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 1433-38. [DOI] [PubMed] [Google Scholar]
- 23.Ivashkina, N., B. Wölk, V. Lohmann, R. Bartenschlager, H. E. Blum, F. Penin, and D. Moradpour. 2002. The hepatitis C virus RNA-dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J. Virol. 7613088-13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277570-574. [DOI] [PubMed] [Google Scholar]
- 25.Koradi, R., M. Billeter, and K. Wuthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1451-55. [DOI] [PubMed] [Google Scholar]
- 26.Laskowski, R. A., J. A. Rullmann, M. W. MacArthur, R. Kaptein, and J. M. Thornton. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8477-486. [DOI] [PubMed] [Google Scholar]
- 27.Lundin, M., H. Lindstrom, C. Gronwall, and M. A. Persson. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J. Gen. Virol. 873263-3272. [DOI] [PubMed] [Google Scholar]
- 28.Lundin, M., M. Monne, A. Widell, G. Von Heijne, and M. A. Persson. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 775428-5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merutka, G., H. J. Dyson, and P. E. Wright. 1995. “Random coil” 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR 514-24. [DOI] [PubMed] [Google Scholar]
- 30.Miller, D. J., and P. Ahlquist. 2002. Flock house virus RNA polymerase is a transmembrane protein with amino-terminal sequences sufficient for mitochondrial localization and membrane insertion. J. Virol. 769856-9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyanari, Y., M. Hijikata, M. Yamaji, M. Hosaka, H. Takahashi, and K. Shimotohno. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral RNA replication. J. Biol. Chem. 27850301-50308. [DOI] [PubMed] [Google Scholar]
- 32.Montserret, R., E. Aubert-Foucher, M. J. McLeish, J. M. Hill, D. Ficheux, M. Jaquinod, M. van der Rest, G. Deleage, and F. Penin. 1999. Structural analysis of the heparin-binding site of the NC1 domain of collagen XIV by CD and NMR. Biochemistry 386479-6488. [DOI] [PubMed] [Google Scholar]
- 33.Moradpour, D., E. Bieck, T. Hügle, W. Wels, J. Z. Wu, Z. Hong, H. E. Blum, and R. Bartenschlager. 2002. Functional properties of a monoclonal antibody inhibiting the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 277593-601. [DOI] [PubMed] [Google Scholar]
- 34.Moradpour, D., M. J. Evans, R. Gosert, Z. H. Yuan, H. E. Blum, S. P. Goff, B. D. Lindenbach, and C. M. Rice. 2004. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 787400-7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moradpour, D., P. Kary, C. M. Rice, and H. E. Blum. 1998. Continuous human cell lines inducibly expressing hepatitis C virus structural and nonstructural proteins. Hepatology 28192-201. [DOI] [PubMed] [Google Scholar]
- 36.Moradpour, D., F. Penin, and C. M. Rice. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5453-463. [DOI] [PubMed] [Google Scholar]
- 37.Morgenstern, J. P., and H. Land. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 183587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.National Institutes of Health. 2002. National Institutes of Health consensus development conference statement: management of hepatitis C—2002. Hepatology 36(Suppl. 1)S3-S20. [DOI] [PubMed] [Google Scholar]
- 39.Opella, S. J. 1997. NMR and membrane proteins. Nat. Struct. Biol. 4845-848. [PubMed] [Google Scholar]
- 40.Penin, F., V. Brass, N. Appel, S. Ramboarina, R. Montserret, D. Ficheux, H. E. Blum, R. Bartenschlager, and D. Moradpour. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 27940835-40843. [DOI] [PubMed] [Google Scholar]
- 41.Penin, F., C. Geourjon, R. Montserret, A. Bockmann, A. Lesage, Y. S. Yang, C. Bonod-Bidaud, J. C. Cortay, D. Negre, A. J. Cozzone, and G. Deléage. 1997. Three-dimensional structure of the DNA-binding domain of the fructose repressor from Escherichia coli by 1H and 15N NMR. J. Mol. Biol. 270496-510. [DOI] [PubMed] [Google Scholar]
- 42.Qu, L., L. K. McMullan, and C. M. Rice. 2001. Isolation and characterization of noncytopathic pestivirus mutants reveals a role for nonstructural protein NS4B in viral cytopathogenicity. J. Virol. 7510651-10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinkert, D., R. Bartenschlager, and V. Lohmann. 2005. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 7913594-13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raussens, V., V. Narayanaswami, E. Goormaghtigh, R. O. Ryan, and J. M. Ruysschaert. 1995. Alignment of the apolipophorin-III alpha-helices in complex with dimyristoylphosphatidylcholine. A unique spatial orientation. J. Biol. Chem. 27012542-12547. [DOI] [PubMed] [Google Scholar]
- 45.Robinson, A. B., J. W. Scotchler, and J. H. McKerrow. 1973. Rates of nonenzymatic deamidation of glutaminyl and asparaginyl residues in pentapeptides. J. Am. Chem. Soc. 958156-8159. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt-Mende, J., E. Bieck, T. Hügle, F. Penin, C. M. Rice, H. E. Blum, and D. Moradpour. 2001. Determinants for membrane association of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 27644052-44063. [DOI] [PubMed] [Google Scholar]
- 47.Schweizer, A., M. Ericsson, T. Bachi, G. Griffiths, and H. P. Hauri. 1993. Characterization of a novel 63 kDa membrane protein. Implications for the organization of the ER-to-Golgi pathway. J. Cell Sci. 104671-683. [DOI] [PubMed] [Google Scholar]
- 48.Schwer, B., S. Ren, T. Pietschmann, J. Kartenbeck, K. Kaehlcke, R. Bartenschlager, T. S. Yen, and M. Ott. 2004. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 787958-7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwieters, C. D., J. J. Kuszewski, N. Tjandra, and G. M. Clore. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 16065-73. [DOI] [PubMed] [Google Scholar]
- 50.Simmonds, P., J. Bukh, C. Combet, G. Deleage, N. Enomoto, S. Feinstone, P. Halfon, G. Inchauspe, C. Kuiken, G. Maertens, M. Mizokami, D. G. Murphy, H. Okamoto, J. M. Pawlotsky, F. Penin, E. Sablon, I. T. Shin, L. J. Stuyver, H. J. Thiel, S. Viazov, A. J. Weiner, and A. Widell. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42962-973. [DOI] [PubMed] [Google Scholar]
- 51.Tellinghuisen, T. L., M. J. Evans, T. von Hahn, S. You, and C. M. Rice. 2007. Studying hepatitis C virus: making the best of a bad virus. J. Virol. 818853-8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vuilleumier, S., J. Sancho, R. Loewenthal, and A. R. Fersht. 1993. Circular dichroism studies of barnase and its mutants: characterization of the contribution of aromatic side chains. Biochemistry 3210303-10313. [DOI] [PubMed] [Google Scholar]
- 54.Whitmore, L., and B. A. Wallace. 2004. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32W668-W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3842-848. [DOI] [PubMed] [Google Scholar]
- 56.Wishart, D. S., B. D. Sykes, and F. M. Richards. 1992. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 311647-1651. [DOI] [PubMed] [Google Scholar]
- 57.Wüthrich, K. 1986. NMR of proteins and nucleic acids. John Wiley & Sons, New York, NY.
- 58.Yau, W. M., W. C. Wimley, K. Gawrisch, and S. H. White. 1998. The preference of tryptophan for membrane interfaces. Biochemistry 3714713-14718. [DOI] [PubMed] [Google Scholar]
- 59.Yu, G. Y., K. J. Lee, L. Gao, and M. M. Lai. 2006. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J. Virol. 806013-6023. [DOI] [PMC free article] [PubMed] [Google Scholar]