

Abstract
Naphthalene and close structural analogues have been shown to cause necrosis of bronchiolar epithelial cells in mice by both inhalation exposure and by systemic administration. Cancer bioassays of naphthalene in mice have demonstrated a slight increase in bronchiolar/alveolar adenomas in female mice, and in inflammation and metaplasia of the olfactory epithelium in the nasal cavity. Similar work in rats demonstrated a significant, and concentration-dependent increase in the incidence of respiratory epithelial adenomas and neuroblastomas in the nasal epithelium of both male and female rats. Although the studies on the acute toxicity of the methylnaphthalene derivatives are more limited, it appears that the species selective toxicity associated with naphthalene administration also is observed with methylnaphthalenes. Chronic administration of the methylnaphthalenes, however, failed to demonstrate the same oncogenic potential as that observed with naphthalene. The information available on the isopropylnaphthalene derivatives suggests that they are not cytotoxic. Like the methylnaphthalenes, 1-nitronaphthalene causes lesions in both Clara and ciliated cells. However, the species selective lung toxicity observed in the mouse with both naphthalene and the methylnaphthalenes is not seen with 1-nitronaphthalene. With 1-nitronaphthalene, the rat is far more susceptible to parenteral administration of the compound than mice. The wide-spread distribution of these compounds in the environment and the high potential for low level exposure to humans supports a need for further work on the mechanisms of toxicity in animal models with attention to whether these processes are applicable to humans. Although it is tempting to suppose that the toxicity and mechanisms of toxicity of the alkylnaphthalenes and nitronaphthalenes are similar to naphthalene, there is sufficient published literature to suggest that this may not be the case. Certainly the enzymes involved in the metabolic activation of each of these substrates are likely to differ. The available data showing extensive oxidation of the aromatic nucleus of naphthalene, nitronaphthalene and the methylnaphthalenes (with some oxidation of the methyl group) contrasts with the isopropylnaphthalene derivatives, where the major metabolites involve side chain oxidation. Overall, these data support the view that ring epoxidation is a key step in the process involved in cytotoxicity. Whether the epoxide itself or a downstream metabolite mediates the toxic effects is still not clear even with naphthalene, the best studied of this group of compounds. Additional work is needed in several areas to further assess the potential human health consequences of exposure to these agents. These studies should involve the definition of the extent and severity of methylnaphthalene toxicity after single dose exposures with attention to both the nasal and respiratory epithelia. The cytochromes P450 responsible for the initial activation of these agents in rodents with subsequent complimentary studies in primate models should help determine whether key metabolic processes responsible for toxicity occur also in primates. Finally, the precise involvement of reactive metabolite formation and adduction of cellular proteins in toxicity will be important in not only assessing the potential for human toxicity, but also in developing an understanding of the genetic and environmental factors which could alter the toxicity of these agents.
Keywords: naphthalene, 1-methylnaphthalene, 2-methylnaphthalene, dimethylnaphthelenes, 1-nitronaphthalene, nasal toxicity, Clara cells, cytochrome P450, respiratory tract
Introduction
Chronic obstructive pulmonary diseases are currently the fourth leading cause of death worldwide (Chapman et al., 2006). The incidence of cancer of the lung is second only to cancers of the digestive system. More importantly, because the prognosis for cancers of the lung is so poor, it is the leading cause of cancer-related deaths in both males and females in the US. One of the most clearly established etiologic factors in the incidence of lung diseases is related to the use of tobacco products. While many of the several thousand constituents of tobacco smoke are known to result in both acute injury and in pulmonary cancer in experimental animals, the levels found in cigarette smoke are considerably less than those required to produce effects in experimental animal models (Witschi et al., 1997). Potentially, the overall “burden” of exposure, causing an increased incidence of pulmonary cancer and other lung diseases in smokers is a result of the combined effects of carcinogens and toxicants in cigarette smoke with exposure to other environmental and industrial chemicals. As a class, the volatile and semi-volatile, low molecular weight aromatic hydrocarbons, such as naphthalene, nitronaphthalenes and several of the alkylnaphthalenes, are possible contributors to these processes both because they selectively target the respiratory tract and there is relatively widespread human exposure. Although naphthalene is the best studied of the low molecular weight aromatic hydrocarbons both in terms of human exposure (Li et al., 2007; Kang-Sickel et al., 2008) and in terms of health effects, a number of petroleum products including gasoline, jet fuel and kerosene contain significant quantities of the methylnaphthalenes (Kim et al., 2006; Bagheri and Creaser, 1988). The main emissions from coal tar creosote, which is ranked 23rd in the Superfund/National Priorities List of hazardous chemicals, are the methylnaphthalenes which, in some cases, account for nearly 20% of the volatiles and are present in significantly higher quantities than naphthalene (Gallego et al., 2008). Accordingly, this review focuses on the sources of human exposure as well as on the available data on the toxicology and metabolism of substituted naphthalene derivatives, including 1- and 2-methylnaphthalene, isopropylnaphthalenes and 1-nitronaphthalene. Where appropriate, these will be placed in context with the information on the most studied of the aromatic hydrocarbons, naphthalene. Several recent reviews have been published on this topic and the reader is referred to these for further information (Anon, 2005; Buckpitt et al., 2002; Preuss et al., 2003). There are substantial gaps in our understanding of the metabolism of methylnaphthalene and the relationship of formation of specific metabolites to toxicity- especially when viewed in light of our understanding of naphthalene toxicity and metabolism. Although more information is available regarding specific metabolic pathways for 1-nitronaphthalene, further studies focusing on the identification of protein adducts generated from electrophilic intermediates derived from 1-nitronaphthalene, as well as their possible involvement in the cascade of events which leads to cellular degeneration are needed. Thus, one of the goals of this review is to highlight where additional work is needed to understand the mechanisms of toxicity of these chemicals. Ideally, this additional work might point to the development and implementation of biomarker approaches that would assist in determining whether the data developed in rodent models is applicable to the human. In all cases, we suggest that use of non-human primates can be an excellent intermediate species in which to test key concepts and to validate the approaches which could subsequently be applied to exposed human populations. Only in this fashion can we hope to make well-educated judgments regarding the potential for deleterious human health effects of exposure to these compounds.
Sources of human exposure
Like naphthalene, the methylnaphthalenes are relatively ubiquitous in the environment. They are by-products of various combustion processes and are emitted from both gasoline and diesel-powered vehicles. Naphthalene and close structural congeners (methyl, dimethyl and methylnitro derivatives) are quantitatively the most significant PAH emitted from mobile sources. In contrast to the nitrated aromatics, the amounts of methylnaphthalene emitted from diesel powered trucks are more than 10 fold lower per km than a gasoline powered car (Hampton et al., 1983). The gas phase concentrations of methylnaphthalenes (1- and 2-methyl) emitted are slightly lower than those measured for naphthalene, but are similar to many of the alkanes measured and are several orders of magnitude higher than the larger PAHs (Reisen and Arey, 2005). As a class, the parent naphthalene and methyl/ethyl/dimethylnaphthalenes have been measured in ambient air in Los Angeles at concentrations above 2.5 μg m-3 (Wang et al., 2007). Recent studies have demonstrated the presence of several 2 and 3 ring PAH on wheat in California’s Central Valley (Kobayashi et al., 2008). Although the levels of naphthalene (as high as 12 μg/kg) exceeded those of the methylnaphthalenes by more than 10-fold, the methylnaphthalenes as a group (1- and 2-methyl as well as di- and trimethylnaphthalenes) constituted the second most abundant class of PAH found in grain. The levels of PAH in the grain are thought to reflect levels in ambient air since there was no difference in concentration in wheat that was mechanically harvested vs hand harvested.
The methylnaphthalenes undergo gas phase reactions in the atmosphere which are catalyzed by OH• and NO2 to generate methylnitronaphthalenes and dicarbonyl derivatives (Gupta et al., 1996; Grosovsky et al., 1999; Phousongphouang and Arey, 2002, 2003; Wang et al., 2007; Nishimo et al., 2008). These nitrated naphthalenes and nitromethylnaphthalenes have been reported to account for a substantial fraction of the mutagenicity associated with particulates in the atmosphere (Grosovsky et al., 1999). 1- and 2-Methylnaphthalene are both natural constituents of crude oil and crude oil derivatives, and are common contaminants found in the refining industry (Aislabie et al., 1999). They are present in roofing and asphalt tars, and are the single most prevalent of the aromatic hydrocarbons reported in measurements of vapor phase constituents (NIOSH, 2000). The concentrations of methylnaphthalenes exceed those of naphthalene by two-fold and are 20-30 fold higher than the larger hydrocarbons. Methyl-naphthalenes are major constituents of creosote and high levels have been measured in coal tar derived from former gas works plants (Anon, 1984). One of the major industrial uses of 2-methylnaphthalene is as a starting point in the synthesis of Vitamin K but worker exposures have received relatively little attention. Likewise, 1- and 2-methylnaphthalene are used as intermediates in the synthesis of various pesticides including carbaryl; pesticide synthesis accounts for slightly more than 10% of the industrial use of methylnaphthalenes. Furthermore, methylnaphthalenes are generated during aluminum smelting, and high levels have been measured in the workplace (Bjorseth et al., 1978).
Mainstream and side stream cigarette smoke are likely a significant source of human exposure to methylnaphthalenes. The combined levels of 1- and 2-methylnaphthalene in mainstream cigarette smoke are slightly less than those of naphthalene, but are approximately 30% higher than naphthalene in side stream smoke (Schmeltz et al., 1976). The naphthalenes are present in both fractions of cigarette smoke at concentrations that far exceed those of the less volatile polycylic aromatic hydrocarbons such as benzo(a)pyrene (Witschi et al., 1997).
The primary atmospheric source of human exposure to the nitronaphthalenes is via airborne particles; these appear to be generated from diesel combustion and as an atmospheric reaction product from the parent aromatic hydrocarbons. These products include nitronaphthalene, methylnitronaphthalenes and dimethylnitronaphthalenes (Phousongphouang and Arey, 2003). In addition to the atmospheric exposures, there are potential workplace exposures in the aluminum smelting industry where nitronaphthalene has been measured in the coal tar pitch used to prepare electrodes (Farant and Ogilvie, 2002).
Toxicity of 1 and 2-methylnaphthalenes: Comparison to naphthalene and 1-nitronaphthalene
Acute toxicity-Lung
A number of studies have reported on the dose- and time-dependent cytotoxicity of 2-methylnaphthalene in mice. The times of maximal injury as well as the minimal doses at which injury was observed appear to vary somewhat by strain (ddy, Swiss Webster, C57Bl/6 and DBA) (Griffin et al., 1982; 1983; Rasmussen et al., 1986; Honda et al., 1990). Intraperitoneal administration of methylnaphthalenes to mice caused dose-dependent injury to nonciliated bronchiolar epithelial (Clara) cells, first observed 8 h after administration of 400 mg/kg. Twenty four h post exposure, extensive exfoliation of cells from the airways had occurred, and the epithelium was completely denuded in 20% of the subjects (Griffin et al., 1981; 1982). Similarly, in DBA mice, signs of pulmonary epithelial injury were observed starting at the 100 mg/kg dose, and injury became progressively worse with increasing doses (Griffin et al., 1983). Later studies evaluating the lung toxicity of methyl- and 1-nitronaphthalene derivatives by both light and electron microscopy corroborated these findings and showed that, while the primary target cell for 1- and 2-methylnaphthalene injury was the Clara cell, ciliated cells were affected as well. No discernable injury to alveolar epithelial or capillary endothelial cells was noted in these studies (Rasmussen et al., 1986). The severity of the lung lesion associated with 2-methynaphthalene treatment was reported to be similar to that of naphthalene, and intermediate to that of 1-nitronaphthalene (most toxic) and 1-methylnaphthalene (least toxic). At the highest doses of 2-methylnaphthalene tested (426 mg/kg), ciliated cells appeared flattened and vacuolated and many Clara cells had exfoliated from the bronchiolar epithelium. Signs of bronchiolar injury were noted as early as the 6 hr time point; injury was maximal 24 h after toxicant administration. Alterations in the bronchiolar epithelium 72 h after administration of high doses of 2-methylnaphthalene were similar to those noted at 24 h. By 7 days postreatment, repair was well underway and signs of the severe toxicity noted at 24 h were not evident. There were no discernable differences in the appearance of the bronchiolar epithelium between control and treated animals 14 days after treatment. No evidence of hepatic or renal injury associated with methylnaphthalenes (1- or 2-methyl) was reported (Rasmussen et al., 1986).
After intraperitoneal administration, the doses of methylnaphthalenes used to ellicit a lesion in the airway epithelium are very high and are well above the range of any potential human exposures. Similarly, in the species most sensitive to the airway epithelial toxicity of naphthalene, doses of 50 mg/kg or greater are needed to produce observable structural alterations in Clara cells (Plopper et al., 1992b). However, when exposures are via the inhalation route, 4 h exposures of 2 ppm (well below the current occupational exposure standard of 10 ppm for 8 h) detectable alterations in the morphology of Clara cells were observed (West et al., 2001). The rat nasal epithelium appears even more sensitive. Whereas 200 mg/kg doses of naphthalene administered intraperitoneally cause necrosis of olfactory epithelium of rats (Plopper et al., 1992a), low ppm or even sub-ppm inhaled concentrations are sufficient to produce detectable lesions (Lee et al., 2005; Dodd et al., 2008). Although the various pathologic endpoints associated with methylnaphthalene inhalation have been studied following long-term exposure, there is no information available on changes in the epithelium of the respiratory tract after inhalation. There is a need for additional, detailed studies in this area to define levels below which there are no detectable adverse effects.
In contrast to the lung injury associated with the methylnaphthalenes, which resembles naphthalene in that it is species selective, 1-nitronaphthalene produces lung injury in both rats (Dinsdale and Verschoyle, 1987, Verschoyle et al., 1993; Sauer et al., 1995, 1997; Paige et al., 1997, Fanucchi et al., 2004) and mice (Rasmussen et al., 1986; Fanucchi et al., 1997). Airway epithelial cells of adult rats are injured at lower doses than in mice. There was a significant decrease in the percentage of ciliated cells and an increase in vacuolated cells in midlevel airways of adult rats treated with 50 mg/kg 1-nitronaphthalene ip. In comparison, only 100 mg/kg doses resulted in an increase in vacuolated cells in either terminal or midlevel airways of mice. Significant decreases in ciliated cells were not observed at any dose of 1-nitronaphthalene examined (Fanucchi et al., 2004). In rats, at doses of 25 mg/kg, toxicity is confined to the Clara cells of intermediate airways whereas higher doses resulted in nearly complete exfoliation of nonciliated epithelial cells of both large (trachea) and small airways (including the terminal bronchiole). At lethal or near-lethal (100-150 mg/kg) doses of 1-nitronaphthalene, ciliated cells were vacuolated and there were areas of denuded basement membrane. In contrast to naphthalene and the methylnaphthalenes where parenteral administration of high doses causes no measurable hepatotoxicity, 1-nitronaphthalene is a hepatotoxicant in both mice (Rasmussen et al., 1986) and rats (Verschoyle et al., 1993; Sauer et al., 1995, 1997).
Later studies comparing the pulmonary toxicity of naphthalene with 2-methyl-, isopropyl and diisopropylnaphthalene confirmed earlier work with naphthalene and 2-methylnaphthalene and, further, showed that administration of either the isopropyl or diisopropylnaphthalene, even at doses of 3000 mg/kg, did not result in any detectable lung injury. Four h inhalation exposures of both mice and rats to 1- or 2-methylnaphthalene at concentrations which varied from 5-70 ppm resulted in a concentration-dependent decrease in respiratory rate (Korsak et al., 1998). Although no alterations were noted in rotarod performance, decrements in pain response were observed at the higher inhalation concentrations for both compounds. Tissues were not assessed by histopathology and the effects reported could well be due to decrements in CNS function.
A number of factors have been identified that alter the susceptibility of respiratory tissues to low molecular weight aromatic hydrocarbons. Female mice are more susceptible than males to the acute cytotoxic effects of naphthalene (Van Winkle et al., 2002). Likewise, despite the fact that the pulmonary cytochrome P450 monooxygenases are not fully developed, neonatal rats and mice are more susceptible to the acute toxicity of both naphthalene and 1-nitronaphthalene than are adult animals (Fanucchi et al., 1997, 2004).
Acute Toxicity- Nasal epithelium
The nasal epithelium is a target for naphthalene in both rats and mice after both parenteral administration (Plopper et al., 1992a) and inhalation exposure (Lee et al., 2005). The volatility of methylnaphthalenes is only slightly less than that reported for naphthalene. The fact that naphthalene and methylnaphthalenes share the same species and cell selective toxicities and that these compounds are metabolized via similar pathways suggests that the naal epithelium might be an important target for the methylnaphthalenes. This deserves additional experimental investigation.
Chronic toxicity
Tolerance
A number of years ago, our laboratories reported that lungs from mice treated daily with necrogenic doses of naphthalene (200 mg/kg/day for 7 days) not only appear normal at the end of 7 days but are remarkably resistant to high challenge doses of the compound (O’Brien at al., 1989; Lakritz et al., 1996). The tolerance is not restricted to naphthalene administered parenterally but can be induced by inhalation of concentrations as low as 10 ppm (West et al., 2003). It is now clear that alterations in glutathione metabolism account, at least in part, for the observed tolerance (West et al., 2000, 2002). Whether rodents develop tolerance to methyl- or nitronaphthalenes is not known, but this would appear to be an important issue for further consideration based on the fact that humans are exposed to small concentrations on a continuous basis.
Tumor formation
Both 1- and 2-methylnaphthalene have been tested for oncogenic effects in mouse feeding studies. The levels in the diet were established based on 13 week feeding studies which showed that dietary intake of concentrations higher than 0.15% resulted in a > 20% decrease in weight gain. Accordingly, both 1-and 2-methylnaphthalene were fed to B6C3F1 mice at concentrations of 0.075% or 0.15% for a period of 81 weeks (Murata et al., 1993; 1997). The incidence of adenomas in the bronchiolar/alveolar regions of the lung was elevated from control at both exposure concentrations of 1-methylnaphthalene in the diet, but only in male animals. Moreover, this effect was not concentration-dependent as the incidence of adenomas was identical in both treatment groups (Murata et al., 1993). No statistically significant increases in adenomas were noted in either male or female mice fed 2-methylnaphthalene in the diet. No other significant increases in tumors at other locations were noted in these studies leading to the conclusion that neither of the methylnaphthalenes studied possess unequivocal oncogenic potential. These findings are consistent with the short term mutagenicity studies which indicate that sister chromatid exchange frequencies were less than two fold higher than control at all concentrations of 1-and 2-methylnaphthalene examined. 2-Methylnaphthalene, in the presence of S9, results in cytotoxicity in the human lymphocyte test system but only at very high concentrations (Kulka et al., 1988). These findings are remarkeably similar to those with naphthalene where short term mutagenicity assays were generally negative (see Brusick, 2008 for review).
The cancer bioassays for naphthalene have been performed in both mice and rats. In mice, (a slight increase in bronchiolar/alveolar adenomas was observed in mice at the highest exposure level tested but only in females (Abdo et al., 1992). In rats, dose-dependent increases in adenomas of the respiratory epithelium and a significant increase in olfactory epithelial neuroblastomas were observed in males and females, respectively. In several animals, nasal masses, some of which had begun invading the central nervous system, were observed (Abdo, 2001). In the olfactory epithelium, the incidence of hyperplasia and chronic inflammation was nearly 100% even at the lowest concentration tested (10 ppm). Based on the similarities between naphthalene and methylnaphthalenes, additional short term exposures which focus on injury to the nasal epithelium are warranted.
Other non-oncogenic lesions were associated with feeding of both 1- and 2-methylnaphthalene. These lesions included marked increases in alveolar proteinosis (40-60% in both studies) in both males and females (Murata et al., 1993;1997). Likewise, skin painting studies in mice using total doses of 2-methylnaphthalene of 7 g/kg distributed over 30 weeks resulted in 100% incidence of alveolar proteinosis. This rare lesion also occurs in humans and appears to be related to marked accumulation of lipids and proteins in the alveolar regions of the lung (for review see Ioachimescu and Kavuru, 2006). The etiology of the disease in humans is not well- established but has been associated with inhalation exposures to particles including cement, silica and aluminum. Since the Clara cell is responsible for elaborating CC-10, a surfactant protein found in the alveolar regions of the lung (Ryerse et al., 2001), and overexpression of IL-4 using the CC-10 promotor construct results in alveolar proteinosis (Ikegami et al., 2000), it is possible that long term exposures to 1- and 2-methylnaphthalene affect the secretion and/or catabolism of surfactant proteins elaborated by cells residing in or near the terminal airways.
Metabolism of alkylnaphthalenes and nitronaphthalene
Initial metabolic step (regiochemistry, trapping with glutathione)
The first step in the metabolism of methylnaphthalenes can occur either via ring epoxidation or via oxidation of the alkylmethyl to generate an alcohol. Both processes are catalyzed by the cytochrome P450 monooxygenases. The role of these enzymes in the initial oxidation of methylnaphthalenes is relatively well-established experimentally (Griffin et al, 1981; Shultz et al., 2001). What is not well-documented is whether the cytochrome P450 monooxygenases are responsible for generating metabolites that mediate the lung toxicity of the methylnaphthalenes. The methylnaphthalenes selectively target Clara cells and, since Clara cells are an important locus of cytochrome P450 monooxygenases in the lung (Serabjit-Singh et al., 1980; Plopper, 1993), the tacit assumption is that in situ metabolic activation by the cytochrome P450 monooxygenases is associated with the mechanism of toxicity. This assumption appears to be correct with naphthalene (Warren et al., 1982) and 1-nitronaphthalene (Verschoyle and Dinsdale, 1990; Verschoyle et al., 1993) where pretreatment of animals with various inhibitors of the cytochrome P450 monooxygenases blocks the toxicity associated with subsequent administration of the aromatic hydrocarbon. In contrast, studies in which mice were pretreated with the cytochrome P450 monooxygenase inhibitors, SKF 525A or piperonyl butoxide, failed to demonstrate any effect of either inhibitor on 2-methylnaphthalene-induced lung toxicity (Griffin et al., 1982). Both SKF 525A and piperonyl butoxide form metabolite inhibitor complexes and thus require metabolism to exert their inhibitory effects on the cytochrome P450 complex (Halpert, 1995). Accordingly, they show some selectivity towards inhibition of specific cytochromes P450. One of the possibilities that must be considered is that different cytochrome P450s are responsible for 2-methylnaphthalene metabolism as compared to naphthalene. This view is partially supported by data showing that addition of either piperonyl butoxide or SKF 525A to microsomal incubations containing 2-methylnaphthalene decreased the rates of reactive metabolite formation by only 40% (Griffin et al., 1982) while, with naphthalene, inhibition was greater than 70% (Buckpitt et al., 1984).
The presence of the epoxides as intermediates in 2-methylnaphthalene metabolism is presumptive and is based on both mass spectral and NMR characterization of the 3,4-, 5,6-, and 7,8-dihydrodiols generated from 2-methylnaphthalene by microsomal enzymes (Griffin et al., 1982; Breger et al., 1983; Melancon et al., 1985). Further support for epoxides as intermediates in the cytochrome P450-dependent metabolism of 2-methylnaphthalene comes from studies demonstrating the presence of multiple dihydrohydroxyglutathione metabolites in incubations with glutathione and glutathione transferases using both microsomal enzymes and recombinant protein as methods for generating reactive metabolites (Teshima et al., 1983; Shultz et al., 2001). Although the identity of all of the glutathione conjugates was confirmed by mass spectrometry, the regio- and stereochemistry of conjugation were not determined. Liver microsomal enzymes produced nine conjugates which could be separated by HPLC while recombinant CYP2F2 generated only five of these. This suggests that metabolism occurs with a reasonable amount of either regio- or stereoselectivity. Earlier work using lung and liver microsomes demonstrated that the 7,8-diol was the predominant metabolite generated from 2-methylnaphthalene followed by the 5,6-diol. The 3,4-diol was generated at the lowest rates from 6 to 13 % of the total amounts of diol generated in the incubations (Griffin et al., 1981) (Figure 1). Similar regioselectivity was observed in lung microsomal incubations with 1-nitronaphthalene where epoxidation at the 7,8- position predominated (Watt et al., 1999; Watt and Buckpitt, 2000).
Figure 1.
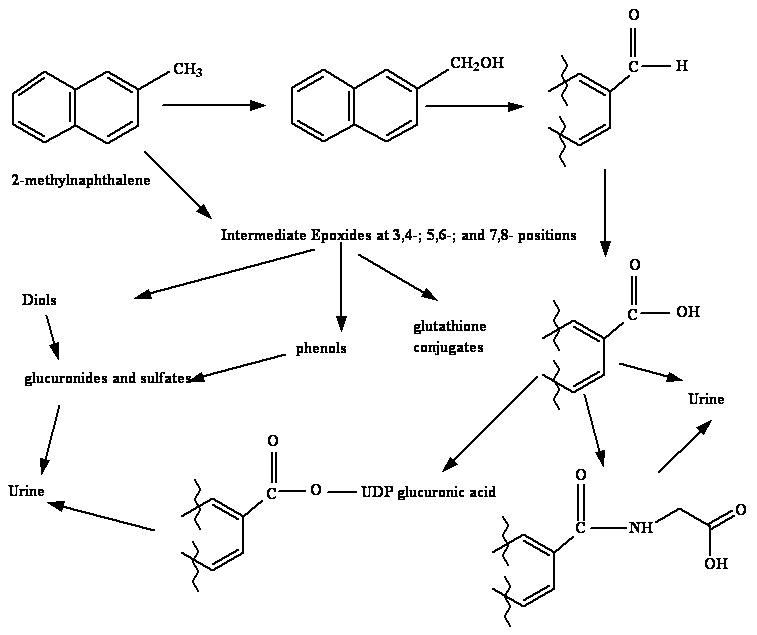
Enzymes involved in naphthalene/naphthalene congener metabolism
Comprehensive studies investigating the role of individual human cytochrome 450s have been reported with the metabolism of naphthalene but not any of the substituted naphthalenes (Cho et al., 2006). These investigators showed that the most catalytically active proteins involved in naphthalene metabolism (as assessed by Vmax/Km) were CYP1A2 and CYP2E1. CYP1A2 is localized primarily in the liver whereas CYP2E1 is found in a number of organs including respiratory tissue (Ding and Kaminsky, 2003). More recently, Fukami and coworkers (2008) have shown that CYP2A13 metabolizes naphthalene with relatively high turnover and low Km. Since this protein is expressed in human lung, albeit with a high degree of variability, it is a potential candidate for catalyzing the initial metabolism of naphthalene in human respiratory tissue (Zhang et al., 2007). Other data available come from work conducted with a single recombinant protein, CYP2F2 (Shultz et al., 2001). Although this protein appears to be abundant in airways of the mouse, available evidence suggests that the rat and Rhesus macaque orthologues are present in far smaller amounts in the lung (Baldwin et al., 2004, 2005; Buckpitt et al., 1995). This protein metabolizes naphthalene, 2-methylnaphthalene and 1-nitronaphthalene, all with relatively low Km and high Vmax (Table 2), and, based on inhibition studies with 5-phenyl-1-pentyne appears to play a major role in the epoxidation of closely related substrates, styrene (Cruzan et al., 2002) and coumarin (Born et al., 2002) . These data suggest that this protein plays a quantitatively important role in the metabolic activation of these substrates at least in the mouse. The presence of large quantities of this protein in target cells may, in part, explain the species differences in susceptibility to naphthalene and 2-methylnaphthalene in mouse but not in rat. Based on the effects of various inhibitors and inducers used in vivo, Verschoyle et al. (1993) concluded that CYP2B1 was the most likely cytochrome P450 involved in 1-nitronaphthalene activation in the lung. The fact that the cytotoxicity of 1-nitronaphthalene is not species selective, while naphthalene and the methylnaphthalenes are, is consistent with the view that cytochromes P450 other than CYP2F are responsible for the metabolic activation of 1-nitronaphthalene.
Table 2.
Comparison of Kinetics of Naphthalene, 2-methylnaphthalene and 1-Nitronaphthalene by Cytochrome P450 2F2
| Substrate | Kcat (min-1) | Km (μM) |
|---|---|---|
| naphthalene | 104 | 3 |
| 2-methylnaphthalene | 67.6 | 3.7 |
| 1-nitronaphthalene | 17.1 | 21.5 |
Data taken from Shultz et al., 2001.
Urinary metabolites
The most prominent metabolites isolated in rat urine after treatment with low doses of 2-methylnaphthalene originated from initial oxidation of the parent hydrocarbon on the methyl moiety (Melancon et al., 1982). Thirty to 35% of a dose of 14C-2-methylnaphthalene was recovered as a glycine conjugate of 2-naphthoic acid. Six to eight percent of the dose was represented by dihydrodiols and 3-5% of the dose was recovered as parent hydrocarbon. Other polar metabolites appeared to account for 35-45% of the radioactivity in the urine. Later work, by Teshima et al. (1983), showed that approximately 75% of the radioactive metabolites eliminated in the urine of guinea pigs administered a low dose of 3H-2-methylnaphthalene resulted from oxidation of the methyl group. These metabolites included free naphthoic acid, the glucuronide of naphthoic acid as well as the glycine conjugate. In these studies, a cysteine derivative, accounting for approximately 10% of the total urinary radioactivity, was reported in the urine. Finally, small percentages of sulfate and glucuronide conjugates of 8-hydroxy-2-methylnaphthalene (<10% of total urinary radioactivity) were measured.
Comparison of the metabolism of diisopropylnaphthalene with naphthalene, 2-methylnaphthalene and 1-nitronaphthalene provides a plausible mechanistic explanation for the lack of lung toxicity associated with both isopropyl and diisopropylnaphthalene (Hoke and Zellerhoff, 1998). In contrast to naphthalene, 2-methyl-, and 1-nitronaphthalene, diisopropyl naphthalene is metabolized primarily by side chain oxidation with little evidence for extensive formation of arene oxides (Kojima et al., 1982; 1985). Urinary metabolites derived from side chain oxidation of isopropylnaphthalene accounted for more than 40% of the dose of compound administered to rats whereas only 1.5% of the dose was eliminated as a dihydrodiol derivative (Kojima et al., 1985).
More recent studies on the disposition and metabolism of 3H-1,2-dimethylnaphthalene (28 mg/kg) in rats showed that the radioactive parent compound was rapidly absorbed after ip administration, reaching peak levels within 4 h (Kilanowicz and Sapota, 1998). Sixty five % of the administered radioactivity was recovered in the excreta within 24 h, with roughly equal amounts eliminated in the urine and feces. Greater than 95% of the administered radioactivity was recovered in the excreta within 72 h of administration. The highest tissue concentrations of radioactivity were observed in fat, but these fell rapidly to very low levels within 48 h. This compound apparently distributes rapidly to the fat but redistributes easily due to the rapid clearance of the compound. Urinary metabolites were identified in ether extracts of acidified (pH 1) urine. The parent compound (representing roughly 30% of the ether-extractable metabolites from urine), several dimethylthionaphthols, at least 2 dimethylmethylthionaphthalene derivatives as well as several derivatives generated from oxidation of the methyl groups to the alcohol and subsequently to the acid were measured in the urine following dimethylnaphthalene administration. The most prominent metabolites were the dimethylthionaphthol derivatives and the metabolites generated from side chain oxidation. We note that the 30% of the radioactivity unextracted by ether at pH 1 may include a number of conjugated metabolites including glucuronides, sulfates and mercapturic acids. The results from more recent studies of the metabolism and distribution of radioactivity from 3H-1,4-dimethylnaphthalene and 1,6-dimethylnaphthalene are nearly identical to those with the 1,2-dimethylnaphthalene derivative. Again, radioactivity is rapidly absorbed reaching peak plasma concentrations within 4 h of administration. Metabolites which were derived from both oxidation of the methyl groups and the aromatic nucleus (Kilanowicz and Sapota, 1998; Kilanowicz et al., 2000; 2002) were isolated from the urine of treated rats. These metabolites included methylnaphthoic acid as well as the intermediates leading to this derivative (methylhydroxymethyl, methylnaphthaldehyde). Trace quantities of a methylthio metabolite were observed; these metabolites have been measured in the urine of naphthalene-treated rodents as well (Stillwell et al., 1978, 1982).
Phenolic metabolites, excreted as glucuronides, sulfates and N-acetylcysteine conjugates, were eliminated in the urine after administration of 1-nitronaphthalene to rats. 1-Naphthylamine, a product of reductive metabolism of 1-nitronaphthalene accounts for less than 2% of the administered dose (Halladay et al., 1999). In contrast, human studies focusing on a larger nitroaromatic component of diesel exhaust (1-nitropyrene) showed that a third of the metabolites recovered in urine were reduced 1-amino derivatives (Toriba et al., 2007). This reductive metabolic step appears to be catalyzed by microflora in the gastrointestinal tract (El-Bayoumy et al., 1984).
Studies on the rates of metabolism of naphthalene and 1-nitronaphthalene in well-defined segments of the lung have been reported and provide some interesting contrasts. Neither compound results in any detectable injury to the alveolar epithelium (Plopper et al., 1992b) and, with naphthalene, this is consistent with the lack of metabolism of this substrate in the parenchyma (Plopper et al., 1991). In contrast, glutathione is depleted from the parenchymal subcompartment of rat lung 2 h after administration of 1-nitronaphthalene suggesting that either metabolism occurs in this subcompartment or that glutathione-depleting metabolites are diffusing from the liver or from other metabolically active segments of the lung. Subsequent work showed that the rates of 1-nitronaphthalene metabolism were as high in microsomes isolated from the parenchyma as from airway subcompartments (Watt et al., 1999), thus supporting the in situ generation of reactive metabolites from 1-nitronaphthalene. Thus, there is a mismatch between the capability to conduct substrate turnover and susceptibility to injury. Glutathione levels are slightly higher in the parenchymal subcompartment and this may, in part, explain the relative resistance to injury. Other factors, including differences in the amounts of protein adduct generated or the nature of proteins adducted by reactive metabolites, could account for the apparent differences in susceptibility of different regions of the respiratory tract which show similar ability to catalyze the formation of reactive epoxides. This concept has been supported recently by experimental data showing that adducts of reactive 1-nitronaphthalene metabolites with calreticulin occurred in airway epithelium from ozone-tolerant but not filtered-air exposed, control rats. Although it is plausible that the high degree of adduction of calreticulin is related to the substantial increase in susceptibility of ozone-tolerant compared to filtered air exposed animals treated with 1-nitronaphthalene, additional experimental support is necessary.
The role of glutathione in metabolism and toxicity of substituted naphthalenes
Not surprisingly, glutathione is depleted from the liver by approximately 50% after oral doses of 500 mg/kg 2-methylnaphthalene in guinea pigs (Teshima et al., 1983), and in a dose-dependent manner in the lungs of mice treated with doses varying from 144 to 432 mg/kg (Honda et al., 1990). An approximate loss of 50-65% of pulmonary glutathione was observed at the highest doses of 2-methylnaphthalene tested 6 h after treatment (Griffin et al., 1983; Honda et al., 1990). This contrasts with a loss of 40 and 35% for identical doses (on a molar basis) of isopropyl and diisopropylnaphthalene and is likely due to the fact that the primary metabolites of the isopropylnaphthalene are derived from side chain oxidation rather than via epoxidation. Prior treatment with glutathione depletors (diethyl maleate) markedly enhanced the subsequent Clara cell toxicity of 2-methylnaphthalene; Clara cells in mice treated with diethyl maleate followed by high doses of either isopropyl or diisopropyl naphthalene were unaffected by treatment (Honda et al., 1990).
N-Acetylcysteine derivatives of naphthalene and 1-nitronaphthalene are found in the urine of mice and rats treated with their respective parent hydrocarbons and these metabolites account for a substantial portion of the total urinary metabolites (Chen and Dorough, 1979; Pakenham et al., 2002; Halladay et al., 1999). Quantitative estimates of the percentages of dose of the alkylnaphthalenes eliminated as N-acetylcysteine conjugates in the urine of rodents have not been published, but side chain oxidation likely draws a significant percentage of the metabolites away from mercapturic acids.
Formation of reactive metabolites
There are extensive data showing that reactive metabolites of naphthalene, 2-methylnaphthalene and 1-nitronaphthalene are generated in vivo and become bound covalently to tissue proteins (see Buckpitt et al., 2002 for a review on naphthalene, Franklin et al., 1991 for review on 2-methylnaphthalene and Rasmussen, 1986 and Wheelock et al., 2005 for 1-nitronaphthalene). With all three compounds, the formation of reactive metabolites precedes any signs of tissue injury and the amounts of metabolite generated are related to the dose given. Tissue susceptibility to cytotoxicity does not correlate with the levels of bound metabolite in whole tissue for either naphthalene or 2-methylnaphthalene (Table 3). The overall covalent binding of reactive metabolites in the liver, a non-target tissue, is 30 to 250% higher than in the target tissue with naphthalene and 2-methylnaphthalene, respectively (Table 3). While this might be interpreted as indicating that reactive metabolite binding is not associated with toxicity, there are other possibilities which could potentially explain these data. The heterogeneity of lung tissue combined with the high degree of localization of the cytochrome P450 monooxygenases in airway epithelial cells likely means that reactive metabolites are bound covalently in a relatively small subset of the total cell population of the lung. Recent work in our laboratory with naphthalene supports this viewpoint. The overall levels of reactive naphthalene metabolite binding to airway epithelial cell proteins recovered by lysis lavage (Wheelock et al., 2004) were 2-4 fold higher than in residual tissue . In addition, with the strains of mice used in this study, the levels of bound metabolite in airway epithelium also were 2 to 4 fold higher than in the liver.
Table 3.
Approximate levels of covalently bound radioactivity associated with target and non-target tissues after administration of naphthalene, 1-nitronaphthalene or 2-methylnaphthalene
| Toxicant | Dose (mmoles/kg) | Lung (nmoles/mg protein) | Liver (nmoles/mg protein) |
|---|---|---|---|
| Naphthalenea | 1.6 | 0.6 | 0.8 |
| 1-Nitronaphthaleneb | 0.29 | 5.0 | 1.3 |
| 2-Methylnaphthalenec | 2.8 | 0.6 | 1.5 |
Covalent binding data were derived from experiments conducted in susceptible rodent species (naphthalene and 2-methylnaphthalene-mice and 1-nitronaphthalene-rats) 2-4 hrs after intraperitoneal administration of a toxic dose of compound.
data taken from Buckpitt and Warren, 1983
data taken from Wheelock et al., 2005; values were with samples obtained by lysis lavage
data taken from Griffin et al., 1982
As a fraction of the dose, much higher percentages of 1-nitronaphthalene are bound covalently to tissue proteins than with naphthalene and this is greater than for 2-methylnaphthalene (Table 3). The levels of covalent binding for nitronaphthalene, naphthalene and 2-methylnaphthalene correlate moderately well with the relative potency of these agents as airway epithelial cell toxicants. Doses of 1-nitronaphthalene as low as 12.5 mg/kg resulted in decreased epithelial thickness in terminal bronchioles of adult rats (Fanucchi et al., 2004). In contrast, much higher doses of 50 mg/kg and 144 mg/kg of naphthalene and 2-methylnaphthalene, respectively were the lowest doses reported to produce detectable alterations in the airways of mice. Although measurement of the total levels of covalent binding is expected to provide a good assessment of the amounts of reactive metabolite generated, binding to specific critical proteins in the cell is likely to be a much better marker for cellular events critical to the toxicity of these agents. The ‘critical’ protein target(s) are not established with any of these pulmonary cytotoxic PAH but, in our view, those low abundance proteins with high levels of bound metabolite are more likely to be important than the abundant proteins with relatively low levels of adduct formation.
Future Directions
There are several major questions which require further work with all of these cytotoxic aromatic hydrocarbons. The assessment of human risk to exposure is still an open question particularly with naphthalene and methylnaphthalenes where there are dramatic species differences in susceptibility to these agents. The levels of exposure used in rodent studies far exceeds those likely to be encountered in the environment. However, the vast majority of the toxicological work has been conducted as single exposures whereas human exposure is long term and at low concentrations. The relatively high incidence of human lung disease and the myriad of chemical exposures likely to be contributing to the adverse health outcomes make it unlikely that epidemiological approaches will be useful in identifying (or exonerating) these compounds as hazards. PBPK models have been developed for naphthalene (Ghanem et al, 2000 and Quick et al., 1999) and these have been useful in describing parameters critical to the cytotoxic response. While these models fit the data in rodents quite well, rates of metabolism of the parent compound and primary metabolites are not established in human tissues and this remains a data gap for all three compounds. Similar PBPK models for the methyl- and nitro-naphthalenes have not been reported. In our view, understanding those reactive metabolite protein interactions which are closely linked to toxicity would allow the development of biomarkers which are based on reactive metabolite protein adducts obtained from nasal lavage or as peptide adducts in the urine. Careful validation of these markers in susceptible and non susceptible rodent models and in non-human primates would provide a basis for determining whether the toxicity of these compounds in the respiratory tract of rodents is relevant for exposed human populations.
A further critical issue which needs to be addressed with all three compounds is related to susceptible populations. Studies have already shown the heightened susceptibility of young animals to naphthalene and nitronaphthalene compared to adults and to female animals compared to males for naphthalene but understanding the precise mechanisms for these differences is incomplete. Whether similar differences occur with methylnaphthalene has not been reported. Multiple exposures to naphthalene result in marked tolerance to subsequent high level exposures. Whether this same phenomenon would be apparent if the exposures were more intermittent or whether the repeated cycle of injury and repair followed by re-injury would lead to long-term decrements in the structure and function of the lungs is unknown. While the metabolic pathways for naphthalene have been well-worked out, it is still not clear what the specific contributions of the putative reactive metabolites (epoxide and quinones) of this compound are to toxicity. Understanding this and knowing which enzymes are responsible for the formation and detoxication of these small aromatic hydrocarbons will lead to a better appreciation of where genetic differences in the proteins responsible for the metabolic disposition of these compounds may lead to enhanced or diminished susceptibility. For example, Ding and his colleagues have elegantly demonstrated the importance of CYP2A13 in the metabolic activation of NNK, a tobacco specific lung carcinogen (Zhang et al., 2007) and showed that only a very small percentage of the human population studied had detectable levels of protein in their lung tissue. These differences may, in part, explain the substantial interindividual differences in susceptibility to tobacco smoke. Recently published epidemiologic studies focusing on the link between children exposed to mobile source air pollution and asthma have provided intriguing information which associates PAH exposure and asthma (Salam et al., 2007). Higher odds ratios for asthma were observed for those children with polymorphisms in two of the major enzymes involved in PAH metabolism which lead to increases in microsomal epoxide hydrolase and decreases in glutathione transferase Pi. These enzymes produce key metabolic intermediates in the metabolism and metabolic disposition of PAH. Much more extensive studies are needed with the methyl and nitronaphthalenes to determine whether the metabolites that have been reported comprise the full list of metabolites generated.
Finally, there is a clear need to understand the influence of exposure to multiple pulmonary toxicants to discern whether such exposures lead to additive, synergistic or antagonistic actions. A case in point is the work showing that 1-nitronaphthalene is substantially more toxic in ozone-tolerant rats (Paige et al., 2000; Wheelock et al, 2005; Schmelzer et al, 2006). Although the lung of ozone-tolerant animals is more susceptible to 1-nitronaphthalene, the nasal epithelium is less so (Lee et al., 2008). Several sources of human exposure including ambient air and cigarette smoke contain mixtures of all three compounds discussed in this review yet there is no information about whether these chemicals would produce synergistic effects in the respiratory tract. The fact that there are synergistic and antagonistic effects of exposure to multiple pulmonary toxicants underscores the need to understand the factors important to the toxicity of these chemicals. Defining these mechanisms in animal models is key to our ability to determine whether these chemicals are a human health hazard and for determining which factors are likely to influence interindividual susceptibility.
Figure 2.
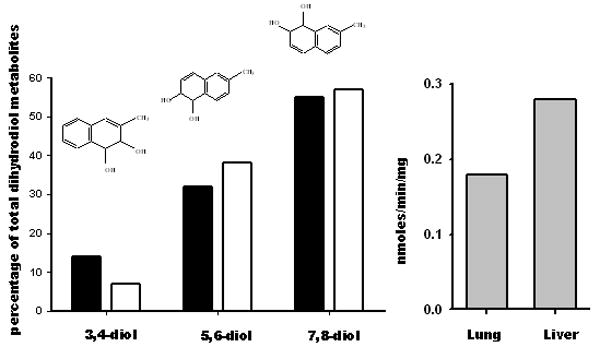
Table 1.
Comparison of the toxicities of naphthalene with methyl and nitro derivatives
| Structure | Dose mg/kg (mmoles/kg) | Species | Toxicity | Comments | Reference | ||
|---|---|---|---|---|---|---|---|
| Clara | ciliated | hepatic | |||||
 |
50 (0.39) | mouse | ± | 0 | 0 | murine specific toxicant | Mahavi et al., 1977; Plopper et al, 1992 |
| 200 (1.56) | +++ | 0 | 0 | ||||
| 1600 (12.5) | rat | 0 | 0 | 0 | |||
 |
173 (1.0) | mouse | ++ | + | + | results in toxicity in both mice and rats |
Dinsdale and Verschoyle, 1987; Rasmussen et al., 1986; Verschoyle et al., 1993; Sauer et al., 1995, 1997; Paige et al.,1997 |
| 25 (0.14) | rat | + | + | 0 | |||
| 100 (0.57) | rat | +++ | 0 | + | |||
| 173 (1.0) | rat | + | + | + | |||
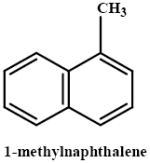 |
144 (1.0) | mouse | ± | 0 | 0 | only mice reported | Rasmussen et al., 1986 |
| 288 (2.0) | mouse | + | 0 | 0 | |||
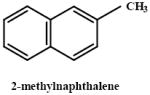 |
0.1, 1, 10, 100, 200, 400, 600, 800, 1000 (0.0007-6.94 mmoles/kg) | mouse | Dose dependent toxicity in C57Bl and DBA mice depletion of GSH by DEM increases toxicity |
Griffin et al., 1983; Rasmussen et al., 1986; Honda et al.,1990 Rasmussen et al., 1986 |
|||
| 144 (1.0) | |||||||
| 288 (2.0) | |||||||
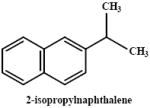 |
3000 mg/kg (17.6) | mouse | 0 | 0 | nd | no cytotoxicity noted even with prior depletion of GSH | Honda et al., 1990 |
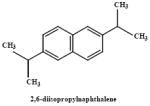 |
3000 mg/kg (14.2) mmoles/kg | mouse | 0 | 0 | nd | no cytotoxicity noted even with prior depletion of GSH | Honda et al., 1990 |
Acknowledgments
Work in the authors laboratories has been supported from grants from NIEHS (04311, 04699 and 06700). This publication was aided by a gift from ExxonMobil Biomedical Sciences, Clinton, NJ.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdo K, Eustis S, McDonald M, Joiken M, Adkins B, Haseman J. Naphthalene: A respiratory tract toxicant and carcinogen for mice. Inhal Toxicol. 1992;4:393–409. [Google Scholar]
- Abdo KM, Grumbein S, Chou BJ, Herbert R. Toxicity and carcinogenicity study in f344 rats following 2 years of whole-body exposure to naphthalene vapors. Inhal Toxicol. 2001;13:931–950. doi: 10.1080/089583701752378179. [DOI] [PubMed] [Google Scholar]
- Aislabie J, Balks M, Astori N, Stevenson G, Symons R. Polycyclic aromatic hydrocarbons in fuel-oil contaminated soils. Antarctica Chemosphere. 1999;39:2201–2207. doi: 10.1016/s0045-6535(99)00144-7. [DOI] [PubMed] [Google Scholar]
- Anon. Toxicological profile for naphthalene, 1-methylnaphthalene and 2-methylnaphthalene. Agency for Toxic Substances and Disease Registry. 2005 http://www.atsdr.cdc.gov/toxprofiles/tp67.pdf.
- Anon. Prepared for utility solid waste activities group, Superfund committee. Project # P-D215. Environmental Research and Technology, Inc.; 1984. Handbook on manufactured gas plant sites. [Google Scholar]
- Bagheri H, Creaser CS. Determination of some two- and three-ring aromatic hydrocarbons by combined gas chromatography and fluorescence spectrometry. Analyst. 1988;113:1175–1178. [Google Scholar]
- Baldwin RM, Jewell WT, Fanucchi MV, Plopper CG, Buckpitt AR. Comparison of pulmonary/nasal CYP2F expression levels in rodents and rhesus macaque. J Pharmacol Exp Ther. 2004;309:127–136. doi: 10.1124/jpet.103.062901. [DOI] [PubMed] [Google Scholar]
- Baldwin RM, Shultz MA, Buckpitt AR. Bioactivation of the pulmonary toxicants naphthalene and 1-nitronaphthalene by rat CYP2F4. J Pharmacol Exp Ther. 2005;312:857–865. doi: 10.1124/jpet.104.075440. [DOI] [PubMed] [Google Scholar]
- Bjorseth A, Bjorseth O, Fjeldstad PE. Polycyclic aromatic hydrocarbons in the work atmosphere. I. Determination in an aluminum reduction plant. Scand J Work Environ Health. 1978;4:212–23. doi: 10.5271/sjweh.2704. [DOI] [PubMed] [Google Scholar]
- Born SL, Caudill DK, Fliter L, Purdon MP. Identification of the cytochromes P450 that catalyze coumarin 3,4-epoxidation and 3-hydroxylation. Drug Metab Dispos. 2002;30:483–487. doi: 10.1124/dmd.30.5.483. [DOI] [PubMed] [Google Scholar]
- Breger RK, Franklin RB, Lech JJ. Metabolism of 2-methylnaphthalene to isomeric dihydrodiols by hepatic microsomes of rat and rainbow trout. Drug Metab Dispos. 1981;9:88–93. [PubMed] [Google Scholar]
- Breger R, Novak R, Franklin R, Rickert D, Lech J. Further structural analysis of rat liver microsomal metabolites of 2-methylnaphthalene. Drug Metab Dispos. 1983;11:319–323. [PubMed] [Google Scholar]
- Brusick D. Critical assessment of the genetic toxicity of naphthalene. Regul Toxicol Pharmacol. 2008;51:S37–S42. doi: 10.1016/j.yrtph.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Buckpitt AR, Bahnson LS, Franklin RB. Hepatic and pulmonary microsomal metabolism of naphthalene to glutathione adducts: factors affecting the relative rates of conjugate formation. J Pharmacol Exp Ther. 1984;231:291–300. [PubMed] [Google Scholar]
- Buckpitt A, Boland B, Isbell M, Morin D, Shultz M, Baldwin R, Chan K, Karlsson A, Lin C, Taff A, West J, Fanucchi M, Van Winkle L, Plopper C. Naphthalene-induced respiratory tract toxicity: metabolic mechanisms of toxicity. Drug Metab Rev. 2002;34:791–820. doi: 10.1081/dmr-120015694. [DOI] [PubMed] [Google Scholar]
- Buckpitt A, Chang A, Weir A, Van Winkle L, Duan X, Philpot RM, Plopper CG. Relationship of cytochrome P450 activity to Clara cell cytotoxicity. IV. Metabolism of naphthalene and naphthalene oxide in microdissected airways from mice, rats and hamsters. Mol Pharmacol. 1995;47:74–81. [PubMed] [Google Scholar]
- Chapman KR, Mannino DM, Soriano JB, Vermeire PA, Buist AS, Thun MJ, Connell C, Jemal A, Lee TA, Miravitlles M, Aldington S, Beasley R. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J. 2006;27:188–207. doi: 10.1183/09031936.06.00024505. [DOI] [PubMed] [Google Scholar]
- Chen K, Dorough H. Glutathione and mercapturic acid conjugations in the metabolism of naphthalene and 1-naphthyl N-methylcarbamate (carbaryl) Drug Chem Tox. 1979;2:331–354. doi: 10.3109/01480547909016028. [DOI] [PubMed] [Google Scholar]
- Cho TM, Rose RL, Hodgson E. In vitro metabolism of naphthalene by human liver microsomal cytochrome P450 enzymes. Drug Metab Dispos. 2006;34:176–183. doi: 10.1124/dmd.105.005785. [DOI] [PubMed] [Google Scholar]
- Cruzan G, Carlson GP, Johnson KA, Andrews LS, Banton MI, Bevan C, Cushman JR. Styrene respiratory tract toxicity and mouse lung tumors are mediated by CYP2F-generated metabolites. Regul Toxicol Pharmacol. 2002;35:308–319. doi: 10.1006/rtph.2002.1545. [DOI] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: Function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- Dinsdale D, Verschoyle RD. Pulmonary toxicity of naphthalene derivatives in the rat. Arch Toxicol Suppl. 1987;11:288–291. doi: 10.1007/978-3-642-72558-6_54. [DOI] [PubMed] [Google Scholar]
- Dodd DE, James RA, Gross EA, Miller RA, Piccirillo VJ, Wong BA. Nasal epithelial lesions in rats following an acute inhalation exposure to naphthalene vapor at low concentrations. The Toxicologist. 2008 510 abs. [Google Scholar]; el Bayoumy K, Reddy B, Hecht SS. Identification of ring oxidized metabolites of 1-nitropyrene in the feces and urine of germfree F344 rats. Carcinogenesis. 1984;5:1371–1373. doi: 10.1093/carcin/5.10.1371. [DOI] [PubMed] [Google Scholar]
- Fanucchi MV, Buckpitt AR, Murphy ME, Plopper CG. Naphthalene cytotoxicity of differentiating Clara cells in neonatal mice. Toxicol Appl Pharmacol. 1997;144:96–104. doi: 10.1006/taap.1997.8119. [DOI] [PubMed] [Google Scholar]
- Fanucchi MV, Day KC, Clay CC, Plopper GC. Increased vulnerability of neonatal rats and mice to 1-nitronaphthalene-induced pulmonary injury. Toxicol Appl Pharmacol. 2004;201:53–65. doi: 10.1016/j.taap.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Farant JP, Ogilvie D. Investigation of the presence of amino and nitro polycyclic aromatic hydrocarbons in a Soderberg primary aluminum smelter. AIHA J. 2002;63:721–725. doi: 10.1080/15428110208984761. [DOI] [PubMed] [Google Scholar]
- Franklin R, Plopper C, Buckpitt A. Naphthalene and 2-methylnaphthalene-induced pulmonary bronchiolar epithelial cell necrosis: metabolism and relationship to toxicity. In: Gram T, editor. Metabolic Activation and Toxicity of Chemical Agents to Lung Tissue and Cells. Pergamon Press; New York: 1993. pp. 123–144. [Google Scholar]
- Fukami T, Katoh M, Yamazaki H, Yokoi T, Nakajima M. Human cytochrome P450 2A13 efficiently metabolizes chemicals in air pollutants: naphthalene, styrene, and toluene. Chem Res Toxicol. 2008;21:720–725. doi: 10.1021/tx700325f. [DOI] [PubMed] [Google Scholar]
- Gallego E, Roca FJ, Perales JF, Guardino X, Berenguer MJ. VOCs and PAHs emissions from creosote-treated wood in a field storage area. Sci Total Environ. 2008;402:130–138. doi: 10.1016/j.scitotenv.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Ghanem A, Shuler ML. Combining cell culture analogue reactor designs and PBPK models to probe mechanisms of naphthalene toxicity. Biotechnol Prog. 2000;16:334–345. doi: 10.1021/bp9901522. [DOI] [PubMed] [Google Scholar]
- Griffin KA, Johnson CB, Breger RK, Franklin RB. Effects of inducers and inhibitors of cytochrome P-450-linked monooxygenases on the toxicity, in vitro metabolism and in vivo irreversible binding of 2-methylnaphthalene in mice. J Pharmacol Exp Ther. 1982;221:517–524. [PubMed] [Google Scholar]
- Griffin KA, Johnson CB, Breger RK, Franklin RB. Pulmonary toxicity, hepatic, and extrahepatic metabolism of 2-methylnaphthalene in mice. Toxicol Appl Pharmacol. 1981;61:185–96. doi: 10.1016/0041-008x(81)90408-7. [DOI] [PubMed] [Google Scholar]
- Griffin KA, Johnson CB, Breger RK, Franklin RB. Pulmonary toxicity of 2-methylnaphthalene: lack of a relationship between toxicity, dihydrodiol formation and irreversible binding to cellular macromolecules in DBA/2J mice. Toxicol. 1983;26:213–30. doi: 10.1016/0300-483x(83)90083-5. [DOI] [PubMed] [Google Scholar]
- Grosovsky AJ, Sasaki JC, Arey J, Eastmond DA, Parks KK, Atkinson R. Evaluation of the potential health effects of the atmospheric reaction products of polycyclic aromatic hydrocarbons. Health Effects Institute; Boston, MA: 1999. pp. 1–29. Research Report # 84. [PubMed] [Google Scholar]
- Gupta P, Harger W, Arey J. The contribution of nitro and methylnitronaphthalenes to the vapor phase mutagenicity of ambient air samples. Atmos Env. 1996;30:3157–3166. [Google Scholar]
- Halladay J, Sauer J, Sipes I. The metabolism and disposition of 1-nitronaphthalene in the male Sprague Dawley rat. Drug Metab Dispos. 1999;27:1456–1465. [PubMed] [Google Scholar]
- Halpert JR. Structural basis of selective cytochrome P450 inhibition. Ann Rev Pharmacol Toxicol. 1995;35:29–53. doi: 10.1146/annurev.pa.35.040195.000333. [DOI] [PubMed] [Google Scholar]
- Hampton CV, Pierson WR, Schuetzie D, Harver TM. Hydrocarbon gases emitted from vehicles on the road 2. Determination of emission rates from diesel and spark-ignition vehicles. Env Sci Technol. 1983;17:699–708. doi: 10.1021/es00118a003. [DOI] [PubMed] [Google Scholar]
- Honda T, Fukada A, Kiyozumi M, Kojima S. Identification and determination of urinary metabolites of 2-isopropylnaphthalene in rabbits. Eur J Drug Metab Pharmacokin. 1987;12:11–16. doi: 10.1007/BF03189856. [DOI] [PubMed] [Google Scholar]
- Honda T, Kiyozumi M, Kojima S. Alkylnaphthalene. XI. Pulmonary toxicity of naphthalene, 2-methylnaphthalene, and isopropylnaphthalenes in mice. Chem Pharm Bull (Tokyo) 1990;38:3130–3135. doi: 10.1248/cpb.38.3130. [DOI] [PubMed] [Google Scholar]
- Hoke H, Zellerhoff R. Metabolism and toxicity of diisopropylnaphthalene as compared to naphthalene and monoalkyl naphthalenes: a minireview. Toxicol. 1998;126:1–7. doi: 10.1016/s0300-483x(97)00187-x. [DOI] [PubMed] [Google Scholar]
- Ikegami M, Whitsett JA, Chroneos ZC, Ross GF, Reed JA, Bachurski CJ, Jobe AH. IL-4 increases surfactant and regulates metabolism in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278:L75–L80. doi: 10.1152/ajplung.2000.278.1.L75. [DOI] [PubMed] [Google Scholar]
- Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis. Chron Respir Dis. 2006;3:149–159. doi: 10.1191/1479972306cd101rs. [DOI] [PubMed] [Google Scholar]
- Kang-Sickel JC, Fox DD, Nam TG, Jayaraj K, Ball LM, French JE, Klapper DG, Gold A, Nylander-French LA. S-Arylcysteine-keratin adducts as biomarkers of human dermal exposure to aromatic hydrocarbons. Chem Res Toxicol. 2008;21:852–858. doi: 10.1021/tx7003773. [DOI] [PubMed] [Google Scholar]
- Kilanowicz A, Sapota A, Darago A. Disposition and metabolism of 1,4-dimethylnaphthalene in rat. Int J Occup Med Environ Health. 2000;13:325–334. [PubMed] [Google Scholar]
- Kilanowicz A, Sapota A, Czerski B. Disposition and metabolism of 1,6-dimethylnaphthalene in rats. Toxicol Lett. 2002;134:227–235. doi: 10.1016/s0378-4274(02)00170-4. [DOI] [PubMed] [Google Scholar]
- Kilanowicz A, Sapota A. Disposition and metabolism of 1,2-dimethylnaphthalene in rats. Int J Occup Med Environ Health. 1998;11:305–317. [PubMed] [Google Scholar]
- Kim D, Andersen ME, Nylander-French LA. Dermal absorption and penetration of jet fuel components in humans. Toxicol Lett. 2006;165:11–21. doi: 10.1016/j.toxlet.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Kobayashi R, Okamoto RA, Maddalena RL, Kado NY. Polycyclic aromatic hydrocarbons in edible grain: a pilot study of agricultural crops as a human exposure pathway for environmental contaminants using wheat as a model crop. Environ Res. 2008;107:145–151. doi: 10.1016/j.envres.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Kojima ST, Honda M, Nakagawa M, Kiyozumi M, Takadate A. Urinary metabolites of 2,6-diisopropylnaphthalene in rats. Drug Metab Dispos. 1982;10:429–433. [PubMed] [Google Scholar]
- Kojima S, Honda T, Kiyozumi M. Biliary metabolites of 2,6-diisopropylnaphthalene in rats. Bull Environ Contam Toxicol. 1985;35:745–749. doi: 10.1007/BF01636583. [DOI] [PubMed] [Google Scholar]
- Korsak Z, Majcherek W, Rydzynski K. Toxic effects of acute inhalation exposure to 1-methylnaphthalene and 2-methylnaphthalene in experimental animals. Int J Occup Med Environ Health. 1998;11:335–342. [PubMed] [Google Scholar]
- Kulka U, Schmid E, Huber R, Bauchinger M. Analysis of the cytogenetic effect in human lymphocytes induced by metabolically activated 1- and 2-methylnaphthalene. Mutat Res. 1988;208:155–158. doi: 10.1016/0165-7992(88)90052-8. [DOI] [PubMed] [Google Scholar]
- Lakritz J, Chang A, Weir A, Nishio S, Hyde D, Philpot R, Buckpitt A, Plopper C. Cellular and metabolic basis of Clara cell tolerance to multiple doses of cytochrome P450-activated cytotoxicants. I: Bronchiolar epithelial reorganization and expression of cytochrome P450 monooxygenases in mice exposed to multiple doses of naphthalene. J Pharmacol Exp Ther. 1996;278:1408–1418. [PubMed] [Google Scholar]
- Lee MG, Phimister A, Morin D, Buckpitt A, Plopper C. In situ naphthalene bioactivation and nasal airflow cause region-specific injury patterns in the nasal mucosa of rats exposed to naphthalene by inhalation. J Pharmacol Exp Ther. 2005;314:103–110. doi: 10.1124/jpet.105.084517. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wheelock AM, Boland B, Plopper CG. Long-term ozone exposure attenuates 1-nitronaphthalene-induced cytotoxicity in nasal mucosa. Am J Respir Cell Mol Biol. 2008;38:300–309. doi: 10.1165/rcmb.2005-0416OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Sandau CD, Romanoff LC, Caudill SP, Sjodin A, Needham LL, Patterson DG., Jr Concentration and profile of 22 urinary polycyclic aromatic hydrocarbon metabolites in the US population. Environ Res. 2008;107:320–331. doi: 10.1016/j.envres.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Melancon MJ, Williams DE, Buhler DR, Lech JJ. Metabolism of 2-methylnaphthalene by rat and rainbow trout hepatic microsomes and purified cytochromes P-450. Drug Metab Dispos. 1985;13:542–547. [PubMed] [Google Scholar]
- Melancon MJ, Rickert DE, Lech JJ. Metabolism of 2-methylnaphthalene in the rat in vivo. I. Identification of 2-naphthoylglycine. Drug Metab Dispos. 1982;10:128–133. [PubMed] [Google Scholar]
- Murata Y, Denda A, Maruyama H, Konishi Y. Chronic toxicity and carcinogenicity studies of 1- methylnaphthalene in B6C3F1 mice. Fundam Appl Toxicol. 1993;21:44–51. doi: 10.1006/faat.1993.1070. [DOI] [PubMed] [Google Scholar]
- Murata Y, Denda A, Maruyama H, Nakae D, Tsutsumi M, Tsujiuchi T, Konishi Y. Chronic toxicity and carcinogenicity studies of 2-methylnaphthalene in B6C3F1 mice. Fundam Appl Toxicol. 1997;36:90–93. doi: 10.1006/faat.1996.2283. [DOI] [PubMed] [Google Scholar]
- Murata Y, Emi Y, Denda A, Konishi Y. Ultrastructural analysis of pulmonary alveolar proteinosis induced by methylnaphthalene in mice. Exp Toxicol Pathol. 1992;44:47–54. doi: 10.1016/S0940-2993(11)80137-5. [DOI] [PubMed] [Google Scholar]
- NIOSH: Hazard Review. Health effects of occupational exposure to asphalt. 2000 DHHS publication # 2001-110 http://www.cdc.gov/niosh/docs/2001-110/.
- Nishino N, Atkinson R, Arey J. Formation of nitro products from the gas-phase OH radical-initiated reactions of toluene, naphthalene, and biphenyl: effect of NO2 concentration. Environ Sci Technol. 2008;42:9203–9209. doi: 10.1021/es802046m. [DOI] [PubMed] [Google Scholar]
- O’Brien KA, Suverkropp C, Kanekal S, Plopper CG, Buckpitt AR. Tolerance to multiple doses of the pulmonary toxicant, naphthalene. Toxicol Appl Pharmacol. 1989;99:487–500. doi: 10.1016/0041-008x(89)90156-7. [DOI] [PubMed] [Google Scholar]
- Paige R, Wong V, Plopper CG. Dose-related airway specific epithelial toxicity of 1-nitronaphthalene in rats. Toxicol Appl Pharmacol. 1997;147:224–233. doi: 10.1006/taap.1997.8297. [DOI] [PubMed] [Google Scholar]
- Paige RC, Wong V, Plopper CG. Long-term exposure to ozone increases acute pulmonary centriacinar injury by 1-nitronaphthalene: II. Quantitative histopathology. J Pharmacol Exp Ther. 2000;295:942–950. [PubMed] [Google Scholar]
- Pakenham G, Lango J, Buonarati M, Morin D, Buckpitt A. Urinary naphthalene mercapturates as biomarkers of exposure and stereoselectivity of naphthalene epoxidation. Drug Metab Dispos. 2002;30:247–253. doi: 10.1124/dmd.30.3.247. [DOI] [PubMed] [Google Scholar]
- Phousongphouang PT, Arey J. Rate constants for the gas-phase reactions of a series of alkylnaphthalenes with the OH radical. Environ Sci Technol. 2002;36:1947–1952. doi: 10.1021/es011434c. [DOI] [PubMed] [Google Scholar]
- Phousongphouang PT, Arey J. Rate constants for the gas-phase reactions of a series of alkylnaphthalenes with the nitrate radical. Environ Sci Technol. 2003;37:308–313. doi: 10.1021/es026015+. [DOI] [PubMed] [Google Scholar]
- Plopper CG. Pulmonary bronchiolar epithelial cytotoxicity: Microanatomical considerations. In: Gram TE, editor. Metabolic Activation and Toxicity of Chemical Agents to Lung Tissue and Cells. Pergamon Press; New York: 1993. pp. 1–24. [Google Scholar]
- Plopper CG, Chang A, Pang A, Buckpitt A. Use of microdissected airways to better define metabolism and cytotoxicity in murine bronchiolar epithelium. Exp Lung Res. 1991;17:181–196. doi: 10.3109/01902149109064411. [DOI] [PubMed] [Google Scholar]
- Plopper CG, Suverkropp C, Morin D, Nishio S, Buckpitt A. Relationship of cytochrome P-450 activity to Clara cell cytotoxicity. I. Histopathologic comparison of the respiratory tract of mice, rats and hamsters after parenteral administration of naphthalene. J Pharmacol Exp Ther. 1992a;261:353–363. [PubMed] [Google Scholar]
- Plopper CG, Macklin J, Nishio S, Hyde D, Buckpitt A. Relationship of cytochrome P450 activity to Clara cell cytotoxicity. III. Morphometric comparison of changes in the epithelial populations of terminal bronchioles and lobar bronchi in mice, hamsters and rats after parenteral administration of naphthalene. Lab Invest. 1992b;67:553–565. [PubMed] [Google Scholar]
- Preuss R, Angerer J, Drexler H. Naphthalene-an environmental and occupational toxicant. Int Arch Occup Environ Health. 2003;76:556–576. doi: 10.1007/s00420-003-0458-1. [DOI] [PubMed] [Google Scholar]
- Quick DJ, Shuler ML. Use of in vitro data for construction of a physiologically based pharmacokinetic model for naphthalene in rats and mice to probe species differences. Biotechnol Prog. 1999;15:540–555. doi: 10.1021/bp990057t. [DOI] [PubMed] [Google Scholar]
- Rasmussen R, Do D, Kim T, Dearden L. Comparative cytotoxicity of naphthalene and its monomethyl- and mononitro-derivatives in the mouse lung. J Appl Toxicol. 1986;6:13–20. doi: 10.1002/jat.2550060104. [DOI] [PubMed] [Google Scholar]
- Rasmussen RE. Metabolism and macromolecular binding of 1-nitronaphthalene in the mouse. Toxicology. 1986;41:233–247. doi: 10.1016/0300-483x(86)90202-7. [DOI] [PubMed] [Google Scholar]
- Reisen F, Arey J. Atmospheric reactions influence seasonal PAH and nitro-PAH concentrations in the Los Angeles basin. Environ Sci Technol. 2005;39:64–73. [PubMed] [Google Scholar]
- Ryerse JS, Hoffmann JW, Mahmoud S, Nagel BA, deMello DE. Immunolocalization of CC10 in Clara cells in mouse and human lung. Histochem Cell Biol. 2001;115:325–332. doi: 10.1007/s004180100251. [DOI] [PubMed] [Google Scholar]
- Salam MT, Lin PC, Avol EL, Gauderman WJ, Gilliland FD. Microsomal epoxide hydrolase, glutathione S-transferase P1, traffic and childhood asthma. Thorax. 2007;62:1050–1057. doi: 10.1136/thx.2007.080127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J, Eversole R, Lehmann C, Johnson D, Beuving L. An ultrastructural evaluation of acute 1-nitronaphthalene induced hepatic and pulmonary toxicity in the rat. Toxicol Lett. 1997;90:19–27. doi: 10.1016/s0378-4274(96)03817-9. [DOI] [PubMed] [Google Scholar]
- Sauer JM, Hooser SB, Sipes IG. All-trans-retinol alteration of 1-nitronaphthalene-induced pulmonary and hepatic injury by modulation of associated inflammatory responses in the male Sprague-Dawley rat. Toxicol Appl Pharmacol. 1995;133:139–149. doi: 10.1006/taap.1995.1135. [DOI] [PubMed] [Google Scholar]
- Schmeltz I, Tosk J, Hoffman D. Formation and determination of naphthalenes in cigarette smoke. Anal Chem. 1976;48:645–650. doi: 10.1021/ac60368a031. [DOI] [PubMed] [Google Scholar]
- Schmelzer KR, Wheelock AM, Dettmer K, Morin D, Hammock BD. The role of inflammatory mediators in the synergistic toxicity of ozone and 1-nitronaphthalene in rat airways. Environ Health Perspect. 2006;114:1354–1360. doi: 10.1289/ehp.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz MA, Morin D, Chang AM, Buckpitt A. Metabolic capabilities of CYP2F2 with various pulmonary toxicants and its relative abundance in mouse lung subcompartments. J Pharmacol Exp Ther. 2001;296:510–519. [PubMed] [Google Scholar]
- Serabjit-Singh C, Wolf C, Philpot R, Plopper C. Cytochrome P450: Localization in rabbit lung. Science. 1980;207:1469–1470. doi: 10.1126/science.6767272. [DOI] [PubMed] [Google Scholar]
- Stillwell W, Bouwsma O, Thenot J, Horning M, Griffin G, Ishikawa K, Takaku M. Methylthio metabolites of naphthalene excreted by the rat. Res Comm Chem Path Pharmacol. 1978;20:509–530. [PubMed] [Google Scholar]
- Stillwell W, Horning M, Griffin G, Tsang W. Identification of new sulfur-containing metabolites of napthalene in mouse urine. Drug Metab Dispos. 1982;10:624–631. [PubMed] [Google Scholar]
- Teshima R, Nagamatsu K, Ikebuchi H, Kido Y, Terao T. In vivo and in vitro metabolism of 2-methylnaphthalene in the guinea pig. Drug Metab Dispos. 1983;11:152–157. [PubMed] [Google Scholar]
- Toriba A, Kitaoka H, Dills RL, Mizukami S, Tanabe K, Takeuchi N, Ueno M, Kameda T, Tang N, Hayakawa K, Simpson CD. Identification and quantification of 1-nitropyrene metabolites in human urine as a proposed biomarker for exposure to diesel exhaust. Chem Res Toxicol. 2007;20:999–1007. doi: 10.1021/tx700015q. [DOI] [PubMed] [Google Scholar]
- Van Winkle LS, Gunderson AD, Shimizu JA, Baker GL, Brown CD. Gender differences in naphthalene metabolism and naphthalene-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1122–L1134. doi: 10.1152/ajplung.00309.2001. [DOI] [PubMed] [Google Scholar]
- Verschoyle RD, Dinsdale D. Protection against chemical-induced lung injury by inhibition of pulmonary cytochrome P-450. Environ Health Perspect. 1990;85:95–100. doi: 10.1289/ehp.85-1568337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschoyle RD, Carthew P, Wolf CR, Dinsdale D. 1-Nitronaphthalene toxicity in rat lung and liver: Effects of inhibiting and inducing cytochrome P450 activity. Toxicol Appl Pharmacol. 1993;122:208–213. doi: 10.1006/taap.1993.1189. [DOI] [PubMed] [Google Scholar]
- Wang L, Atkinson R, Arey J. Dicarbonyl products of the OH radical-initiated reactions of naphthalene and the C1- and C2-alkylnaphthalenes. Environ Sci Technol. 2007;41:2803–2810. doi: 10.1021/es0628102. [DOI] [PubMed] [Google Scholar]
- Warren DL, Brown D, Jr, Buckpitt A. Evidence for cytochrome P450 mediated metabolism in the bronchiolar damage by naphthalene. Chem -Biol Interact. 1982;40:287–303. doi: 10.1016/0009-2797(82)90152-1. [DOI] [PubMed] [Google Scholar]
- Watt KC, Buckpitt AR. Species differences in the regio- and stereoselectivity of 1-nitronaphthalene metabolism. Drug Metab Dispos. 2000;28:376–378. [PubMed] [Google Scholar]
- Watt K, Morin D, Kurth MJ, Mercer RS, Plopper CG, Buckpitt A. Glutathione conjugation of electrophilic metabolites of 1-nitronaphthalene in rat tracheobronchial airways and liver: Identification by mass spectrometry and proton nuclear magnetic resonance spectroscopy. Chem Res Toxicol. 1999;12:831–839. doi: 10.1021/tx990023v. [DOI] [PubMed] [Google Scholar]
- West JA, Buckpitt AR, Plopper CG. Elevated airway GSH resynthesis confers protection to Clara cells from naphthalene injury in mice made tolerant by repeated exposures. J Pharmacol Exp Ther. 2000;294:516–523. [PubMed] [Google Scholar]
- West J, Pakenham G, Morin D, Fleschner C, Buckpitt A, Plopper C. Inhaled naphthalene causes dose-dependent Clara cell toxicity in mice but not in rats. Tox Appl Pharmacol. 2001;173:114–119. doi: 10.1006/taap.2001.9151. [DOI] [PubMed] [Google Scholar]
- West JA, Van Winkle LS, Morin D, Fleschner CA, Forman HJ, Plopper CG. Repeated inhalation exposures to the bioactivated cytotoxicant naphthalene (NA) produce airway-specific Clara cell tolerance in mice. Toxicol Sci. 2003;75:161–168. doi: 10.1093/toxsci/kfg156. [DOI] [PubMed] [Google Scholar]
- West JA, Williams KJ, Toskala E, Nishio SJ, Fleschner CA, Forman HJ, Buckpitt AR, Plopper CG. Induction of tolerance to naphthalene in Clara cells is dependent on a stable phenotypic adaptation favoring maintenance of the glutathione pool. Am J Pathol. 2002;160:1115–1127. doi: 10.1016/S0002-9440(10)64932-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock A, Zhang L, Tran Mai-Uyen, Morin D, Penn S, Buckpitt A, Plopper CG. Isolation of rodent airway epithelial cell proteins facilitates in vivo proteomics studies of lung toxicity. Am J Physiol Lung Cell Mol Physiol. 2004;286:L399–L410. doi: 10.1152/ajplung.00072.2003. [DOI] [PubMed] [Google Scholar]
- Wheelock AM, Boland BC, Isbell M, Morin D, Wegesser TC, Plopper CG, Buckpitt AR. In Vivo effects of ozone exposure on protein adduct formation by 1-nitronaphthalene in rat lung. Am J Respir Cell Mol Biol. 2005;33:130–137. doi: 10.1165/rcmb.2005-0047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witschi H, Espiritu I, Maronpot RR, Pinkerton KE, Jones AD. The carcinogenic potential of the gas phase of environmental tobacco smoke. Carcinogenesis. 1997;18:2035–2042. doi: 10.1093/carcin/18.11.2035. [DOI] [PubMed] [Google Scholar]
- Zhang X, D’Agostino J, Wu H, Zhang QY, von Weymarn L, Murphy SE, Ding X. CYP2A13: variable expression and role in human lung microsomal metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. J Pharmacol Exp Ther. 2007;323:570–578. doi: 10.1124/jpet.107.127068. [DOI] [PubMed] [Google Scholar]