

Abstract
With the advent of dense maps of human genetic variation, it is now possible to detect positive natural selection across the human genome. Here we report an analysis of over 3 million polymorphisms from the International HapMap Project Phase 2 (HapMap2)1. We used ‘long-range haplotype’ methods, which were developed to identify alleles segregating in a population that have undergone recent selection2, and we also developed new methods that are based on cross-population comparisons to discover alleles that have swept to near-fixation within a population. The analysis reveals more than 300 strong candidate regions. Focusing on the strongest 22 regions, we develop a heuristic for scrutinizing these regions to identify candidate targets of selection. In a complementary analysis, we identify 26 non-synonymous, coding, single nucleotide polymorphisms showing regional evidence of positive selection. Examination of these candidates highlights three cases in which two genes in a common biological process have apparently undergone positive selection in the same population: LARGE and DMD, both related to infection by the Lassa virus3, in West Africa; SLC24A5 and SLC45A2, both involved in skin pigmentation4,5, in Europe; and EDAR and EDA2R, both involved in development of hair follicles6, in Asia.
An increasing amount of information about genetic variation, together with new analytical methods, is making it possible to explore the recent evolutionary history of the human population. The first phase of the International Haplotype Map, including ~1 million single nucleotide polymorphisms (SNPs)7, allowed preliminary examination of natural selection in humans. Now, with the publication of the Phase 2 map (HapMap2)1 in a companion paper, over 3 million SNPs have been genotyped in 420 chromosomes from three continents (120 European (CEU), 120 African (YRI) and 180 Asian from Japan and China (JPT + CHB)).
In our analysis of HapMap2, we first implemented two widely used tests that detect recent positive selection by finding common alleles carried on unusually long haplotypes2. The two, the Long-Range Haplotype (LRH)8 and the integrated Haplotype Score (iHS)9 tests, rely on the principle that, under positive selection, an allele may rise to high frequency rapidly enough that long-range association with nearby polymorphisms—the long-range haplotype8—will not have time to be eliminated by recombination. These tests control for local variation in recombination rates by comparing long haplotypes to other alleles at the same locus. As a result, they lose power as selected alleles approach fixation (100% frequency), because there are then few alternative alleles in the population (Supplementary Fig. 2 and Supplementary Tables 1–2).
We next developed, evaluated and applied a new test, Cross Population Extended Haplotype Homozogysity (XP-EHH), to detect selective sweeps in which the selected allele has approached or achieved fixation in one population but remains polymorphic in the human population as a whole (Methods, and Supplementary Fig. 2 and Supplementary Tables 3–6). Related methods have recently also been described10-12.
Our analysis of recent positive selection, using the three methods, reveals more than 300 candidate regions1(Supplementary Fig. 3 and Supplementary Table 7), 22 of which are above a threshold such that no similar events were found in 10 Gb of simulated neutrally evolving sequence (Methods). We focused on these 22 strongest signals (Table 1), which include two well-established cases, SLC24A5 and LCT2,5,13, and 20 other regions with signals of similar strength.
Table 1.
The twenty-two strongest candidates for natural selection
| Region | Chr:position (MB, HG17) |
Selected population | Long Haplotype Test | Size (Mb) | Total SNPs with Long Haplotype Signal |
Subset of SNPs that fulfil criteria 1 |
Subset of SNPs that fulfil criteria 1 and 2 |
Subset of SNPs that fulfil criteria 1, 2 and 3 |
Genes at or near SNPs that fulfil all three criteria |
|---|---|---|---|---|---|---|---|---|---|
| 1 | chr1:166 | CHB+JPT | LRH, iHS | 0.4 | 92 | 39 | 30 | 2 | BLZF1, SLC19A2 |
| 2 | chr2:72.6 | CHB+JPT | XP-EHH | 0.8 | 732 | 250 | 0 | 0 | |
| 3 | chr2:108.7 | CHB+JPT | LRH, iHS, XP-EHH | 1.0 | 972 | 265 | 7 | 1 | EDAR |
| 4 | chr2:136.1 | CEU | LRH, iHS, XP-EHH | 2.4 | 1,213 | 282 | 24 | 3 | RAB3GAP1, R3HDM1, LCT |
| 5 | chr2:177.9 | CEU,CHB+JPT | LRH, iHS, XP-EHH | 1.2 | 1,388 | 399 | 79 | 9 | PDE11A |
| 6 | chr4:33.9 | CEU,YRI, CHB+JPT | LRH, iHS | 1.7 | 413 | 161 | 33 | 0 | |
| 7 | chr4:42 | CHB+JPT | LRH, iHS, XP-EHH | 0.3 | 249 | 94 | 65 | 6 | SLC30A9 |
| 8 | chr4:159 | CHB+JPT | LRH, iHS, XP-EHH | 0.3 | 233 | 67 | 34 | 1 | |
| 9 | chr10:3 | CEU | LRH, iHS, XP-EHH | 0.3 | 179 | 63 | 16 | 1 | |
| 10 | chr10:22.7 | CEU, CHB+JPT | XP-EHH | 0.3 | 254 | 93 | 0 | 0 | |
| 11 | chr10:55.7 | CHB+JPT | LRH, iHS, XP-EHH | 0.4 | 735 | 221 | 5 | 2 | PCDH15 |
| 12 | chr12:78.3 | YRI | LRH, iHS | 0.8 | 151 | 91 | 25 | 0 | |
| 13 | chr15:46.4 | CEU | XP-EHH | 0.6 | 867 | 233 | 5 | 1 | SLC24A5 |
| 14 | chr15:61.8 | CHB+JPT | XP-EHH | 0.2 | 252 | 73 | 40 | 6 | HERC1 |
| 15 | chr16:64.3 | CHB+JPT | XP-EHH | 0.4 | 484 | 137 | 2 | 0 | |
| 16 | chr16:74.3 | CHB+JPT, YRI | LRH, iHS | 0.6 | 55 | 35 | 28 | 3 | CHST5, ADAT1, KARS |
| 17 | chr17:53.3 | CHB+JPT | XP-EHH | 0.2 | 143 | 41 | 0 | 0 | |
| 18 | chr17:56.4 | CEU | XP-EHH | 0.4 | 290 | 98 | 26 | 3 | BCAS3 |
| 19 | chr19:43.5 | YRI | LRH, iHS, XP-EHH | 0.3 | 83 | 30 | 0 | 0 | |
| 20 | chr22:32.5 | YRI | LRH | 0.4 | 318 | 188 | 35 | 3 | LARGE |
| 21 | chr23:35.1 | YRI | LRH, iHS | 0.6 | 50 | 35 | 25 | 0 | |
| 22 | chr23:63.5 | YRI | LRH, iHS | 3.5 | 13 | 3 | 1 | 0 | |
| Total SNPs | 16.74 | 9,166 | 2,898 | 480 | 41 |
Twenty-two regions were identified at a high threshold for significance (Methods), based on the LRH, iHS and/or XP-EHH test. Within these regions, we examined SNPs with the best evidence of being the target of selection on the basis of having a long haplotype signal, and by fulfilling three criteria: (1) being a high-frequency derived allele; (2) being differentiated between populations and common only in the selected population; and (3) being identified as functional by current annotation. Several candidate polymorphisms arise from the analysis including well-known LCT and SLC24A5 (ref. 2), as well as intriguing new candidates.
The challenge is to sift through genetic variation in the candidate regions to identify the variants that were the targets of selection. Our candidate regions are large (mean length, 815 kb; maximum length, 3.5 Mb) and often contain multiple genes (median, 4; maximum, 15). A typical region harbours ~400–4,000 common SNPs (minor allele frequency >5%), of which roughly three-quarters are represented in current SNP databases and half were genotyped as part of HapMap2 (Supplementary Table 8).
We developed three criteria to help highlight potential targets of selection (Supplementary Fig. 1): (1) selected alleles detectable by our tests are likely to be derived (newly arisen), because long-haplotype tests have little power to detect selection on standing (pre-existing) variation14; we therefore focused on derived alleles, as identified by comparison to primate outgroups; (2) selected alleles are likely to be highly differentiated between populations, because recent selection is probably a local environmental adaptation2; we thus looked for alleles common in only the population(s) under selection; (3) selected alleles must have biological effects. On the basis of current knowledge, we therefore focused on non-synonymous coding SNPs and SNPs in evolutionarily conserved sequences. These criteria are intended as heuristics, not absolute requirements. Some targets of selection may not satisfy them, and some will not be in current SNP databases. Nonetheless, with ~50% of common SNPs in these populations genotyped in HapMap2, a search for causal variants is timely.
We applied the criteria to the regions containing SLC24A5 and LCT, each of which already has a strong candidate gene, mutation and trait. At SLC24A5, the 600 kb region contains 914 genotyped SNPs. Applying filters progressively (Table 1 and Fig. 1a–d), we found that 867 SNPs are associated with the long-haplotype signal, of which 233 are high-frequency derived alleles, of which 12 are highly differentiated between populations, and of which only 5 are common in Europe and rare in Asia and Africa. Among these five SNPs, there is only one implicated as functional by current knowledge; it has the strongest signal of positive selection and encodes the A111T polymorphism associated with pigment differences in humans and thought to be the target of positive selection5. Our criteria thus uniquely identify the expected allele.
Figure 1. Localizing SLC24A5 and EDAR signals of selection.

a–d, SLC24A5. a, Strong evidence for positive selection in CEU samples at a chromosome 15 locus: XP-EHH between CEU and JPT + CHB (blue), CEU and YRI (red), and YRI and JPT + CHB (grey). SNPs are classified as having low probability (bordered diamonds) and high probability (filled diamonds) potential for function. SNPs were filtered to identify likely targets of selection on the basis of the frequency of derived alleles (b), differences between populations (c) and differences between populations for high-frequency derived alleles (less than 20% in non-selected populations) (d). The number of SNPs that passed each filter is given in the top left corner in red. The threonine to alanine candidate polymorphism in SLC24A5 is the clear outlier. e–h, EDAR. e, Similar evidence for positive selection in JPT + CHB at a chromosome 2 locus: XP-EHH between CEU and JPT + CHB (blue), between YRI and JPT + CHB (red), and between CEU and YRI (grey); iHS in JPT + CHB (green). A valine to alanine polymorphism in EDAR passes all filters: the frequency of derived alleles (f), differences between populations (g) and differences between populations for high-frequency derived alleles (less than 20% in non-selected populations) (h). Three other functional changes, a D→E change in SULT1C2 and two SNPs associated with RANBP2 expression (Methods), have also become common in the selected population.
At the LCT locus, we found similar degrees of filtration. Within the 2.4 Mb selective sweep, 24 polymorphisms fulfil the first two criteria (Table 1, and Supplementary Fig. 4), with the polymorphism thought to confer adult persistence of lactase among them. However, this SNP was only identified as functional after extensive study of the LCT gene15. Thus LCT shows both the utility and the limits of the heuristics.
Given the encouraging results for SLC24A5 and LCT, we performed a similar analysis on all 22 candidate regions (Table 1). Filtering the 9,166 SNPs associated with the long-haplotype signal, we found that 480 satisfied the first two criteria. We identified 41 out of the 480 SNPs (0.2% of all SNPs genotyped in the regions) as possibly functional on the basis of a newly compiled database of polymorphisms in known coding elements, evolutionarily conserved elements and regulatory elements (Methods; B.F., unpublished), together containing ~ 5.5% of all known SNPs.
Eight of the forty-one SNPs encode non-synonymous changes (Table 1 and Supplementary Table 9). Apart from the well-known case of SLC24A5, they are found in EDAR, PCDH15, ADAT1, KARS, HERC1, SLC30A9 and BLFZ1. The remaining 33 potentially functional SNPs lie within conserved transcription factor motifs, introns, UTRs and other non-coding regions.
To identify additional candidates, we reversed the process by taking non-synonymous coding SNPs with highly differentiated high-frequency derived alleles; these SNPs comprise a tiny fraction of all SNPs and have a higher a priori probability of being targets of selection. Of the 15,816 non-synonymous SNPs in HapMap2, 281 (Supplementary Table 10) have both a high derived-allele frequency (frequency >50%) and clear differentiation between populations (FST is in the top 0.5 percentile). We examined these 281 SNPs to identify those embedded within long-range haplotypes16, and identified 26 putative cases of positive selection. These include the eight non-synonymous SNPs identified in the genome-wide analysis above.
Interestingly, analysis of the top regions and the non-synonymous SNPs together revealed three cases of two genes in the same pathway both having strong evidence of selection in a single population.
In the European sample, there is strong evidence for two genes already shown to be associated with skin pigment differences among humans. The first is SLC24A5, described above. We further examined the global distribution (Fig. 2) and the predicted effect on protein activity of the SLC24A5 A111T polymorphism (Supplementary Fig. 5, 6). The second, SLC45A2, has an important role in pigmentation in zebrafish, mouse and horse4. An L374F substitution in SLC45A2 is at 100% frequency in the European sample, but absent in the Asian and African samples. A recent association study has shown that the Phe-encoding allele is correlated with fair skin and non-black hair in Europeans4. Together, the data support SLC45A2 as a target of positive selection in Europe10,17.
Figure 2. Global distribution of SLC24A5 A111T and EDAR V370A.

Worldwide allele-frequency distributions for candidate polymorphisms with the strongest evidence for selection20. a, SLC24A5 A111T is common in Europe, Northern Africa and Pakistan, but rare or absent elsewhere. b, EDAR V370A is common in Asia and the Americas, but absent in Europe and Africa.
In the African sample (Yoruba in Ibadan, Nigeria), there is evidence of selection for two genes with well-documented biological links to the Lassa fever virus. The strongest signal in the genome, on the basis of the LRH test, resides within a 400 kb region that lies entirely within the gene LARGE. The LARGE protein is a glycosylase that post-translationally modifies α-dystroglycan, the cellular receptor for Lassa fever virus (as well as other arenaviruses), and the modification has been shown to be critical for virus binding3. The virus name is derived from Lassa, Nigeria, where the disease is endemic, with 21% of the population showing signs of exposure18. We also noted that the DMD locus is on our larger candidate list of regions, with the signal of selection again in the Yoruba sample. DMD encodes a cytosolic adaptor protein that binds to α-dystroglycan and is critical for its function. We hypothesize that Lassa fever created selective pressure at LARGE and DMD12. This hypothesis can be tested by correlating the geographical distribution of the selected haplotype with endemicity of the Lassa virus, studying infection of genotyped cells in vitro, and searching for an association between the selected haplotype and clinical outcomes in infected patients.
In the Asian samples, we found evidence of selection for non-synonymous polymorphisms in two genes in the ectodysplasin (EDA) pathway, which is involved in development of hair, teeth and exocrine glands6. The genes are EDAR and EDA2R, which encode the key receptors for the ligands EDA A1 and EDA A2, respectively. Notably, the EDA signalling pathway has been shown to be under positive selection for loss of scales in multiple distinct populations of freshwater stickleback fish19. A mutation encoding a V370A substitution in EDAR is near fixation in Asia and absent in Europe and Africa (Fig. 1e–h). An R57K substitution in EDA2R has derived-allele frequencies of 100% in Asia, 70% in Europe and 0% in Africa.
The EDAR polymorphism is notable because it is highly differentiated between the Asian and other continental populations (the 3rd most differentiated among 15,816 non-synonymous SNPs), and also within Asian populations (in the top 1% of SNPs differentiated between the Japanese and Chinese HapMap samples). Genotyping of the EDAR polymorphism in the CEPH (Centre d'Etudie du Polymorphisme Humain) global diversity panel20 shows that it is at high but varying frequency throughout Asia and the Americas (for example, 100% in Pima Indians and in parts of China, and 73% in Japan) (Fig. 2, and Supplementary Fig. 7). Studying populations like the Japanese, in which the allele is still segregating, may provide clues to its biological significance.
EDAR has a central role in generation of the primary hair follicle pattern, and mutations in EDAR cause hypohidrotic ectodermal dysplasia (HED) in humans and mice, characterized by defects in the development of hair, teeth and exocrine glands6. The V370A polymorphism, proposed to be the target of selection, lies within EDAR's highly conserved death domain (Supplementary Fig. 8), the location of the majority of EDAR polymorphisms causing HED21. Our structural modelling predicts that the polymorphism lies within the binding site of the domain (Fig. 3).
Figure 3. Structural model of the EDAR death domain.
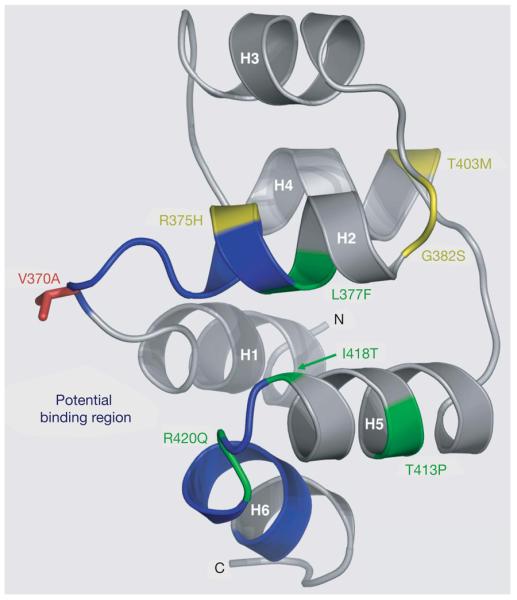
Ribbon representation of a homology model of the EDAR death domain (DD), based on the alignment of the EDAR DD amino acid sequence (EDAR residues 356–431), with multiple known DD structures. The helices are labelled H1 to H6. Residues in blue (the H1–H2 and H5–H6 loops, residues 370–376 and 419–425, respectively) correspond to the homologous residues in Tube that interact with Pelle in the Tube-DD–Pelle-DD structure24. These EDAR-DD residues therefore form a potential region of interaction with a DD-containing EDAR-interacting protein, such as EDARADD. The V370A polymorphic residue (red) is located prominently within this potential binding region in the H1–H2 loop. Seven of the thirteen known mis-sense mutations in EDAR that lead to hypohidrotic ectodermal dysplasia (HED) in humans are located in the EDAR-DD: the only four mutations in EDAR that lead to the dominant transmission of HED (green) and three recessive mutations (yellow)21. Four of these mutations, R375H, L377F, R420Q and I418T are located in the vicinity of the predicted interaction interface.
Our analysis only scratches the surface of the recent selective history of the human genome. The results indicate that individual candidates may coalesce into pathways that reveal traits under selection, analogous to the alleles of multiple genes (for example, HBB, G6PD and DARC) that arose and spread in Africa and other tropical populations as a result of the partial protection they confer against malaria2,12. Such endeavours will be enhanced by continuing development of analytical methods to localize signals in candidate regions, generation of expanded data sets, advances in comparative genomics to define coding and regulatory regions, and biological follow-up of promising candidates. True understanding of the role of adaptive evolution will require collaboration across multiple disciplines, including molecular and structural biology, medical and population genetics, and history and anthropology.
METHODS SUMMARY
Genotyping data
Phase 2 of the International Haplotype Map (HapMap2) (www.hapmap.org) contains 3.1 million SNPs genotyped in 420 chromosomes in 3 continental populations (120 European (CEU), 120 African (YRI) and 180 Asian (JPT+CHB))1. We further genotyped our top HapMap2 functional candidates in the HGDR-CEPH Human Genome Diversity Cell Line Panel20.
LRH, iHS and XP-EHH tests
The Long-Range Haplotype (LRH), integrated Haplotype Score (iHS) and Cross Population EHH (XP-EHH) tests detect alleles that have risen to high frequency rapidly enough that long-range association with nearby polymorphisms—the long-range haplotype—has not been eroded by recombination; haplotype length is measured by the EHH8,9. The first two tests detect partial selective sweeps, whereas XP-EHH detects selected alleles that have risen to near fixation in one but not all populations. To evaluate the tests, we simulated genomic data for each HapMap population in a range of demographic scenarios—under neutral evolution and twenty scenarios of positive selection—developing the program Sweep (www.broad.mit.edu/mpg/sweep) for analysis. For our top candidates by the three tests, we tested for haplotype-specific recombination rates and copy-number polymorphisms, possible confounders.
Localization
We calculated FST and derived-allele frequency for all SNPs within the top candidate regions. We developed a database for those regions to annotate all potentially functional DNA changes (B.F., unpublished), including non-synonymous variants, variants disrupting predicted functional motifs, variants within regions of conservation in mammals and variants previously associated with human phenotypic differences, as well as synonymous, intronic and untranslated region variants.
Structural model
We generated a homology model of the EDAR death domain (DD) from available DD structures using Modeller 9v1 (ref. 22). The distribution of conserved residues, built using ConSurf23 with an EDAR sequence alignment from 22 species, shows a bias to the protein core in helices H1, H2 and H5, supporting our model.
METHODS
Genotyping data
The chromosomes examined in HapMap 2 were phased by the consortium using PHASE25.
The HGDR-CEPH Human Genome Diversity Cell Line Panel20 consists of 1,051 individuals from 51 populations across the world. We obtained DNA for the panel from the Foundation Jean Dausset (CEPH) and genotyped our top functional candidates for selection in the panel.
LRH, iHS, and XP-EHH tests
The Long-Range Haplotype (LRH) and the integrated Haplotype Score (iHS) tests have been previously described8,9 and our methods are given in Supplementary Methods.
EHH between two SNPs, A and B, is defined as the probability that two randomly chosen chromosomes are homozygous at all SNPs between A and B, inclusive8; it is usually calculated using a sample of chromosomes from a single population. Explicitly, if the N chromosomes in a sample form G homozygous groups, with each group i having ni elements, EHH is defined as
The XP-EHH test detects selective sweeps in which the selected allele has risen to high frequency or fixation in one population, but remains polymorphic in the human population as a whole; for this purpose it is more powerful than either iHS or LRH (Supplementary Fig. 2 and Supplementary Tables 3-6). XP-EHH uses cross-population comparison of haplotype lengths to control for local variation in recombination rates. Such cross-population comparison is complicated by the fact that haplotype lengths also depend on population history, such as bottlenecks and expansions26. The XP-EHH test normalizes for genome-wide differences in haplotype length between populations.
We define the XP-EHH test with respect to two populations, A and B, a given core SNP and a given direction (centromere distal or proximal). EHH is calculated for all SNPs in population A between the core SNP and X, and the value integrated with respect to genetic distance, with the result defined as IA. IB is defined analogously for population B. The statistic ln(IA/IB) is then calculated; an unusually positive value suggests selection in population A, a negative value selection in B. For identifying outliers, the log-ratio is normalized to have zero mean and unit variance. Details are given in Supplementary Methods.
We developed a computer program, Sweep, to implement these tests (LRH, iHS and XP-EHH) for positive selection, (Supplementary Methods; www.broad.mit.edu/mpg/sweep). In identifying the 22 strongest candidate regions, we considered regions with signals in at least two of five tests (LRH, iHS and XP-EHH in the three pairwise comparisons among the three populations), as well as those that had the strongest signal for each individual test. With this threshold we found no events in 10 Gb of simulated neutrally evolving sequence. For the top candidates by the three tests, we have taken additional steps to rule out the effects of recombination rate variation and copy number polymorphisms (Supplementary Methods).
Simulations and power calculations
We simulated the evolution of 1 MB sections of 120 chromosomes from each of the three continental HapMap populations, using a previously validated demographic model27, under neutrality and under twenty scenarios of positive selection. We studied the effects of demo-graphy by further simulating recent bottlenecks with a range of intensity. Details of simulations and power calculations are given in Supplementary Methods.
Functional annotation
We developed an annotation database for our candidate regions to identify all DNA changes with potential functional consequence (B.F., unpublished). We first examined candidates most likely to be functional, including non-synonymous mutations, variants that disrupt predicted functional motifs (transcription factor motifs in conserved regions up to 10-kb 5′ of known genes and miRNA binding-site motifs in conserved 3′ untranslated regions of known genes), and variations reported to be associated with human phenotypic differences. For the last category, we identified variations associated with a clinical state (for example, malaria resistance) by a review of the published literature and those associated with changes to gene expression in lymphoblastoid cell lines from the HapMap individuals. The annotation included insertion/deletion mutations of all sizes. We also examined candidates with lower probability of being functional, including synonymous, intronic and untranslated variations and those that occur within regions of conservation in mammalian species. These methods are described in greater detail in Supplementary Methods.
Structural model of EDAR's death domain
We generated a homology model for EDAR's death domain (DD) using six solved DD structures: p75 NGFR-DD, RAIDD-DD, Pelle-DD, FADD-DD, Fas-DD and IRAK4-DD24,28-32. We aligned the corresponding protein sequences using SALIGN33. We then added the amino acid sequence of EDAR's DD (residues 356-431) to this structural alignment using Modeller 9v1 (ref. 22). The resulting alignment was used as the input to Modeller 9v1 to build ten EDAR-DD structure models, and the best model was selected based on the Objective Function Score. Owing to the high DOPE scores in the H1-H2 loop we performed a loop refinement using Modeller9v1, significantly reducing the energy of this region. We further evaluated the model by examining the distribution of conserved residues using ConSurf23 with an alignment of EDAR-DD sequences from 22 species. We observed a bias of conserved residues to the protein core in H1, H2 and H5, which supports our EDAR-DD model. To identify potential binding regions of EDAR-DD, we used LSQMAN34 to superimpose the model to the Tube-DD-Pelle-DD complex structure24. The H1-H2 and H5-H6 loops of the EDAR-DD correspond to Tube residues interacting with Pelle, and H2-H3 and H4-H5 loops to Pelle residues interacting with Tube. We focused our analysis on the residues corresponding to the interacting region in Tube because our EDAR-DD model is most similar to Tube. Figures were generated with PyMOL35.
Other analysis
Description of methods for calculating FST, derived-allele frequency, alignment of the SLC24 amino acids, species alignments, conservation graphs, and estimation of the fraction of SNPs genotyped in HapMap2 and identified in dbSNP, are given in Supplementary Methods.
Supplementary Material
Acknowledgements
P.C.S. is funded by a Burroughs Wellcome Career Award in the Biomedical Sciences and has been funded by the Damon Runyon Cancer Fellowship and the L'Oreal for Women in Science Award. We thank A. Schier, B. Voight, R. Roberts, M. Kreiger, A. Abzhanov, D. Degusta, M. Burnette, E. Lieberman, M. Daly, D. Altshuler, D. Reich, D. Lieberman and I. Woods for helpful discussions on our analysis and results. We also thank L. Ziaugra, D. Tabbaa and T. Rachupka for experimental assistance. This work was funded in part by grants from the National Human Genome Research Institute (to E.S.L.) and from the Broad Institute of MIT and Harvard.
The International HapMap Consortium
The International HapMap Consortium (Participants are arranged by institution and then alphabetically within institutions except for Principal Investigators and Project Leaders, as indicated.)
Genotyping centres: Perlegen Sciences Kelly A. Frazer (Principal Investigator)1, Dennis G. Ballinger2, David R. Cox2, David A. Hinds2, Laura L. Stuve2; Baylor College of Medicine and ParAllele BioScience Richard A. Gibbs (Principal Investigator)3, John W. Belmont3, Andrew Boudreau4, Paul Hardenbol5, Suzanne M. Leal3, Shiran Pasternak6, David A. Wheeler3, Thomas D. Willis4, Fuli Yu7; Beijing Genomics Institute Huanming Yang (Principal Investigator)8, Changqing Zeng (Principal Investigator)8, Yang Gao8, Haoran Hu8, Weitao Hu8, Chaohua Li8, Wei Lin8, Siqi Liu8, Hao Pan8, Xiaoli Tang8, Jian Wang8, Wei Wang8, Jun Yu8, Bo Zhang8, Qingrun Zhang8, Hongbin Zhao8, Hui Zhao8, Jun Zhou8; Broad Institute of Harvard and Massachusetts Institute of Technology Stacey B. Gabriel (Project Leader)7, Rachel Barry7, Brendan Blumenstiel7, Amy Camargo7, Matthew Defelice7, Maura Faggart7, Mary Goyette7, Supriya Gupta7, Jamie Moore7, Huy Nguyen7, Robert C. Onofrio7, Melissa Parkin7, Jessica Roy7, Erich Stahl7, Ellen Winchester7, Liuda Ziaugra7, David Altshuler (Principal Investigator)7,9; Chinese National Human Genome Center at Beijing Yan Shen (Principal Investigator)10, Zhijian Yao10; Chinese National Human Genome Center at Shanghai Wei Huang (Principal Investigator)11, Xun Chu11, Yungang He11, Li Jin12, Yangfan Liu11, Yayun Shen11, Weiwei Sun11, Haifeng Wang11, Yi Wang11, Ying Wang11, Xiaoyan Xiong11, Liang Xu11; Chinese University of Hong Kong Mary M. Y. Waye (Principal Investigator)13, Stephen K. W. Tsui13; Hong Kong University of Science and Technology Hong Xue (Principal Investigator)14, J. Tze-Fei Wong14; Illumina Luana M. Galver (Project Leader)15, Jian-Bing Fan15, Kevin Gunderson15, Sarah S. Murray1, Arnold R. Oliphant16, Mark S. Chee (Principal Investigator)17; McGill University and Génome Québec Innovation Centre Alexandre Montpetit (Project Leader)18, Fanny Chagnon18, Vincent Ferretti18, Martin Leboeuf18, Jean-Franc¸ois Olivier4, Michael S. Phillips18, Stéphanie Roumy15, Clémentine Sallée19, Andrei Verner18, Thomas J. Hudson (Principal Investigator)20; University of California at San Francisco and Washington University Pui-Yan Kwok (Principal Investigator)21, Dongmei Cai21, Daniel C. Koboldt22, Raymond D. Miller22, Ludmila Pawlikowska21, Patricia Taillon-Miller22, Ming Xiao21; University of Hong Kong Lap-Chee Tsui (Principal Investigator)23, William Mak23, You Qiang Song23, Paul K. H. Tam23; University of Tokyo and RIKEN Yusuke Nakamura (Principal Investigator)24,25, Takahisa Kawaguchi25, Takuya Kitamoto25, Takashi Morizono25, Atsushi Nagashima25, Yozo Ohnishi25, Akihiro Sekine25, Toshihiro Tanaka25, Tatsuhiko Tsunoda25; Wellcome Trust Sanger Institute Panos Deloukas (Project Leader)26, Christine P. Bird26, Marcos Delgado26, Emmanouil T. Dermitzakis26, Rhian Gwilliam26, Sarah Hunt26, Jonathan Morrison27, Don Powell26, Barbara E. Stranger26, Pamela Whittaker26, David R. Bentley (Principal Investigator)28
Analysis groups: Broad Institute Mark J. Daly (Project Leader)7,9, Paul I. W. de Bakker7,9, Jeff Barrett7,9, Yves R. Chretien7, Julian Maller7,9, Steve McCarroll7,9, Nick Patterson7, Itsik Pe'er29, Alkes Price7, Shaun Purcell9, Daniel J. Richter7, Pardis Sabeti7, Richa Saxena7,9, Stephen F. Schaffner7, Pak C. Sham23, Patrick Varilly7, David Altshuler (Principal Investigator)7,9; Cold Spring Harbor Laboratory Lincoln D. Stein (Principal Investigator)6, Lalitha Krishnan6, Albert Vernon Smith6, Marcela K. Tello-Ruiz6, Gudmundur A. Thorisson30; Johns Hopkins University School of Medicine Aravinda Chakravarti (Principal Investigator)31, Peter E. Chen31, David J. Cutler31, Carl S. Kashuk31, Shin Lin31; University of Michigan Gonc¸alo R. Abecasis (Principal Investigator)32, Weihua Guan32, Yun Li32, Heather M. Munro33, Zhaohui Steve Qin32, Daryl J. Thomas34; University of Oxford Gilean McVean (Project Leader)35, Adam Auton35, Leonardo Bottolo35, Niall Cardin35, Susana Eyheramendy35, Colin Freeman35, Jonathan Marchini35, Simon Myers35, Chris Spencer7, Matthew Stephens36, Peter Donnelly (Principal Investigator)35; University of Oxford, Wellcome Trust Centre for Human Genetics Lon R. Cardon (Principal Investigator)37, Geraldine Clarke38, David M. Evans38, Andrew P. Morris38, Bruce S. Weir39; RIKEN Tatsuhiko Tsunoda (Principal Investigator)25, Todd A. Johnson25; US National Institutes of Health James C. Mullikin40; US National Institutes of Health National Center for Biotechnology Information Stephen T. Sherry41, Michael Feolo41, Andrew Skol42
Community engagement/public consultation and sample collection groups: Beijing Normal University and Beijing Genomics Institute Houcan Zhang43, Changqing Zeng8, Hui Zhao8; Health Sciences University of Hokkaido, Eubios Ethics Institute, and Shinshu University Ichiro Matsuda (Principal Investigator)44, Yoshimitsu Fukushima45, Darryl R. Macer46, Eiko Suda47; Howard University and University of Ibadan Charles N. Rotimi (Principal Investigator)48, Clement A. Adebamowo49, Ike Ajayi49, Toyin Aniagwu49, Patricia A. Marshall50, Chibuzor Nkwodimmah49, Charmaine D. M. Royal48; University of Utah Mark F. Leppert (Principal Investigator)51, Missy Dixon51, Andy Peiffer51
Ethical, legal and social issues: Chinese Academy of Social Sciences Renzong Qiu52; Genetic Interest Group Alastair Kent53; Kyoto University Kazuto Kato54; Nagasaki University Norio Niikawa55; University of Ibadan School of Medicine Isaac F. Adewole49; University of Montréal Bartha M. Knoppers19; University of Oklahoma Morris W. Foster56; Vanderbilt University Ellen Wright Clayton57; Wellcome Trust Jessica Watkin58
SNP discovery: Baylor College of Medicine Richard A. Gibbs (Principal Investigator)3, John W. Belmont3, Donna Muzny3, Lynne Nazareth3, Erica Sodergren3, George M. Weinstock3, David A. Wheeler3, Imtaz Yakub3; Broad Institute of Harvard and Massachusetts Institute of Technology Stacey B. Gabriel (Project Leader)7, Robert C. Onofrio7, Daniel J. Richter7, Liuda Ziaugra7, Bruce W. Birren7, Mark J. Daly7,9, David Altshuler (Principal Investigator)7,9; Washington University Richard K. Wilson (Principal Investigator)59, Lucinda L. Fulton59; Wellcome Trust Sanger Institute Jane Rogers (Principal Investigator)26, John Burton26, Nigel P. Carter26, Christopher M. Clee26, Mark Griffiths26, Matthew C. Jones26, Kirsten McLay26, Robert W. Plumb26, Mark T. Ross26, Sarah K. Sims26, David L. Willey26
Scientific management: Chinese Academy of Sciences Zhu Chen60, Hua Han60, Le Kang60; Genome Canada Martin Godbout61, John C. Wallenburg62; Génome Québec Paul L'Archevêque63, Guy Bellemare63; Japanese Ministry of Education, Culture, Sports, Science and Technology Koji Saeki64; Ministry of Science and Technology of the People's Republic of China Hongguang Wang65, Daochang An65, Hongbo Fu65, Qing Li65, Zhen Wang65; The Human Genetic Resource Administration of China Renwu Wang66; The SNP Consortium Arthur L. Holden15; US National Institutes of Health Lisa D. Brooks67, Jean E. McEwen67, Mark S. Guyer67, Vivian Ota Wang67,68, Jane L. Peterson67, Michael Shi69, Jack Spiegel70, Lawrence M. Sung71, Lynn F. Zacharia67, Francis S. Collins72; Wellcome Trust Karen Kennedy61, Ruth Jamieson58, John Stewart58
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Reprints and permissions information is available at www.nature.com/reprints.
The Scripps Research Institute, 10550 North Torrey Pines Road MEM275, La Jolla, California 92037, USA.
Perlegen Sciences, 2021 Stierlin Court, Mountain View, California 94043, USA.
Baylor College of Medicine, Human Genome Sequencing Center, Department of Molecular and Human Genetics, 1 Baylor Plaza, Houston, Texas 77030, USA.
Affymetrix, 3420 Central Expressway, Santa Clara, California 95051, USA.
Pacific Biosciences, 1505 Adams Drive, Menlo Park, California 94025, USA.
Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, New York 11724, USA.
The Broad Institute of Harvard and Massachusetts Institute of Technology, 1 Kendall Square, Cambridge, Massachusetts 02139, USA.
Beijing Genomics Institute, Chinese Academy of Sciences, Beijing 100300, China.
Massachusetts General Hospital and Harvard Medical School, Simches Research Center, 185 Cambridge Street, Boston, Massachusetts 02114, USA.
Chinese National Human Genome Center at Beijing, 3-707 N. Yongchang Road, Beijing Economic-Technological Development Area, Beijing 100176, China.
Chinese National Human Genome Center at Shanghai, 250 Bi Bo Road, Shanghai 201203, China.
Fudan University and CAS-MPG Partner Institute for Computational Biology, School of Life Sciences, SIBS, CAS, Shanghai, 201203, China.
The Chinese University of Hong Kong, Department of Biochemistry, The Croucher Laboratory for Human Genetics, 6/F Mong Man Wai Building, Shatin, Hong Kong.
Hong Kong University of Science and Technology, Department of Biochemistry and Applied Genomics Center, Clear Water Bay, Knowloon, Hong Kong.
Illumina, 9885 Towne Centre Drive, San Diego, California 92121, USA.
Complete Genomics, 658 North Pastoria Avenue, Sunnyvale, California 94085, USA.
Prognosys Biosciences, 4215 Sorrento Valley Boulevard, Suite 105, San Diego, California 92121, USA.
McGill University and Génome Québec Innovation Centre, 740 Dr Penfield Avenue, Montréal, Québec H3A 1A4, Canada.
University of Montréal, The Public Law Research Centre (CRDP), PO Box 6128, Downtown Station, Montréal, Québec H3C 3J7, Canada.
Ontario Institute for Cancer Research, MaRS Centre, South Tower, 101 College Street, Suite 500, Toronto, Ontario,M5G 1L7, Canada.
University of California, San Francisco, Cardiovascular Research Institute, 513 Parnassus Avenue, Box 0793, San Francisco, California 94143, USA.
Washington University School of Medicine, Department of Genetics, 660 S. Euclid Avenue, Box 8232, St Louis, Missouri 63110, USA.
University of Hong Kong, Genome Research Centre, 6/F, Laboratory Block, 21 Sassoon Road, Pokfulam, Hong Kong.
University of Tokyo, Institute of Medical Science, 4-6-1 Sirokanedai, Minatoku, Tokyo 108-8639, Japan.
RIKEN SNP Research Center, 1-7-22 Suehiro-cho, Tsurumi-ku Yokohama, Kanagawa 230-0045, Japan.
Wellcome Trust Sanger Institute, The Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK.
University of Cambridge, Department of Oncology, Cambridge CB1 8RN, UK.
Solexa, Chesterford Research Park, Little Chesterford, nr SaffronWalden, Essex CB10 1XL, UK.
Columbia University, 500 West 120th Street, New York, New York 10027, USA.
University of Leicester, Department of Genetics, Leicester LE1 7RH, UK.
Johns Hopkins University School of Medicine, McKusick-Nathans Institute of Genetic Medicine, Broadway Research Building, Suite 579, 733 N. Broadway, Baltimore, Maryland 21205, USA.
University of Michigan, Center for Statistical Genetics, Department of Biostatistics, 1420 Washington Heights, Ann Arbor, Michigan 48109, USA.
International Epidemiology Institute, 1455 Research Boulevard, Suite 550, Rockville, Maryland 20850, USA.
Center for Biomolecular Science and Engineering, Engineering 2, Suite 501, Mail Stop CBSE/ITI, UC Santa Cruz, Santa Cruz, California 95064, USA.
University of Oxford, Department of Statistics, 1 South Parks Road, Oxford OX1 3TG, UK.
University of Chicago, Department of Statistics, 5734 S. University Avenue, Eckhart Hall, Room 126, Chicago, Illinois 60637, USA.
Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue North, Seattle, Washington 98109, USA.
University of Oxford/Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7BN, UK.
University of Washington Department of Biostatistics, Box 357232, Seattle, Washington 98195, USA.
US National Institutes of Health, National Human Genome Research Institute, 50 South Drive, Bethesda, Maryland 20892, USA.
US National Institutes of Health, National Library of Medicine, National Center for Biotechnology Information, 8600 Rockville Pike, Bethesda, Maryland 20894, USA.
University of Chicago, Department of Medicine, Section of Genetic Medicine, 5801 South Ellis, Chicago, Illinois 60637, USA.
Beijing Normal University, 19 Xinjiekouwai Street, Beijing 100875, China.
Health Sciences University of Hokkaido, Ishikari Tobetsu Machi 1757, Hokkaido 061-0293, Japan.
Shinshu University School of Medicine, Department of Medical Genetics, Matsumoto 390-8621, Japan.
United Nations Educational, Scientific and Cultural Organization (UNESCO Bangkok), 920 Sukhumwit Road, Prakanong, Bangkok 10110, Thailand.
University of Tsukuba, Eubios Ethics Institute, PO Box 125, Tsukuba Science City 305-8691, Japan.
Howard University, National Human Genome Center, 2216 6th Street, NW, Washington, District of Columbia 20059, USA.
University of Ibadan College of Medicine, Ibadan, Oyo State, Nigeria.
Case Western Reserve University School of Medicine, Department of Bioethics, 10900 Euclid Avenue, Cleveland, Ohio 44106, USA.
University of Utah, Eccles Institute of Human Genetics, Department of Human Genetics, 15 North 2030 East, Salt Lake City, Utah 84112, USA.
Chinese Academy of Social Sciences, Institute of Philosophy/Center for Applied Ethics, 2121, Building 9, Caoqiao Xinyuan 3 Qu, Beijing 100067, China.
Genetic Interest Group, 4D Leroy House, 436 Essex Road, London N130P, UK.
Kyoto University, Institute for Research in Humanities and Graduate School of Biostudies, Ushinomiya-cho, Sakyo-ku, Kyoto 606-8501, Japan.
Nagasaki University Graduate School of Biomedical Sciences, Department of Human Genetics, Sakamoto 1-12-4, Nagasaki 852-8523, Japan.
University of Oklahoma, Department of Anthropology, 455 W. Lindsey Street, Norman, Oklahoma 73019, USA.
Vanderbilt University, Center for Genetics and Health Policy, 507 Light Hall, Nashville, Tennessee 37232, USA.
Wellcome Trust, 215 Euston Road, London NW1 2BE, UK.
Washington University School of Medicine, Genome Sequencing Center, Box 8501, 4444 Forest Park Avenue, St Louis, Missouri 63108, USA.
Chinese Academy of Sciences, 52 Sanlihe Road, Beijing 100864, China.
Genome Canada, 150 Metcalfe Street, Suite 2100, Ottawa, Ontario K2P 1P1, Canada.
McGill University, Office of Technology Transfer, 3550 University Street, Montréal, Québec H3A 2A7, Canada.
Génome Québec, 630, boulevard René-Lévesque Ouest, Montréal, Québec H3B 1S6, Canada.
Ministry of Education, Culture, Sports, Science, and Technology, 3-2-2 Kasumigaseki, Chiyodaku, Tokyo 100-8959, Japan.
Ministry of Science and Technology of the People's Republic of China, 15 B. Fuxing Road, Beijing 100862, China.
The Human Genetic Resource Administration of China, b7, Zaojunmiao, Haidian District, Beijing 100081, China.
US National Institutes of Health, National Human Genome Research Institute, 5635 Fishers Lane, Bethesda, Maryland 20892, USA.
US National Institutes of Health, Office of Behavioral and Social Science Research, 31 Center Drive, Bethesda, Maryland 20892, USA.
Novartis Pharmaceuticals Corporation, Biomarker Development, One Health Plaza, East Hanover, New Jersey 07936, USA.
US National Institutes of Health, Office of Technology Transfer, 6011 Executive Boulevard, Rockville, Maryland 20852, USA.
University of Maryland School of Law, 500 W. Baltimore Street, Baltimore, Maryland 21201, USA.
US National Institutes of Health, National Human Genome Research Institute, 31 Center Drive, Bethesda, Maryland 20892, USA.
References
- 1.The International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. doi: 10.1038/nature06258. doi:10.1038/nature06258 (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabeti PC, et al. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 3.Kunz S, et al. Posttranslational modification of α-dystroglycan, the cellular receptor for arenaviruses, by the glycosyltransferase LARGE is critical for virus binding. J. Virol. 2005;79:14282–14296. doi: 10.1128/JVI.79.22.14282-14296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graf J, Hodgson R, van Daal A. Single nucleotide polymorphisms in the MATP gene are associated with normal human pigmentation variation. Hum. Mutat. 2005;25:278–284. doi: 10.1002/humu.20143. [DOI] [PubMed] [Google Scholar]
- 5.Lamason RL, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 6.Botchkarev VA, Fessing MY. Edar signaling in the control of hair follicle development. J. Investig. Dermatol. Symp. Proc. 2005;10:247–251. doi: 10.1111/j.1087-0024.2005.10129.x. [DOI] [PubMed] [Google Scholar]
- 7.The International Haplotype Map Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 9.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura R, Fujimoto A, Tokunaga K, Ohashi J. A practical genome scan for population-specific strong selective sweeps that have reached fixation. PLoS ONE. 2007;2:e286. doi: 10.1371/journal.pone.0000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang K, Thornton KR, Stoneking M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007;5:e171. doi: 10.1371/journal.pbio.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williamson SH, et al. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bersaglieri T, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teshima KM, Coop G, Przeworski M. How reliable are empirical genomic scans for selective sweeps? Genome Res. 2006;16:702–712. doi: 10.1101/gr.5105206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuokkanen M, et al. Transcriptional regulation of the lactase–phlorizin hydrolase gene by polymorphisms associated with adult-type hypolactasia. Gut. 2003;52:647–652. doi: 10.1136/gut.52.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller RG. Simultaneous statistical inference. XVI. Springer; New York: 1981. p. 299. [Google Scholar]
- 17.Soejima M, Tachida H, Ishida T, Sano A, Koda Y. Evidence for recent positive selection at the human AIM1 locus in a European population. Mol. Biol. Evol. 2006;23:179–188. doi: 10.1093/molbev/msj018. [DOI] [PubMed] [Google Scholar]
- 18.Richmond JK, Baglole DJ. Lassa fever: epidemiology, clinical features, and social consequences. Br. Med. J. 2003;327:1271–1275. doi: 10.1136/bmj.327.7426.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colosimo PF, et al. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science. 2005;307:1928–1933. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg NA, et al. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 21.Chassaing N, Bourthoumieu S, Cossee M, Calvas P, Vincent MC. Mutations in EDAR account for one-quarter of non-ED1-related hypohidrotic ectodermal dysplasia. Hum. Mutat. 2006;27:255–259. doi: 10.1002/humu.20295. [DOI] [PubMed] [Google Scholar]
- 22.Marti-Renom MA, et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 23.Landau M, et al. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao T, Towb P, Wasserman SA, Sprang SR. Three-dimensional structure of a complex between the death domains of Pelle and Tube. Cell. 1999;99:545–555. doi: 10.1016/s0092-8674(00)81542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crawford DC, et al. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nature Genet. 2004;36:700–706. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- 27.Schaffner SF, et al. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berglund H, et al. The three-dimensional solution structure and dynamic properties of the human FADD death domain. J. Mol. Biol. 2000;302:171–188. doi: 10.1006/jmbi.2000.4011. [DOI] [PubMed] [Google Scholar]
- 29.Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature. 1996;384:638–641. doi: 10.1038/384638a0. [DOI] [PubMed] [Google Scholar]
- 30.Lasker MV, Gajjar MM, Nair SK. Cutting edge: molecular structure of the IL-1R-associated kinase-4 death domain and its implications for TLR signaling. J. Immunol. 2005;175:4175–4179. doi: 10.4049/jimmunol.175.7.4175. [DOI] [PubMed] [Google Scholar]
- 31.Liepinsh E, Ilag LL, Otting G, Ibanez CF. NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J. 1997;16:4999–5005. doi: 10.1093/emboj/16.16.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park HH, Wu H. Crystal structure of RAIDD death domain implicates potential mechanism of PIDDosome assembly. J. Mol. Biol. 2006;357:358–364. doi: 10.1016/j.jmb.2005.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marti-Renom MA, Madhusudhan MS, Sali A. Alignment of protein sequences by their profiles. Protein Sci. 2004;13:1071–1087. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 35.DeLano WL. MacPyMOL: A PyMOL-based Molecular Graphics Application for MacOS X. DeLano Scientific LLC; Palo Alto, California, USA: 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.