

Abstract
A characterization of the genetic variation of recently admixed populations may reveal historical population events, and is useful for the detection of single nucleotide polymorphisms (SNPs) associated with diseases through association studies and admixture mapping. Inference of locus-specific ancestry is key to our understanding of the genetic variation of such populations. While a number of methods for the inference of locus-specific ancestry are accurate when the ancestral populations are quite distant (e.g. African–Americans), current methods incur a large error rate when inferring the locus-specific ancestry in admixed populations where the ancestral populations are closely related (e.g. Americans of European descent).
Results: In this work, we extend previous methods for the inference of locus-specific ancestry by the incorporation of a refined model of recombination events. We present an efficient dynamic programming algorithm to infer the locus-specific ancestries in this model, resulting in a method that attains improved accuracies; the improvement is most significant when the ancestral populations are closely related. An evaluation on a wide range of scenarios, including admixtures of the 52 population groups from the Human Genome Diversity Project demonstrates that locus-specific ancestry can indeed be accurately inferred in these admixtures using our method. Finally, we demonstrate that imputation methods can be improved by the incorporation of locus-specific ancestry, when applied to admixed populations.
Availability: The implementation of the WINPOP model is available as part of the LAMP package at http://lamp.icsi.berkeley.edu/lamp
Contact: heran@icsi.berkeley.edu
1 INTRODUCTION
Recent advances in genotyping technologies have opened up unprecedented opportunities to improve our understanding of complex diseases through disease association studies. Most of these association studies have been performed on Caucasian populations of cases and controls. To gain additional insight, studies are often replicated on other populations, some of which are recently admixed. Recently admixed populations are formed by the mixing of two or more ancestral populations for a small number of generations. For instance, African Americans are a recently admixed population, where the ancestral populations are West Africans and Caucasians. Even the Caucasian population in the USA is in fact a recently admixed population, where the original ancestral populations are different European populations that immigrated to the USA over the last few centuries.
Admixed populations have been extensively used to detect associations in diseases that differ in prevalence across populations through admixture mapping (Reich et al., 2005; Zhu et al., 2005). The technique of admixture mapping is based on the observation that the cases in such an admixed population will have enhanced ancestry from the higher risk population near loci associated with the disease. In order to perform such studies successfully, it is crucial to be able to accurately infer the locus-specific ancestry of each individual. Moreover, accurate estimates of the locus-specific ancestry may reveal patterns of selection (Tang et al., 2007) as well as recent recombination events (Sankararaman et al., 2008b). Particularly, in this work we demonstrate that locus-specific ancestry may also play an important role in the problem of genotype imputation, in which, genotypes left untyped in case–control studies are reliably inferred by leveraging the single nucleotide polymorphism (SNP) correlation information from large repositories of human SNP variation such as the HapMap project (The International HapMap Consortium, 2005).
While many methods have been proposed for the inference of locus-specific ancestry (Hoggart et al., 2004; Patterson et al., 2004; Pritchard et al., 2000; Sankararaman et al., 2008a; b; Sundquist et al., 2008; Tang et al., 2006, more recent works have focused on developing methods that are scalable to whole-genome datasets (Sankararaman et al., 2008a, b; Sundquist et al., 2008; Tang et al., 2006). These methods have been shown to incur low error rates in admixtures that originated from ancestral populations with a high fixation index (Fst), such as African Americans. However, when the ancestral populations are closely related (e.g. the Japanese and Chinese populations), their accuracies have been shown to be quite low (<70%, for populations that have been mixing for seven generations or more) (Sankararaman et al., 2008b).
In contrast to locus-specific ancestry, when considering the averaged genome-wide ancestry of each individual, it has been recently shown that principal component analysis can be used to detect differences between populations that are as close as a few 100 km away from each other (Novembre et al., 2008). However, it is not clear that such high resolution can be achieved by methods that seek to infer the locus-specific ancestry. In particular, it is an open question whether locus-specific ancestry can be accurately inferred on very close populations such as mixtures of Asians, or mixtures of Europeans (e.g. Americans of European descent).
We present here an efficient and accurate method for the inference of locus-specific ancestry. Our method, called WINPOP, is unique in that it achieves high accuracy on admixtures of closely related populations, including mixtures of European populations or mixtures of Asian populations (e.g. JPT-CHB from the HapMap populations). To achieve this, we partition the genome into overlapping, contiguous windows of SNPs, and we optimize a likelihood model over each of the windows. We then glue the solutions together by casting a majority vote for each SNP.
The basic framework in which overlapping windows are used for the inference of local ancestry has been previously suggested in our previously reported method LAMP (Sankararaman et al., 2008a). LAMP is a highly efficient method, that has been shown to be accurate on admixtures of distant populations. The basic idea behind LAMP lies in making predictions in each window using a likelihood model that assumes no recombinations. In contrast to LAMP, our method uses an improved modeling of the recombination events, and it chooses the window size adaptively at each location in the genome, according to the local genetic structure of the ancestral populations. These two new ideas result in a substantial improvement in accuracy.
Extensive simulation results demonstrate that WINPOP achieves improved inference of locus-specific ancestries on both distant and closely related admixtures. The improvements in accuracy across the closely related populations range from 13% to 35%. Further, we examined the utility of locus-specific ancestry on the task of imputing missing genotypes. We show that exploiting accurate methods for locus-specific ancestry leads to lower error in imputation, and that the imputation accuracy critically depends on the accuracy of the ancestral inference.
2 METHODS
In this work, we consider the inference of locus-specific ancestry in recently admixed populations. Recently admixed populations arise from K ancestral populations A1,…,AK that have been mixing for g generations. We focus on the analysis of SNP data in these populations. For a given set of genotypes from the admixed population, we describe each individual genotype as a vector gi, where gij∈{0, 1, 2} is the minor allele count of individual i at position j. At position j, the two alleles of individual i have descended from one or two of the K ancestral populations. We are interested in estimating these ancestral population(s) for each SNP of a genotype. We will assume that the SNP allele frequencies of the ancestral populations are given; e.g. in African-Americans, the ancestral populations can be described as Europeans and West-Africans, and the allele frequencies for those are known.
Mathematically, we model the recently admixed populations as a set of K independent populations that have come together at some point in history and have been mixing (through random mating) for g generations. In each generation, we model the transmission of a chromosome from a parent to a child as a random walk along the chromosome from the 5′-end to the 3′-end, with crossovers between chromosomes occurring as a Poisson process with rate (g − 1)ϕ, where ϕ is the recombination rate (for simplicity of the exposition, we will assume in this article a constant recombination rate, although the discussion can be easily extended to account for variable recombination rates).
2.1 The LAMP framework
LAMP is a highly efficient method, that has been previously shown to accurately infer locus-specific ancestry, particularly on admixed populations with distant ancestral populations (Sankararaman et al., 2008a). It is based on the following idea: the genome of an admixed individual is a mosaic of subregions, where each subregion originates from exactly one population. The typical length of these subregions is a function of the number of generations for which the ancestral populations have been mixing, as well as the recombination rate in the region. LAMP partitions the genome into short, contiguous windows of size l, and assumes that in each window there has not been any recombination event since the original ancestral populations started mixing. Intuitively, if l is small enough, and the number of generations g is not too large, a typical window of length l will have almost no recombination events throughout history, and therefore almost no breakpoints (i.e. recombination event that also have led to a change in the ancestry). LAMP infers the ancestry in each window based on a likelihood model that leverages the assumption of no recombination events, and then uses a majority vote across all overlapping windows to decide on the ancestry of each nucleotide base.
Although LAMP achieves very high accuracy rates, especially for admixtures from distant ancestral population, the current framework has two inherent shortcomings which lead to lower accuracies on admixtures of closely related populations. First, the window length depends on the number of generations of admixture g, and on the recombination rates, but not on the allele frequencies. Thus, the same window length is used for admixtures of close populations or distant populations. For instance, African American populations and Japanese Chinese admixtures are treated similarly. Second, the assumption of no recombination events within each window is limiting and may incur errors.
2.2 A new model for ancestry inference within a window
We propose a new method (WINPOP) for locus-specific ancestry that uses the LAMP framework as a starting point. Particularly, WINPOP works in windows, however we assume at most one recent recombination within each window. In order to find the ancestry estimates that maximize the probability in the new model, we devised a dynamic programming algorithm that enumerates over the positions in the window, and for each position computes the likelihood of having one ancestry upstream and another downstream of that position. Moreover, as opposed to the window length computation of LAMP that depends only on the number of generations and the recombination rates, we introduce here a new procedure that chooses the window length differently at every position, by taking into account the local genetic distance between the two ancestral populations in that window. Finally, our method will assume that the SNPs are uncorrelated. To ensure this, we first search for SNPs that are in linkage disequilibrium (LD) (r2 > 0.1), and we remove the less informative SNP (i.e. the allele frequencies difference between the two populations is lower).
2.2.1 Modeling recombinations
WINPOP assumes exactly one recent recombination event in each window. We are seeking for the recombination location R and for two classification functions θ1, θ2 representing the ancestry of the SNPs upstream and downstream of R in the window. We will denote by 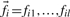 the minor allele frequencies of l independent SNPs in the ancestral population Ai in a given window of length L. We estimate the maximum aposteriori ancestry of the upstream and downstream SNPs in the window, As1At1(As2At2), as well as the index R, by finding the argument that maximize the following probability function:
the minor allele frequencies of l independent SNPs in the ancestral population Ai in a given window of length L. We estimate the maximum aposteriori ancestry of the upstream and downstream SNPs in the window, As1At1(As2At2), as well as the index R, by finding the argument that maximize the following probability function:
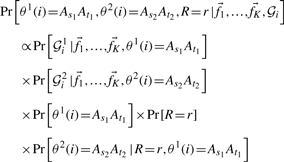 |
(1) |
where 𝒢i1 denotes the first r genotypes (gi1,…, gir) of the individual i in the window and 𝒢i2 denotes the last (l − r) genotypes (gi(r+1),…,gil).
The two terms, 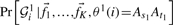 and
and 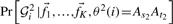 in Equation (1) are estimated, assuming Hardy–Weinberg equilibrium within the admixed population, as follows:
in Equation (1) are estimated, assuming Hardy–Weinberg equilibrium within the admixed population, as follows:
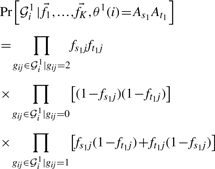 |
(2) |
The admixture fraction, αi refers to the fraction of population Ai in the admixed population (hence ∑iαi = 1). In this work, we assume that the admixture fractions α1,…, αK are known; they can be easily be estimated by other methods e.g. Frappe (Tang et al., 2005). Under the assumption of random mating, the term Pr[θ1(i)=As1At1] is estimated as
| (3) |
where δ(x, y) is 1 iff x=y and 0 otherwise.
The term Pr[R = r] is the probability that a recombination occurs between SNPs r and r + 1 (r ∈ {1,…, l − 1}). Using the Haldane map function (Haldane, 1919):
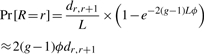 |
(4) |
where dr,r+1 is the physical distance in base pairs between SNPs r and r + 1 and L denotes the window length in base pairs.
The final term in Equation (1), namely the probability of the downstream ancestry given the upstream ancestry and the recombination event between SNPs r and r + 1 is given by the following transition matrix:
| (5) |
Note that in the above equation we implicitly assume that s1 = s2, and so the recombination occurs on the chromosome carrying the ancestries t1, t2.
We implemented a dynamic programming algorithm that finds the maximum estimates for the ancestry in the window using the above equations. We first define 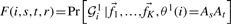 as the likelihood of the first r SNPs in individual i given that their ancestral state is AsAt. From Equation (2) it follows that:
as the likelihood of the first r SNPs in individual i given that their ancestral state is AsAt. From Equation (2) it follows that:
| (6) |
The quantity Pr[gi(r+1)|fs1(r+1)ft1(r+1)] is easily computable, by taking into account the standard Hardy–Weinberg genotype proportions from the given allele frequencies at SNP r + 1.
Similar to F(i,s,t,r), we can define B(i,s,t,r) as the probability of having the ancestry AsAt for the region starting with the (r + 1)th SNP. B(i,s,t,r) is computed from B(i,s,t,r+1) in a similar manner to Equation (6).
For each individual i and each window, WINPOP starts by computing the F(i,s,t,r) and B(i,s,t,r) values for each s,t,r. This is done in time proportional to O(lK2). In a second step, WINPOP loops over all locations and finds the location r and the pair of ancestries As1At1,As1At2 that maximize the probability function of Equation (1), which now can be rewritten as:
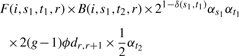 |
(7) |
Finally, we compare the posterior probabilities of these estimates to the estimates obtained assuming no recombination events within the window and choose the maximum of the two. The posterior probability assuming no recombination is given by:
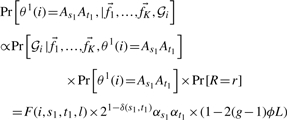 |
(8) |
It is easy to see that our algorithm runs in time proportional to O(lK3), where l is the number of SNPs considered in that window and K is the number of ancestries, which in practice will usually be smaller than 4.
2.2.2 Adaptive window size
In order to decide on the window length l, we devised a new window length computation that takes into account the ‘local fixation index’ between the two ancestral populations in that region. The local fixation index measures the genetic divergence between the two ancestral populations in that window and can be used as a predictor of how much information, in terms of number of SNPs, is required for an accurate prediction of the ancestry.
We first estimate a window length so that the probability that a window will have more than one recombination that changes the ancestry (termed breakpoints) is bounded by a constant ϵ (we use ϵ=0.1 for all the experiments presented in this article). While the recombinations in a window of length L are generated by a Poisson process with parameter 2(g − 1)ϕ, the number of breakpoints correspond to a ‘thinned’ version of this process with parameter λ = 2(g−1)ϕ(1−∑i=1K αi2). For a window of length L, the probability that the window has more than one recombination is given by
| (9) |
We thus choose 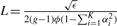 .
.
Starting from the above estimate of the window length, we perform a local search on the length of the window with the goal of obtaining the highest gain in prediction accuracy. The local search is performed in an iterative fashion by either increasing or decreasing the window length with t=20 SNPs provided the new window length shows a gain in accuracy over the current window size. The gain in accuracy from using different window sizes is quantified by testing our model's accuracy on a simulated sample of M=500 admixed individuals: starting from the ancestral allele frequencies and the global admixture proportions, ancestries for admixed individuals are generated by a random walk along the chromosomes, with recombination events occurring as a Poisson process with rate (g−1)ϕ, and then genotypes are generated from the ancestry-specific allele frequencies. When a recombination event occurs, a new ancestry is picked from the distribution given by the global admixture proportions α1,···,αK.
2.3 Upper bound on WINPOP's approach
It is important to understand the limitation of future improvements that will be based on this method. We therefore estimated the best possible accuracy that can be achieved by any method that works on SNPs in linkage equilibrium. We consider the case where the positions of the recent ancestral recombination events are known for each individual. Obviously, methods that are not provided with such information cannot do better than the optimal method that does exploit this information. Particularly, in this case, the optimal method for ancestry detection between any two recombination events is the maximum likelihood approach:
| (10) |
We thus applied the maximum likelihood model for every region defined by two recombination events to obtain an upper bound on the accuracy of both LAMP and WINPOP.
2.4 Hidden Markov model-based methods for inferring locus-specific ancestry
Many methods for locus-specific ancestry use a hidden Markov model (HMM) to model the locus-specific ancestry, where the states in each position correspond to the possible ancestral populations, and the transition probabilities depend on the recombination rates. This basic approach has been proposed by (Falush et al., 2003) in the widely used method STRUCTURE. The different methods differ in the exact formulation of the HMM, and in the inference algorithm that is used to estimate the model parameters. These methods are advantageous as they use a detailed model of the data; on the other hand, parameter estimation can be challenging in these models. The combination of a window-based method such as LAMP, together with an HMM can sometimes provide better results than each of the methods (Sankararaman et al., 2008b). We thus use WINPOP as an initialization to an HMM similar to the one used in STRUCTURE (Falush et al., 2003); we estimate the model parameters using an expectation–maximization algorithm, as described in (Sankararaman et al., 2008b). We also made some changes to this implementation by discarding SNPs with low MAFs in the ancestral populations and by explicitly modeling the probability of more than one recombination in the transition matrix—for instance, the transition matrix from a state A1A1 to a state A1A2
| (11) |
where ϕ refers to the local recombination rate and d refers to the physical distance between the SNPs.
3 RESULTS
We evaluated the performance and accuracy of WINPOP, given that it is using the basic windows framework suggested in LAMP. We compared WINPOP to the existing state-of-the-art methods for local ancestry inference. For our experiments, we used simulated admixed populations using as ancestral populations those from the four HapMap panels (The International HapMap Consortium, 2005), the 52 population groups from the Human Genome Diversity Project (Li et al., 2008) as well as the control group from the Wellcome Trust Case Control Consortium (Wellcome Trust Case Control Consortium, 2007), as part of the control group. Unless otherwise noted, we used only the SNPs found in the Affymetrix 500K GeneChip Assay (http://www.affymetrix.com/products/arrays/specific/500k.affx) from Chromosome 1. For a pair of populations, we simulated an admixed population by picking individuals from the two ancestral populations in the ratio α : 1 − α. In each generation, individuals mate randomly and produce offspring. We repeated the mixing process for g generations. The rate of the recombination process is set to 10−8 per bp per generation. These simulations result in an admixed population with known local ancestry; we used this dataset to test the performance of the inference methods. Each method finds an estimate for the true ancestry, for every genotype in every individual. Note that, although WINOPOP and LAMP use only a subset of SNPs to infer the local ancestry, both methods find an ancestry estimate for all the SNPs in the data set. We measure the accuracy of a method as the fraction of all the genotypes in the dataset for which the correct ancestry was inferred.
3.1 Comparison with existing methods
In a first series of experiments, we compare the accuracy obtained by WINPOP to the best existing methods for local ancestry inference such as LAMP (Sankararaman et al., 2008a), SABER (Tang et al., 2006) and HAPAA (Sundquist et al., 2008). For this comparison, we simulated admixtures starting from the four HapMap populations: the Yorubans (YRI), Japanese (JPT), Han Chinese (CHB) and western Europeans (CEU). Using the simulation procedure described above with α=0.8 and g=7, we generated datasets consisting of admixtures of YRI-CEU, CEU-JPT and JPT-CHB populations. We used α=0.8 as it roughly corresponds to the global African admixture proportion of the African American population.
We note that the comparison between the methods is a bit of ‘apples to oranges’ since LAMP and WINPOP only require information about ancestral allele frequencies, while HAPAA uses additional information about the ancestral haplotypes. We trained the HMM of HAPAA on a sample admixture generated using the methods provided in the HAPAA package starting from the ancestral haplotypes over seven generations with α=0.8. Then the trained model was used to estimate the phasing of the genotypes of the admixture. Finally, the HMM was used to estimate the ancestries given the previously obtained phasing of the genotypes in the test admixture. The default parameters were used for all these steps with the exception of the number of generations and α=0.8 that were provided to HAPAA; we also provided HAPAA with the genetic map of the SNPs in the analysis as inferred from HapMap. Only the ancestral allele frequencies were provided to both LAMP and WINPOP. The ancestral genotypes were provided to SABER. Although the correct admixture proportion was provided to all the methods, we note that it can easily be estimated by other methods such as Frappa (Tang et al., 2005).
We first assessed the gain in accuracy of WINPOP over LAMP due to the improved modeling of recombination events and to the adaptive window size. We see from the second part of Table 1 that WINPOP outperforms LAMP, with the biggest gain in accuracy for the JPT-CHB dataset. We also considered an HMM as described in Sankararaman et al. (2008b) with parameters estimated using an expectation-maximization (EM) algorithm starting from the LAMP (WINPOP) solution (see Section 2.4 for details). These methods are denoted by LAMP-EM and WINPOP-EM, respectively. We note that both methods obtain similar accuracies, regardless of whether the EM algorithm is started from the LAMP or WINPOP solutions, with both accuracies being lower than the ones obtained by WINPOP for all three datasets.
Table 1.
Accuracies of ancestry estimates obtained by the compared methods on the HapMap admixtures
| Method | YRI-CEU | CEU-JPT | JPT-CHB |
|---|---|---|---|
| SABER | 89.4 | 85.2 | 68.2 |
| HAPAA | 93.7 | 88.2 | 72.0 |
| LAMP | 94.8 | 93.0 | 65.8 |
| LAMP-EM | 97.8 | 94.8 | 74.8 |
| WINPOP | 98.0 | 95.9 | 82.8 |
| WINPOP-EM | 97.7 | 94.7 | 74.8 |
| Upper bound | 99.9 | 99.6 | 91.9 |
LAMP-EM (WINPOP-EM) uses LAMP (WINPOP) solution as an initialization for an EM algorithm that optimizes an HMM similar to the model proposed in STRUCTURE (see Section 2.4 for details).
In the first part of the table, we compared the accuracy obtained by WINPOP with the accuracies obtained by SABER and HAPAA showing that WINPOP achieves the best accuracy on all datasets. We note that HAPAA obtains consistently higher accuracy than SABER [as previously reported by Sundquist et al. (2008)], but significantly lower accuracy relative to WINPOP. The biggest improvement in accuracy of WINPOP over the existing methods is attained on admixtures of closely related populations such as JPT-CHB where all the previous methods have accuracies slightly <75%. Such low accuracies may be detrimental to the downstream analysis of closely related admixed populations. In contrast, WINPOP, which includes an improved modeling of recombination, achieves an accuracy of 82.8%. Surprisingly, WINPOP outperforms the HMM-based methods even without the ancestral haplotype data. One possible explanation is that the large number of parameters that need to be estimated in the HMM-based methods reduces their accuracy.
Table 1 also reports the upper bound on the accuracy that can be achieved by any method that uses the same set of SNPs without modeling the LD between SNPs (Section 2.3).
Although WINPOP shows a factor of 3 increase in running time when compared to LAMP, it still runs in <20 min on each of the three HapMap admixtures from Table 1 making it scalable to large-scale datasets. This contrasts with the much larger runtime required by the HMM-based methods; e.g. HAPAA takes around 7 h for each of the datasets described above (500 genotypes over 38 k SNPs) while SABER takes a little>2 h for a set of 4 k SNPs. Due to computational considerations we did not run the HMM-based methods for the remaining results reported in this section.
3.2 Limitations of accuracy of ancestry inference as a function of the Fst
We measured the effect of genetic distance between populations on the accuracy of the inference of locus-specific ancestry. We used forward simulations to generate admixed populations starting from populations with varying genetic distances measured by the fixation index (Fst).1 The fixation index compares the genetic variability within and between populations to give a measure of the distance between populations. We used the computation of the Fst that accounts for differences in sample size (The International HapMap Consortium, 2005).
We first used the WTCCC control groups (58BC and UKBS) (Wellcome Trust Case Control Consortium, 2007), which are drawn from a relatively homogeneous Caucasian British population. To generate simulated populations with different Fst values, we selected two disjoint sets of 500 individuals each, and we simulated g generations of random mating within each population separately (with the population size amplified to 2000 individuals). Clearly, the Fst between the two populations increases with g (e.g. in our experiments the Fst is 0.0012 and 0.0391 after 5 and 200 generations, respectively). For each g, we simulated 10 generations of admixture by random mating of individuals from the two populations using α=0.8. Figure 1 plots the accuracy of WINPOP estimates as a function of the Fst. As a baseline, we have also plotted the upper bound on the accuracy (Section 2.3). Notably, for admixtures of two very closely related ancestral populations (e.g. Fst of 0.0012), no method that does not model the LD can achieve a reasonable accuracy, since the upper bound we calculate for the accuracy is very low (64.2% in this example). As expected, the accuracy of the ancestry estimates increases with the number of generations in the simulation, as the reproductive isolation of the two populations increases the genetic divergence in terms of Fst. Particularly, for Fst values of 0.01 and 0.04, WINPOP achieves an accuracy of 80% and 92%, respectively, a significant increase over LAMP that achieves accuracies of only 59% and 83%, respectively. Most importantly, the accuracy of WINPOP is always within 15% of the best possible accuracy given by the upper bound.
Fig. 1.

Accuracy of ancestry estimates of LAMP and WINPOP, as well as the upper bound on all possible methods as a function of the Fst between the simulated ancestral populations. The accuracy of WINPOP on the JPT-CHB HapMap admixture is also provided as a single point.
Evolutionary forces, such as selection and new mutations, tend to increase the divergence of independently evolving populations; it is therefore possible that the accuracy of the inference methods on real admixed populations may slightly differ from our analysis so far, depending on their specific genetic variation structure. To account for this, we simulated admixed populations for every pair of populations from the Human Genome Diversity Panel (HGDP-CEPH) data (Li et al., 2008). These data consist of 938 unrelated individuals typed at 650 000 SNPs loci spanning 52 populations from sub-Saharan Africa, North Africa, Europe, Middle East, South/Central/East Asia, Oceania and the Americas. We used only the SNPs located on chromosome 1 for our analysis. We simulated admixed populations from every pair of populations from this dataset and measured the accuracy of WINPOP as a function of the Fst of the original populations (Fig. 2). The results are consistent with the WTCCC experiment described above; we notice a considerable improvement of accuracy of WINPOP over LAMP, in particular for the pairs of close populations (e.g. Fst≤0.05). Furthermore, we observe that the upper bound is again very close to WINPOP's accuracy, and thus further improvements in accuracy will be expected to exploit more information (e.g. the background LD patterns).
Fig. 2.

Accuracies of ancestry estimates for admixtures of pairs of populations from HGDP as a function of the Fst.
3.3 A map of accuracy on the HGDP admixtures
It is interesting to characterize the accuracy of the inference methods as a function of the geographic distance between the populations. As shown in Novembre et al. (2008), the inference of genome-wide average ancestry is possible even for pairs of populations that are only a few 100 km away from each other. We observe that the same holds when applying WINPOP to the HGDP data (Fig. 3). As expected, the accuracies obtained by LAMP and WINPOP cluster according to the continental groups, which is an evidence of the fact that admixtures within a continental group are harder to infer. However, WINPOP shows a substantial improvement, and even the closest populations can be inferred with at least 81% accuracy. The lowest accuracy attained by WINPOP on this dataset was 81.49% on the admixture of the Bedouin and the Druze populations. In contrast, LAMP attained only 53.59% accuracy on this admixture. More generally, from Figure 3, it can be seen that the admixtures involving the Middle Eastern populations (Mozabite, Bedouin, Palestinian and Druze) are the most challenging.
Fig. 3.

Comparison of the accuracy of LAMP and WINPOP on admixtures created from the HGDP populations. Red denotes high accuracy, while blue denotes low accuracy. The upper half of the matrix denotes accuracies attained by WINPOP, while the lower half denotes LAMP. While LAMP attains accuracies of<70% on 116 pairs, WINPOP has a minimum accuracy of 81.49%. The populations on the two axes ordered by continental groups.
3.4 A potential pitfall: misspecified ancestral populations
Our analysis so far assumes that the allele frequencies of the ancestral populations are given to the algorithm. In practice, however, the true ancestral populations may not be found, and even if they are found, the genetic background of these populations may have drifted from that of the original ancestral populations. We have studied the effect of such a drift on the inference accuracy. We started with the two evolved WTCCC populations generated as described above for g=50 100 and 150, with Fst of 0.010, 0.018 and 0.028, respectively. These populations were considered as the ancestral populations in our experiment. We simulated 10 generations of random mixing between these pairs of populations, resulting in a set of admixed populations. To capture the drift from the original ancestral populations, we simulated up to 50 generations of random mating within each ancestral population separately (with the population size amplified to 2000). Thus, the allele frequencies that we get are merely an approximation to the true allele frequencies of the original ancestral populations. The resulting distance between the original ancestral populations and the drifted one ranges from Fst of 0.001 to 0.01, corresponding to 5 and 50 generations of random mating, respectively.
As expected, there is a decrease in accuracy estimation as the population drifts from the ancestral population (Fig. 4). Interestingly, Figure 4 also suggests that the rate of decrease in accuracy is higher when the initial ancestral populations are closer. This has important consequences on the way such methods should be used; when considering an admixed population with closely related ancestral groups, it is crucial to choose the ancestral populations as close as possible to the true ancestral populations. This is less critical for admixed populations with distant ancestral groups.
Fig. 4.

Accuracies of ancestry estimates for admixtures of close populations when WINPOP is provided with drifted ancestral populations.
3.5 Admixture inference on Latino populations
Even though previous methods have been used to infer ancestry in Latinos (Price et al., 2008; Tang et al., 2007), to date there has never been any attempt to evaluate the accuracy of the inference methods on such populations. We therefore simulated a Latino population as an admixture of Europeans, West Africans and Native Americans (Tang et al., 2007). Our simulations follow the ones suggested in (Tang et al., 2007), in which they follow the population history of the Puerto Ricans. In Puerto Rico, native Americans started mixing with people of European descent after 1493, when Christopher Columbus discovered the island and Europeans started settling in. Subsequently, Africans were introduced to the island as slaves (Carrion, 1984) and thus most of the genome of current Puerto Ricans is an admixture of three populations with global proportion content of 0.66:0.18:0.16 for European, West African and Native American ancestries, respectively (Burchard et al., 2005). From the HGDP dataset, we took all the Italian, Sardinian and Tuscan individuals to form a European population, all the Maya and Pima individuals to form a Native American population and all the Yoruba individuals for the West African population. Then, we amplified each of the three obtained populations using the Li and Stephens (2003) model. Essentially, the Li and Stephens model assigns probabilities to un-observed haplotypes, given a current sample of observed haplotypes. As suggested in Tang et al. (2007), following the known history of the Puerto Ricans, we first admixed the native American and European populations for 5 generations and then introduced the West African population to the admixture for another 10 generations (Tang et al., 2007). As shown in Table 2, WINPOP achieves an accuracy of 91% for the resulting population, while LAMP performs poorly on such an admixed population with an accuracy of 68%. These large error rates must be taken into account in any downstream analysis, including admixture mapping, and the identification of regions under selective pressure (Tang et al., 2007).
Table 2.
Accuracies of ancestry estimates for the Puerto Rican simulations averaged over 100 datasets
| Method | Accuracy |
|---|---|
| LAMP | 68.1 |
| WINPOP | 91.3 |
| Upper bound | 98.4 |
3.6 Genotype imputation in admixed populations
In this section, we show results that demonstrate the utility of incorporating locus-specific ancestries in downstream analyses. We focus on an important problem arising in genome-wide case–control association studies. These studies follow a simple methodology of typing a very large number of markers, in individuals having a disease (cases), and in individuals not showing the disease (controls), followed by a statistical test of association to find the markers that show high correlation with the disease. Due to the vast number of markers present across the human genome, it is usually assumed that the true causal SNP will not be typed directly due to the limited coverage of current genotyping platforms. Using the typed markers as ‘predictors’ for the true causal SNP not present on the array has recently emerged as a powerful technique for increasing the power of association studies (Marchini et al., 2007; Pei et al., 2008). The additional information required for imputing SNPs not present in the study comes from the SNP correlation information found in large repositories of variation such as the HapMap project (we term these the reference population).
In this section, we focus on the utility of incorporating locus-specific ancestries in imputing genotypes at untyped SNPs. Multiple methods have been successfully employed to solve the imputation problem, and HMMs have been amongst the most popular. Since the scope of our article is not the imputation problem, we will focus this small scale analysis only on GEDI, a recently developed HMM-based method for genotype imputation (Kennedy et al., 2008). GEDI uses an HMM similar to the one of Kimmel and Shamir (2005), Marchini et al. (2007) and Rastas et al. (2008) trained using a standard EM procedure on the reference population of haplotypes. Similarly to other HMM-based methods, imputation of untyped SNPs in the sampled population is performed based on the conditional probability of the alleles at that SNP given the rest of the observed genotypes for that individual.
Starting with three admixtures generated from the HapMap using admixture ratio of 0.5:0.5 and seven generations, we randomly chose 10% of the SNPs as untyped and we masked them from all the individuals in the admixture. We ran WINPOP on the new admixture (with the masked genotypes removed) and we used the neighboring genotypes as a predictor for the ancestry of the previously masked SNP genotype in each individual. In particular, for an individual i and a masked SNP j, we set the ancestry of the masked genotype gij as the ancestry inferred in the most SNPs in a window of 10 un-masked SNPs centered on SNP j (five downstream and five upstream of SNPs j).
We compared the imputation error rate of GEDI as the percentage of erroneously inferred genotypes from the total imputed genotypes for various scenarios. First, we passed to GEDI only one of the pure populations as reference; we denote the average error rate between these two scenarios as GEDI-1ANC. In the second scenario, we passed both ancestral haplotypes to GEDI as reference population; we denote this by GEDI-2ANC. Finally, we ran GEDI on each SNP genotype independently using as reference the locus-specific ancestries inferred by WINPOP.
Table 3 shows the accuracy obtained by GEDI in the various scenarios. As expected we notice a large decrease in error rate from using only one ancestral population as reference to using both. The results also show that the decrease in error rate is correlated with the genetic distance between the two ancestral populations. In general, using the local ancestries as a guide for choosing the right population to be passed to GEDI as the reference population achieves the lowest error rate.
Table 3.
Imputation error rate (%) obtained by GEDI on the three HapMap simulated admixtures GEDI-1ANC denotes imputation based on only one ancestral reference population, GEDI-2ANC denotes imputation based on both ancestral reference populations, while WINPOP+GEDI denotes imputation of each SNP genotype based on the local ancestry estimated by WINPOP
| Method | YRI-CEU | CEU-JPT | JPT-CHB |
|---|---|---|---|
| GEDI–1ANC | 13.12 | 6.85 | 4.04 |
| GEDI–2ANC | 6.43 | 3.97 | 3.48 |
| WINPOP+GEDI | 5.69 | 3.64 | 3.39 |
Although more sophisticated algorithms that employ local ancestries can be devised for genotype imputation and the results in this section are far from being exhaustive, they suggest that accurate local ancestries can be used to improve the accuracy of genotype imputation methods on recently admixed populations.
4 DISCUSSION
We have presented a new model (WINPOP) for estimation of locus-specific ancestry in recently admixed populations using a sliding window-based framework. Through extensive simulations, we show that WINPOP achieves significant improvements over the best available methods, with the gain in accuracy being largest on admixtures of closely related ancestral populations. These improvements stem from two basic ideas: first, we use an improved modeling of recombination events within each window, and second, we use an adaptive window length that depends on the local genetic distance between the ancestral populations within a window. We show that the WINPOP cannot be improved substantially as long as the framework used is based on overlapping windows of independent SNPs. This suggests that further improvements in the accuracy of methods for the inference of locus-specific ancestry require new ideas that will be able to exploit the LD.
In the case of closely related ancestral populations, WINPOP is more accurate than HMM-based methods (Sankararaman et al., 2008b; Sundquist et al., 2008; Tang et al., 2006). This is particularly striking, since some of these methods explicitly model the background LD structure, and they are often provided with more information than WINPOP (e.g. HAPAA was provided the ancestral haplotypes, while WINPOP only uses the ancestral allele frequencies). It is possible that this difference is mainly due to the large number of parameters that need to be optimized in the HMM-based methods, resulting in failure to converge to the global optimum of the parameter space.
Through extensive simulations, we studied the behavior of WINPOP under various scenarious that might arise in the study of real admixed populations. As expected, the accuracy of WINPOP is correlated to the genetic distance between the ancestral populations, as measured by the Fst and is more sensitive to misspecification in ancestral allele frequencies when the ancestral populations are close.
Accurate inference of locus-specific ancestries may play an important role in admixture mapping, in correcting for population substructure, as well as in studying patterns of selection. As an illustration of the utility of these methods, we show that locus-specific ancestries can be used to improve the accuracy of genotype imputation.
WEB RESOURCES
The URLs for data and software presented herein are as follows:
LAMP and WINPOP: http://lamp.icsi.berkeley.edu
HapMap project: http://www.hapmap.org
WTCCC website: http://www.wtccc.org.uk
ACKNOWLEDGEMENTS
This study makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
Funding: NSF (grant 0713254 to E.H. and B.P.); NSF (grant 0513599 to G.K.); Berkeley Fellowship (to S.S.); Wellcome Trust (under award 076113); E.H. is a faculty fellow of the Edmond J. Safra Bioinformatics program at Tel Aviv University.
Conflict of Interest: none declared.
Footnotes
1This experiment is similar to the one performed in Sundquist et al. (2008); however, we also consider admixtures between populations that have diverged for as low as five generations.
REFERENCES
- Burchard G, et al. Latino populations: a unique opportunity for the study of race, genetics, and social environment in epidemiological research. Am. J. Public Health. 2005;95:2161–2168. doi: 10.2105/AJPH.2005.068668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion AM. Puerto Rico: A Political and Cultural History. New York: Norton; 1984. [Google Scholar]
- Falush D, et al. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane J. The combination of linkage values, and the calculation of distances between the loci of linked factors. J. Genet. 1919;8:299–309. [Google Scholar]
- Hoggart C, et al. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 2004;74:965–978. doi: 10.1086/420855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J, et al. Genotype error detection using hidden markov models of haplotype diversity. J. Comput. Biol. 2008;15:1155–1171. doi: 10.1089/cmb.2007.0133. [DOI] [PubMed] [Google Scholar]
- Kimmel G, Shamir R. gerbil: genotype resolution and block identification using likelihood. Proc. Natl Acad. Sci. USA. 2005;102:158–162. doi: 10.1073/pnas.0404730102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, et al. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- Novembre J, et al. Genes mirror geography within europe. Nature. 2008;456:274. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, et al. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y-F, et al. Analyses and comparison of accuracy of different genotype imputation methods. PLoS ONE. 2008;3:e3551. doi: 10.1371/journal.pone.0003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, et al. Long-range ld can confound genome scans in admixed populations. Am. J. Hum. Genet. 2008;83:132–135. doi: 10.1016/j.ajhg.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard J, et al. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastas P, et al. Phasing genotypes using a hidden Markov model. In: Măndoiu I, Zelikovsky A, editors. Bioinformatics Algorithms: Techniques and Applications. Wiley; 2008. pp. 355–372. [Google Scholar]
- Reich D, et al. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 2005;37:1113–1118. doi: 10.1038/ng1646. [DOI] [PubMed] [Google Scholar]
- Sankararaman S, et al. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 2008a;8:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, et al. On the inference of ancestries in admixed populations. Genome Res. 2008b;18:668–675. doi: 10.1101/gr.072751.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundquist A, et al. Effect of genetic divergence in identifying ancestral origin using HAPAA. Genome Res. 2008;18:676–682. doi: 10.1101/gr.072850.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, et al. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- Tang H, et al. Reconstructing genetic ancestry blocks in admixed individuals. Am. J. Hum. Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, et al. Recent genetic selection in the ancestral admixture of Puerto Ricans. Am. J. Hum. Genet. 2007;81:626–633. doi: 10.1086/520769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium (2005) A haplotype map of the human genome. Nature4371299–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature447661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, et al. Admixture mapping for hypertension loci with genome-scan markers. Nat. Genet. 2005;37:177–181. doi: 10.1038/ng1510. [DOI] [PubMed] [Google Scholar]