

Abstract
Motivation: Genome-wide association studies are commonly used to identify possible associations between genetic variations and diseases. These studies mainly focus on identifying individual single nucleotide polymorphisms (SNPs) potentially linked with one disease of interest. In this work, we introduce a novel methodology that identifies similarities between diseases using information from a large number of SNPs. We separate the diseases for which we have individual genotype data into one reference disease and several query diseases. We train a classifier that distinguishes between individuals that have the reference disease and a set of control individuals. This classifier is then used to classify the individuals that have the query diseases. We can then rank query diseases according to the average classification of the individuals in each disease set, and identify which of the query diseases are more similar to the reference disease. We repeat these classification and comparison steps so that each disease is used once as reference disease.
Results: We apply this approach using a decision tree classifier to the genotype data of seven common diseases and two shared control sets provided by the Wellcome Trust Case Control Consortium. We show that this approach identifies the known genetic similarity between type 1 diabetes and rheumatoid arthritis, and identifies a new putative similarity between bipolar disease and hypertension.
Contact: serafim@cs.stanford.edu
1 INTRODUCTION
Genome-wide association studies (GWAS) are an increasingly popular approach for identifying associations between genotype and phenotype. A large number of such studies have been performed recently to try to identify the genetic basis of a wide variety of diseases, and explore how this genetic basis differs depending on the geographic origin of the studied population. High-throughput genotyping chips are used to obtain the genotype of an individual at several hundreds of thousands of single nucleotide polymorphisms (SNPs). These sets of SNPs are able to represent most of the variability at the single locus level that was identified by the HapMap project (Frazer et al., 2007). In a GWAS study, several thousands of disease individuals, and several thousands of healthy controls are genotyped. Statistical tests are used to identify SNPs that show a strong association with the disease. Strong association between a SNP and a disease can be evidence that the SNP is related to the disease, or that it is in linkage disequilibrium with SNPs that are related to the disease. In both cases significant associations provide promising leads for further experimental investigation into the genetic etiology of diseases. These studies have led to the identification of more than 150 risk loci in more than 60 diseases (Manolio and Collins, 2009). The Wellcome Trust Case-Control Consortium (WTCCC) genotype 500 000 SNPs in seven common diseases: type 1 diabetes (T1D), type 2 diabetes (T2D), coronary artery disease (CAD), Crohn's disease (CD), bipolar disease (BD), hypertension (HT) and rheumatoid arthritis (RA) (WTCCC, 2007). In this article we use the individual genotype data from this study.
Computational methods have been used to identify disease similarities using a variety of data sources, including gene expression in cancer (Rhodes et al., 2004) and known relationships between mutations and phenotypes (Goh et al., 2007). However, while a large number of GWAS focusing on individual diseases have been recently published, the attempts to integrate the results of multiple studies have been limited. Most of these integration approaches focus on combining multiple studies of the same disease in order to increase the statistical power (Zeggini et al., 2008), or use data from other high-throughput measurement modalities to improve the results of GWAS studies (Chen et al., 2008). Comparison between the genetic components of diseases have been done using four different approaches. The first approach is based on the identification of the association between one SNP in two different diseases in two independent studies. The second approach selects a group of SNPs that have been previously associated with some disease and tests if they are also associated with a different disease. An example of this approach is the genotyping of a large number of individuals with T1D at 17 SNPs that have been associated with other autoimmune diseases, which leads to the identification of a locus previously associated with only RA as being significantly associated with T1D as well (Fung et al., 2009). The third approach pools data from individuals with several diseases prior to the statistical analysis, and has been used in the original WTCCC study. Several similar diseases (autoimmune diseases, metabolic and cardiovascular diseases) are grouped in order to increase the statistical power for identifying SNPs that are significantly associated with all the diseases in the pool. The fourth approach compares the results of multiple GWAS, and has been previously applied to the WTCCC dataset (Torkamani et al., 2008). They use the P-values indicating the significance of the association between a SNP and a single disease, and compute the correlations between these P-values in pairs of diseases, as well as the size of the intersection of the 1000 most significant SNPs in pairs of diseases. They identify strong similarities between T1D and RA, between CD and HT, and between BD and T2D.
In this work, we introduce a novel approach to identify similarities in the genetic architecture of diseases. We train a classifier that distinguishes between a reference disease and the control set. We then use this classifier to classify all the individuals that have a query disease. If there is a similarity at the genetic level between the query disease and the reference disease, we expect more individuals with the query disease to be classified as belonging to the disease class than if there is no similarity. We generalize our procedure to multiple disease comparison: given a set of multiple diseases, we use each in turn as the reference disease while treating all others as query diseases.
There are two main differences between our new approach and existing analyses. First, previous approaches [such as Torkamani et al. (2008)] compute a significance score for each SNP, and then use these scores for comparing diseases. In our approach, we first compute a classification for each individual, and then compare diseases using these classifications. Second, we train the classifier using information from all SNPs, and during this learning process select the SNPs that contribute to the classification based on the genotype data only. This genome-wide approach makes it possible to see the classifier as a statistical representation of the differences between the disease set and the control set.
The use of classifiers in the context of GWAS has been limited so far. In particular, attempts at using them for predicting outcome based on genotype have been unsuccessful. For example, a recent prospective study in T2D (Meigs et al., 2008) found that using 18 loci known to be associated with T2D in a logistic regression classifier together with known phenotypic risk factors does not significantly improve the risk classification, and leads to a reclassification in only 4% of the patients. A particular challenge in the context of outcome prediction is that the prevalence of most diseases is relatively low and that it is therefore necessary to achieve high precision in order for the classifier to be usable. Our goal is not predicting individual outcomes, and we only compare predictions made by a single classifier. We can therefore ignore disease prevalence.
A second challenge in the use of a classification approach for finding disease similarities is that the classifier does not explicitly identify genetic features of the disease, but rather learns to distinguish the disease set from the control set. Differences between the two sets that are due to other factors might therefore lead to incorrect results. In most GWAS, a careful choice of matched controls limits this risk. However, when using a classifier trained on one GWAS to classify individuals from a different study, there is a risk that the background distribution of SNPs is very different between the populations in which the datasets have been collected, which could lead to errors, particularly when comparing diseases using datasets from different geographic origins. This risk can be limited by using disease data from a single source. In this work, we use genotype data provided by the WTCCC study, in which all individuals were living in Great Britain and individuals with non-Caucasian ancestry were excluded.
In this article, we first provide a detailed description of the analysis approach. We then show that we are able to train classifiers that achieve a classification error that is clearly below the baseline error for T1D, T2D, BD, HT and CAD. We use these classifiers to identify strong similarities between T1D and RA, as well as between HT and BD, and weak similarities between T1D and both BD and HT. We also show that we are able to train a classifier that distinguishes between the two control sets in the WTCCC data. We use this classifier to identify similarities between some diseases and individual control sets. This finding matches observations made during the quality check phase of the original study. The implications of this finding on our approach are addressed in the Section 5. Finally, we discuss the implications of the similarities we find, and propose extensions of this approach. A detailed description of the dataset used in this work, the data pre-processing, the decision tree classifier and the comparison procedure are provided in Sections 3 and 4, respectively, at the end of the article.
2 APPROACH
In this section, we define the general classifier-based approach to identify genetic similarities between diseases. The approach can be separated into four steps: data collection, preprocessing, classifier training and disease comparison. Figure 1 provides an overview of the training and comparison steps.
Fig. 1.

Overview of the approach. This figure presents the classification and comparison steps of our analysis pipeline. These steps are repeated using a different reference disease each time. The classifier returns a real value between 0.0 and 1.0 which we call disease-class probability. The histograms represent the distribution of the disease-class probability of the individuals with the reference disease (left) and of the controls (right). In the situation depicted in this figure, there is evidence that query disease C is more similar to the reference disease than the other query diseases.
The data collection step consists of collecting samples from individuals with several diseases, as well as matched controls, and genotyping them. Alternatively, existing data can be reanalysed. In both cases, it is important to limit the differences between the disease sets and the control sets that are not related to the disease phenotype. Similarly, differences between the different disease sets should also be limited. In particular, it is recommended to use individuals with the same geographic origin, the same ancestry, and a single genotyping technology for the whole study. In this work, we use existing data from the WTCCC which satisfies these criteria.
In the preprocessing step, the data are filtered and uncertain genotype measurements, as well as individuals and SNPs that do not fit quality requirements are discarded. It is important to develop preprocessing steps that ensure good data quality. Approaches that analyze each SNP individually can afford to have a more stringent, often manual post-processing step on the relatively few SNPs that show strong association. The SNPs that do not pass this quality inspection can be discarded without affecting the results obtained on other SNPs. In our approach however, classifier training is done using genome-wide information, and removing even a single SNP used by the classifier could potentially require retraining the entire classifier. It is therefore impractical to perform any kind of post-processing at the SNP level. The Section 3 of this article describes the data used in this work, as well as the quality control measures we take.
The classifier training and comparison steps are interleaved. We start with a list of diseases and a set of individual genotypes for each disease, as well as at least one set of control genotypes. We pick one disease as reference disease, and refer to the remaining diseases as query diseases. We train a classifier distinguishing the corresponding disease set from the control set. For any individual, this classifier could either return a binary classification (with values 0 and 1 indicating that the classifier believes the individual is part of, respectively, the controls class or the disease class) or a continuous value between 0 and 1. This continuous value can be seen as the probability of the individual to be part of the disease class, as predicted by the classifier. We refer to this value as disease-class probability. For simplicity, we will only use the disease-class probability values for the rest of this section, but the comparison step can be performed similarly using binary classifications. During the comparison step, we classify individuals from the query disease sets using the classifier obtained in the training step, and for each query disease, compute the average disease-class probability. The training and comparison steps are then repeated so that each disease is used once as reference disease.
We can compare the average disease-class probability of the different query diseases to identify similarities between them. Diseases that have a higher average disease-class probability are more likely to be similar to the reference disease than diseases with a lower average disease-class probability. Using cross-validation, we can obtain the average disease-class probability of the reference disease set and the control set used for training the classifier, and compare them with the values of the other diseases. One particular caveat that needs to be considered in this analysis is that while the classifier does distinguish the control set from the disease set, there is no guarantee that it will only identify genetic features of the disease set. It is also possible that it will identify and use characteristics of the training set, especially if there are data quality issues. This case can be identified during the comparison step if the average disease-class probability of most query diseases is close to the average disease-class probability of the reference disease, but very different from the average disease-class probability of the control set. It is therefore important to look at the distribution of the average disease-class probabilities of all query diseases before concluding that an individual disease is similar to the reference disease.
It is important to note that the disease-class probability of a given individual does not correspond to the probability of this individual actually having the disease. The disease frequency is significantly higher in the datasets we use for training the classifier than in the real population. In a machine learning problem in which the test data are class-imbalanced, training is commonly done on class-balanced data, and class priors are then used to correct for the imbalance. Such priors would, however, scale all probabilities linearly, and would not affect the relationships we identify, nor their significance. Estimating the probability of an individual having the disease is not the goal of this project and we can therefore ignore class priors.
A large variety of classifiers can be integrated into the analysis pipeline used in our approach. The Section 4 provides a more formal description of the classification task. In this article, we use a common classifier, decision trees, to show that this approach allows us to identify similarities. The specific details about the decision tree classifier, and how its outputs are used in the analysis step are described in the Section 4.
3 RESULTS
We evaluate the ability of our analysis approach to identify similarities between diseases using the set of seven diseases provided by the WTCCC. In this section, we first evaluate the performance of individual classifiers that distinguish one disease from the joint control set. We then show that these classifiers can identify similarities between diseases. Finally, we use our classifier to identify differences between the two control sets, and provide evidence indicating that these differences do not affect the disease similarities we identify.
3.1 Classifier performance
We first train one classifier for each disease using both the 58C and the UKBS sets as controls. The performance of each classifier is evaluated using cross-validation, and reported in Table 1. We compare our classifier to a baseline classifier that classifies all individuals into one class without using the SNP data at all. The best error such a classifier can achieve during cross-validation is the frequency of the smaller class in the training set. We refer to this value as the baseline error.
Table 1.
Classifier performance (cross-validation)
| Disease | Baseline (%) | Error (%) | Precision (%) | Recall (%) | Δp | Leaves |
|---|---|---|---|---|---|---|
| T1D | 40.05 | 22.93 | 71.65 | 70.71 | 0.383 | 9 |
| RA | 38.43 | 33.45 | 59.12 | 42.09 | 0.130 | 12 |
| BD | 38.24 | 33.59 | 62.60 | 30.18 | 0.087 | 11 |
| HT | 39.92 | 36.77 | 57.98 | 28.64 | 0.080 | 12 |
| CAD | 39.05 | 36.62 | 55.25 | 32.73 | 0.075 | 12 |
| T2D | 39.5 | 38.0 | 54.12 | 25.05 | 0.052 | 14 |
| CD | 36.63 | 36.28 | 29.83 | 18.43 | 0.046 | 11 |
Baseline corresponds to the baseline error; Error, Precision and Recall to the cross-validation performance of the decision tree classifier; Δp to the difference between the average disease-class probability of the control set and the average disease-class probability of the disease set; and Leaves to the maximum number of leaves in the pruned classifiers for this disease.
The disease for which the classifier performs best is T1D, with a classification error of 22.93%, compared with a baseline error of 40.05%. The classification error obtained by the decision tree classifier is also below the baseline error for several other diseases, although by a substantially smaller margin. This is the case for RA (with an error of 33.45% versus 38.43%), BD (33.59% versus 38.24%), HT (36.77% versus 39.92%) and CAD (36.62% versus 39.05%). For two diseases, T2D and CD, the improvement compared with the baseline error is only minimal, and we choose not to use these classifiers in our analysis. While the classifiers that we keep only provide small improvements in terms of classification error (with the exception of T1D), they have a significantly better trade-off between precision (at least 55%) and recall (at least 28%) than the baseline classifier (which would classify all individuals as controls).
We do not use these classifiers in a binary way, but rather use the disease-class probability, which is the conditional probability of an individual to be part of the disease-class given its genotype, under the model of the reference disease learned by the classifier (see Section 7 for a precise definition for decision trees). It is therefore interesting to consider the distributions of the disease-class probability, as obtained during cross-validation. Figure 2 illustrates that these distributions differ significantly for T1D. It can also be seen that there are individuals for which the disease-class probability is close to 50%, meaning that there are leaf nodes in the classifier that represent subsets of the data that cannot be distinguished well. Our approach takes this into account by using disease-class probabilities rather than binary classifications. In order to evaluate the ability of our classifiers to distinguish between the disease set and the control set using the disease-class probability metric, we use the difference Δp of the average disease-class probability between the two sets. The classifiers that we keep all have values of Δp above 0.075. This illustrates that while there are only small improvements in binary classification performance, the classifiers are able to distinguish between the disease set and the control set in the way we intend to use them.
Fig. 2.

Distribution of the disease-class probabilities for the T1D classifier. The two histograms show the distribution of the disease-class probability of the individuals, respectively, in the joint control set (top) and in the T1D set (bottom), as computed during cross-validation. The red lines represent the average disease-class probabilities, and the black line indicates the 0.5 probability cut-off used for binary classification. The plot in between the histograms shows the average disease-class probabilities of the six other diseases on the interval between the average disease-class probabilities of the control set and of the disease set.
3.2 Disease similarities
For each of the five classifiers with sufficiently good performance, we compute the average disease-class probability of each of the six query diseases. In summary, we identify strong symmetrical similarities between T1D and RA, as well as between BD and HT. Furthermore, we find that T1D is closer to both BD and HT than other diseases, even though we did not find the symmetrical relation using the T1D classifier. This section provides a detailed presentation of these results.
For T1D, the average disease-class probability for the control set and the disease set, as computed using cross-validation, are 0.259 and 0.642, respectively. Figure 2 shows the distribution of the average disease-class probabilities for the query diseases. RA, another autoimmune disease, is clearly the closest to T1D (average disease-class probability of 0.337). This result is significant, with P-value <10−5 (see the Section 4 for details on how P-values are obtained). All other diseases have an average disease-class probability that is close to that of the control set, which means that there is no evidence of similarity with T1D.
For RA, the average disease-class probabilities are 0.303 for the control set and 0.433 for the disease set. The distribution of the average disease-class probabilities for the other diseases are shown on Figure 3a. We can observe that T1D (average disease-class probability of 0.397) is closest to RA (P< 10−5), meaning that we find a symmetrical similarity between the two diseases. All other diseases have an average disease-class probability close to the one of the control set.
Fig. 3.

Disease-class probabilities comparisons. The plots represent the interval between the average disease-class probabilities of the control set and of the disease set for RA (a), BD (b), HT (c) and CAD (d), respectively. The average disease-class probabilities for all the query diseases are shown in blue on every plot. Note that while all plots on this figure use the same scale, different scales are used for the central plots of figures 2 and 4.
For BD, the average disease-class probabilities are 0.297 for the control set and 0.384 for the disease set. The distribution of the average disease-class probabilities for the query diseases are shown in Figure 3b. We can observe that there is a wider spread in the average disease-class probabilities, and that there is no cluster of diseases close to the control set. We can also observe that HT (average disease-class probability of 0.359, P < 10−5) is closest to BD, followed by T1D (average disease-class probability of 0.354, P-value of 0.001).
For HT, the average disease-class probabilities are 0.315 for the control set and 0.395 for the disease set. The distribution of the average disease-class probabilities for the other diseases are shown in Figure 3c. We can observe that BD (average disease-class probability of 0.381, P-value < 10−5) is clearly closest to HT. T1D (average disease-class probability of 0.368, P < 10−5) is also closer to HT than the remaining diseases.
For CAD the average differences between the query diseases are smaller than for all the other classifiers (Fig. 3d). Furthermore, the classifier for CAD is the one with the worst performance amongst the ones we use in the comparison phase. Therefore, we believe that the results are not strong enough to report putative similarities identified using this classifier, even though some differences between diseases have significant P-values.
3.3 Differences between control sets
The original WTCCC study found several SNPs that are significantly associated with one of the two control sets. These SNPs are filtered out during preprocessing, both in the WTCCC study and in this work. However, the mere existence of differences between two control sets prompted the question whether a classifier could distinguish the two sets, and if so, what the implications of this finding would be on the validity of results obtained with these control sets.
We perform several experiments using the two control sets separately, and report the results in Table 2. First, we train a control–control classifier that distinguishes the two control sets from each other. This classifier achieves an error of 41.15% compared with a baseline error of 49.62%, and a Δp of 0.093. This shows that we are able to distinguish to some extent between the two control sets. Figure 4 shows the distribution of the 58C class probability (which corresponds to the value called disease-class probability when the classifier distinguishes between one disease and the controls). In order to verify that this result is due to differences between the two specific control set, and not the ability of our classifier to distinguish between any two sets, we randomly split all control individuals into two sets, R1 and R2. We train a classifier to distinguish between these two sets. We find that this classifier does only minimally improves the classification error (error of 49.45%, baseline error of 50.03%, Δp of −0.003).
Table 2.
Separate training set classifier performance
| Experiment | Baseline (%) | Error (%) | Precision (%) | Recall (%) | Δp | Leaves |
|---|---|---|---|---|---|---|
| UKBS/58C | 49.62 | 41.15 | 58.33 | 64.05 | 0.093 | 11 |
| R1/R2 | 50.03 | 49.45 | 50.59 | 46.42 | −0.003 | 11 |
| UKBS/T1D | 42.62 | 23.15 | 79.53 | 80.34 | 0.402 | 8 |
| 58C/T1D | 42.99 | 24.46 | 76.60 | 82.22 | 0.370 | 8 |
| UKBS/RA | 44.29 | 36.42 | 66.21 | 70.72 | 0.144 | 10 |
| 58C/RA | 44.66 | 38.11 | 64.89 | 67.83 | 0.135 | 9 |
Baseline corresponds to the baseline error; Error, Precision and Recall to the cross-validation performance of the decision tree classifier; Δp to the difference between the average disease-class probability of the control set, and the average disease-class probability of the disease set; and Leaves to the maximum number of leaves in the pruned classifiers for this experiment. R1 and R2 represent two random splits of the joint control set.
Fig. 4.

Distribution of the class probabilities for the control–control classifier distinguishing the UKBS control set from the 58C control set. The two histograms show the distribution of the 58C class probability of the individuals, respectively, in the UKBS control set (top) and in the 58C control set (bottom), as computed during cross-validation. The red lines represent the average class probabilities, and the black line indicates the 0.5 probability cut-off used for binary classification. The plot in between the histograms shows the average disease-class probabilities of all seven other diseases on the interval between the average class probabilities of the two control sets.
We apply the comparison step of our pipeline using the control–control classifier in order to identify possible similarities between the disease set and one of the control sets. Figure 4 shows the distribution of the average 58C class probabilities for each disease. The average disease-class probabilities obtained during cross-validation are 0.477 for the UKBS set and 0.561 for the 58C set. Both HT (average 58C class probability of 0.521, P < 10−5) and BD (average 58C class probability of 0.514, P-value of 0.0002) are closer to the 58C control set, whereas both RA (average 58C class probability of 0.487, P < 10−5) and CAD (average 58C class probability of 0.489, P-value of 0.0003) are closer to the UKBS control set.
Given the differences between the control sets, and the unexpected similarities between control sets and diseases, we are interested in verifying that the performance of the disease classifiers used in the analysis is not an artifact caused by these differences. We therefore train two new classifiers for each disease, one using only UKBS as control set, and one using only 58C as control set. The performance of these classifiers for T1D and RA is shown in Table 2, and is similar to the performance of the classifiers that use both control sets together. For the remaining diseases (including HT and BD), the classifiers using only one of the control sets do not achieve a classification error below the baseline error, most likely due to the smaller training set (i.e. overfitting). For each of the classifiers for T1D and RA, we compute the average disease-class probability for the other six diseases as well as the unused control set. The similarities between the two diseases are significant in all four classifiers. Furthermore, the average disease-class probability of the unused control set is similar to the average disease-class probability of the other five diseases, and not significantly closer to T1D or RA. Therefore, we can conclude that the results obtained using the T1D and RA classifiers are not due to differences between the control sets. Furthermore, the results using a single control set provide further evidence indicating that the classifiers do identify relevant features of T1D and RA, respectively, rather than relevant features of the control set.
4 DISCUSSION
In this work, we introduce a novel approach for identifying genetic similarities between diseases using classifiers. We identify genetic similarities between several diseases. In this section, we first discuss the implications of these findings. We then consider challenges in the application of classifiers to GWAS data. Finally, we propose possible extensions of this approach.
We identify a strong similarity between T1D and RA. Genetic factors that are common to these two autoimmune diseases were identified well before the advent of GWAS, and linked to the HLA genes (Torfs et al., 1986, Lin et al., 1998). The original WTCCC study (WTCCC, 2007) identifies several genes that appear to be associated with both diseases. We look at the classifiers corresponding to these two diseases. The SNP with the highest information gain in T1D is rs9273363, which is located on chromosome 6, near MHC class II gene HLA-DQB1, and is also the SNP that is most strongly associated with T1D in the initial analysis of the WTCCC data, with a P-value of 4.29 × 10−298 (Nejentsev et al., 2007). This is the strongest association reported for any disease in the WTCCC study, which explains to a large extent why the T1D classifier so clearly outperforms the classifiers for the other diseases. This SNP is also significantly associated with RA (P-value of 6.74 × 10−11). The SNP with the highest information gain in RA is rs9275418, which is also part of the MHC region, and is strongly associated with both RA (P-value of 1.00 × 10−48) and T1D (P-value of 7.36 × 10−126). This shows that our approach is able to recover a known result, and uses SNPs that have been found to be significantly associated with both diseases in an independent analysis of the same data.
The similarity we identify between HT and BD is interesting, since there does not appear to be previous evidence of a link between the two diseases at the genetic level. However, a recent study identified an increased risk of HT in patients with BD compared with general population, as well as compared to patients with schizophrenia in the Dannish population (Johannessen et al., 2006). The WTCCC study only identified SNPs with moderate association to HT (lowest P-value of 7.85 × 10−6) and a single SNP with strong association with BD (P-value of 6.29 × 10−8). The decision trees for both diseases use a large number of SNPs that have a very weak association with the respective disease. Both classifiers have a classification error that is clearly below the baseline error, and provide evidence of similarity between the two diseases. This indicates that our classifier-based approach is able to use the weak signals of a large number of SNPs to identify evidence for similarities that would be missed by comparing only SNPs that show moderate or strong association with the diseases. Further analyzes are necessary to identify the nature and implications of the similarity we find between HT and BD, as well as the weaker similarity we identified between these two diseases and T1D.
We also show that we can train a classifier that can distinguish the two control sets, and we use it to identify diseases that are more similar to one of the control set than the other. This is not an unexpected finding, since SNPs that were strongly associated with a control set were identified and discarded in the WTCCC study. These SNPs were also removed in the preprocessing step of our study, and the results we obtain when trying to distinguish the two control sets therefore show that the decision tree classifier is able to achieve a classification error below the baseline error even though the SNPs with the strongest association could not be used by the classifier. The similarities between some diseases and one of the control sets can most likely be explained by some subtle data quality issue. During quality control, the authors of the WTCCC study found several hundreds of SNPs in which some datasets exhibited a particular probe intensity clustering [see the Supplementary Material of the original WTCCC study (WTCCC, 2007) for details]. This particular pattern was always observed in 58C, BD, CD, HT, T1D, T2D, but not in UKBS, RA and CAD. This matches the result obtained using our classifier-based approach, in which RA and CAD were predicted to be most similar to UKBS, and could therefore be a possible explanation of the similarities we find.
While we do find several interesting similarities between diseases, we also observe that training a classifier that distinguishes between individuals with a disease and controls using SNP data poses numerous challenges. The first is that whether someone will develop a disease is strongly influenced by environmental factors. The genetic associations that can be identified using GWAS are only predispositions, and it is therefore likely that some fraction of the control set will have the predispositions, but will not develop the disease. Furthermore, depending on the level of screening, the disease might be undiagnosed in some control individuals, and individuals that are part of a disease set might have other diseases as well. This is especially true for high-prevalence diseases like HT.
Obtaining good classifier performance by itself is not, however, the main goal of our approach. We show that we can find similarities even when the classifier performance only shows small improvements compared with the baseline error. In this work, we focus on the comparison approach, not on developing a classifier specially suited for the particular task of GWAS classification. We use decision trees because they are a simple, commonly used classification algorithm.
This work shows that classifiers can be used to identify similarities between diseases. This novel approach can be expanded into several directions. First, classification performance can be potentially improved by using a different generic classifier, or by developing classifiers that do take into account the specific characteristics of SNP data. Second, further analysis methods need to be developed in order to analyze the trained classifiers, and identify precisely the SNPs that do lead to the similarities this approach detects. Such a methodology would be useful, for example, to further analyze the putative similarity between HT and BD. Third, building on the fact that our approach considers the whole genotype of an individual, it could be possible to identify subtypes of diseases, and cluster individuals according to their subtype. Finally, modifying the approach to allow the integration of studies performed in populations of different origins or using different genotyping platforms would allow the comparison of a larger number of diseases.
Our approach identifies similarities between the genetic architecture of diseases. This is, however, only one of the many axes along which disease similarities could be described. In particular, both genetic and environmental factors interact in diseases, and the genetic architecture for two diseases could be similar, but the environmental triggers could be different, leading to low co-occurrence. There is therefore a need for methods that integrate similarities of different kinds that were identified using different measurement and analysis modalities. An example of such an approach is the computation of disease profiles that integrate both environmental ethiological factors and genetic factors (Liu et al., 2009).
5 CONCLUSION
GWAS have been used to identify candidate loci likely to be linked to a wide variety of diseases. In this article, we introduce a novel approach that allows identifying similarities between diseases using GWAS data. Our approach is based on training a classifier that distinguishes between a reference disease and a control set, and then using this classifier for comparing several query diseases to the reference disease. This approach is based on the classification of individuals using their full genotype, and is thus different from previous work in which the independent statistical significance of each SNP is used for comparing diseases.
We apply this approach to the genotype data of seven common diseases provided by the WTCCC, and show that we are able to identify similarities between diseases. We replicate the known finding that there is a common genetic basis for T1D and RA, find strong evidence for genetic similarities between BD and HT, as well as evidence for genetic similarities between T1D and both BD and HT. We also find similarities between one of the control sets used in the WTCCC (UKBS) and two disease sets, RA and CAD. This similarity can possibly be a consequence of the subtle differences in genotyping quality that were observed during the initial quality control performed by the WTCCC.
Our results demonstrate that it is possible to use a classifier-based approach to identify genetic similarities between diseases, and more generally between multiple phenotypes. We expect that this approach can be improved by using classifiers that are more specifically tailored for the analysis of GWAS data, and by the integration of a larger number of disease phenotypes. The ability to compare similarities between diseases at the whole-genome level will likely identify many more currently unknown similarities. Genetic similarities between diseases provide new hypotheses to pursue in the investigation of the underlying biology of the diseases, and have the potential to lead to improvements in how these diseases are treated in the clinical setting.
6 DATA
We use the individual genotypes provided by the WTCCC. These genotypes come from a GWAS (WTCCC, 2007) of seven common diseases: T1D, T2D, CAD, CD, BD, HT, and RA. The data consist of a total of 2000 individuals per disease and 3000 shared controls, with 1500 control individuals from the 1958 British Birth Cohort (58C control set) and 1500 individuals from blood donors recruited specifically for the project (UKBS control set). The genotyping of 500 568 SNPs per individual was performed using the Affymetrix GeneChip 500 K Mapping Array Set. In the original analysis of this dataset by the WTCCC, a total of 809 individuals and 31 011 SNPs that did not pass quality control checks are excluded. In addition, SNPs that appear to have a strong association in the original study have been manually inspected for quality issues, and 578 additional SNPs were removed. In this work, we exclude all individuals and SNPs that were excluded in the WTCCC study, as well as an additional 9881 SNPs that do not appear in the WTCCC summary results.
One concern with these quality control steps is the identification of SNPs for which the genotype calling is of poor quality. In the WTCCC study, this is done after the analysis, which makes it possible to visually inspect the small subset of SNPs that are potentially significant. In a classifier-based approach, it is impractical to perform any kind of visual inspection, and we must try to minimize the errors due to genotype calling prior to the analysis. The WTCCC study only uses genotype calls made by a custom algorithm, Chiamo (Marchini et al., in preparation), but the genotype calls made using the standard Affymetrix algorithm BRLMM are also available. While the study does show that Chiamo has, on average, a lower error rate than BRLMM, there are SNPs that are discarded during the quality control process that show errors in the genotype calls made by Chiamo. We use the two genotype sets to create a consensus dataset in which the genotype of a given individual at a given SNP is used only if there is agreement between the call made by Chiamo and the call made by BRLMM, and is considered to be unknown if the calls are different. This approach individually considers the call made for every individual at every SNP, and does not discard entire SNPs. The handling of SNPs that have a high proportion of unknown genotypes is left to the classification algorithm, and will be discussed in the corresponding section. While this approach does reduce the errors in genotype calling, this comes at the cost of discarding cases in which Chiamo is right but BRLMM is not. Overall, the frequency of unknown genotypes is 2% using the consensus approach, compared with 0.65% using Chiamo and 0.74% using BRLMM. Furthermore, BRLMM genotype calls are entirely missing for a total of 184 individuals, which are thus excluded from our study.
After performing these preprocessing steps, the data set used in this study consists of 459 075 SNPs measured in 2938 control individuals (58C: 1480, UKBS: 1458), 1963 with T1D, 1916 individuals with T2D, 1882 individuals with CAD, 1698 individuals with CD, 1819 individuals with BD, 1952 individuals with HT and 1834 individuals with RA.
7 METHODS
In this section, we first formally define the classification task that is central to our approach, then describe the specific classifier we use in this work and how we evaluate its performance, and finally describe how we use the classification results to infer relationships between diseases.
7.1 Classification Task
The data consist of a list of individuals i, a list of SNPs s ∈ S, and the measurement of the genotype g(s, i) of individual i at SNP s. We use Gi={g(1, i),…,g(|S|, i)} to denote the genotype of individual i at all the SNPs in the study. The genotype measurement is a discrete variable which can take four values: homozygote for the major allele, homozygote for the minor allele, heterozygote and unknown: g(s, i)∈{maj, min, het, unk}. Each individual belongs to one of several disease sets, or to the control set. For the WTCCC data used in this work, we have seven disease sets: T1D, T2D, CAD, CD, BD, RA, HT, and we use the union of the 58C and UKBS sets as control set.
For each disease d, we train a classifier that distinguishes between that disease set and the controls. The individuals that are not part of these sets are ignored during the training of this classifier. For each individual i used during training, a binary class variable ci indicates whether the individual belongs to the disease set (ci==disease) or to the control set (ci==control). The supervised classification task consists of predicting the class ci of an individual i given its genotype Gi. In this work, we use a decision tree classifier, but any algorithm able to solve this classification task can be easily integrated into our analysis pipeline.
7.2 Decision trees
In this section, we describe the decision tree classifier (Breiman et al., 1984). We use cross-validation in order to train the classifier, prune the trained decision tree and evaluate its performance on distinct sets of individuals.
We train a decision tree T by recursively splitting the individuals in each node using maximum information gain for feature selection. We use binary categorical splits, meaning that we find the best rule of the form g(s, i)==γ, where γ∈{maj, min, het}. Binary splits make it possible to handle cases in which only one of the three possible genotypes is associated with the disease without unnecessarily splitting individuals that have the two other genotypes. Unknown values are ignored when computing information gain. This is necessary since there is a correlation between the frequency of unknown values and the quality of the genotyping, which in turn is variable between the different datasets. Counting unknown values during training could therefore lead to classifiers separating the two sets of individuals based on data quality differences, rather than based on genetic differences. However, if a large number of measurements are unknown for a given SNP, the information gain for that SNP will be biased. This is particularly true if the fraction of unknowns is very different between the cases and the controls. In order to avoid this situation, we discard all SNPs that do have >5% of unknown genotypes amongst the training individuals in the node we are splitting. In each leaf node L, we compute the fraction fL of training individuals in that node of that are part of the disease class: 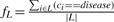 .
.
In order to choose a pruning algorithm, we compare the cross-validation performance obtained using Cost-Complexity Pruning (Breiman et al., 1984), Reduced Error Pruning (Quinlan, 1986), as well as a simple approach consisting of limiting the tree depth. We find that Reduced Error Pruning outperforms Cost-Complexity Pruning, and performs similarly well than limiting the tree depth, but results in smaller decision trees. We therefore use Reduced Error Pruning, which consists of recursively eliminating subtrees that do not improve the classification error on the pruning set (which only contains individuals that were not used during training).
The classification of an individual i using a decision tree T is done by traversing the tree from the root towards a leaf node L(i) according to the genotype of the individual which is classified. If fL(i) >0.5, then the individual is classified as disease, else the individual is classified as control. We can consider the decision tree T as a high-level statistical model of the difference between the disease and the control sets. Under this model, the fraction fL(i) represents the conditional probability of individual i to be part of the disease class given its genotype: PT(ci==disease | Gi)=fL(i). This value is the disease-class probability of individual i. In order to compute the fractions fL over sufficiently large numbers of individuals, we further prune our tree to only have leaf nodes containing at least 100 training individuals. The benefit of using this probability rather than the binary classification is that it allows to distinguish leaf nodes in which there are mainly training individuals from one class from those in which both classes are almost equally represented.
In order to assess the performance of our classifier, we perform 5-fold cross-validation. We start by separating the data into five random sets containing 20% of the individuals each. A decision tree T is trained using four of these sets, while one set is reserved for pruning and testing. The unused set is split randomly into two equal sets. The first of these sets is used to obtain pruned tree T′ from tree T, and the individuals in the second set are used to evaluate the performance of tree T′. The last step is then repeated using the second set for pruning, and the first for testing. Finally, we repeat the training and evaluation four more times, each time leaving out a different set for pruning and testing. This ensures that for every individual in our dataset, there is one pruned decision tree for which the individual was used neither for training nor for pruning. We can therefore evaluate the performance of the classifier on unseen data. We can also compute the average disease-class probability p(C) of the control individuals, and the average disease-class probability p(d) of the individuals with disease d. The difference Δp between those two probabilities indicates how well the classifier is able to distinguish controls from diseases. We use the cross-validation results to compare the performance of the classifier against a baseline classifier which simply assigns the most frequent label amongst the training set to all individuals. Classifiers that do not outperform this baseline classifier, or for which the difference Δp is small, are not used to identify similarities between diseases.
Given the cross-validation scheme used, we end up training not one, but several possibly distinct decision trees. Rather than arbitrarily choosing one, we use the set Td of all decision trees trained during cross-validation for a given disease d. In order to classify a new individual i, we first classify i using each classifier independently, and then return the average classification. Similarly, we average the results of individual classifiers to obtain the average disease-class probability: 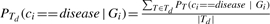 .
.
7.3 Identifying similarities
Once a classifier has been trained to distinguish the set of individuals with reference disease d from the control set, we can use it to identify diseases that are similar to disease d. Using the classifier, we can compute the disease-class probability of an individual with a query disease d′. In order to be able to compare diseases, we are interested in computing the average disease-class probability of all individuals in d′: 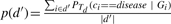 . We expect this average probability to be in, or close to the interval between p(C) and p(d), which were the averages computed on, respectively, the control set and the disease set d during cross-validation. If p(d′) is close to p(C), then d′ is not very different from the control set, whereas a value p(d′) that is close to p(d) indicates similarity between the two diseases. Using this method, we can compare all query diseases to the reference disease d, and identify if there are diseases that are more similar to d than others.
. We expect this average probability to be in, or close to the interval between p(C) and p(d), which were the averages computed on, respectively, the control set and the disease set d during cross-validation. If p(d′) is close to p(C), then d′ is not very different from the control set, whereas a value p(d′) that is close to p(d) indicates similarity between the two diseases. Using this method, we can compare all query diseases to the reference disease d, and identify if there are diseases that are more similar to d than others.
If we find that a query disease d′ is closer to reference disease d than the other query diseases, then we need to assess the significance of this finding. In order to do so, we randomly sample a set r of individuals from all the disease sets except d, such that r is of the same size as d′, and compute p(r). We repeat this procedure 10 000 times. The fraction of random samples r for which p(r) ≥p(d′) indicates how often a random set of individuals would obtain a probability of being part of the disease-class at least as high as the set d′, and is therefore a P-value indicating how significant the similarity between d′ and d is.
ACKNOWLEDGEMENTS
This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
Funding for the project was provided by the Wellcome Trust under award 076113; Richard and Naomi Horowitz Stanford Graduate Fellowship (to M.A.S.).
Conflict of Interest: none declared.
REFERENCES
- Breiman L, et al. Classification and Regression Trees. Wadsworth. CA: Belmont; 1984. [Google Scholar]
- Chen R, et al. FitSNPs: highly differentially expressed genes are more likely to have variants associated with disease. Genome Biol. 2008;9:R170. doi: 10.1186/gb-2008-9-12-r170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung EY, et al. Analysis of 17 autoimmune disease-associated variants in Type 1 diabetes identifies 6q23/tnfaip3 as a susceptibility locus. Genes Immuno. 2009;10:188–191. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- Goh KI, et al. The human disease network. Proc. Natl Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen L, et al. Increased risk of hypertension in patients with bipolar disorder and patients with anxiety compared to background population and patients with schizophrenia. J. Affect. Disord. 2006;95:13–17. doi: 10.1016/j.jad.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Lin J, et al. Familial clustering of rheumatoid arthritis with other autoimmune diseases. Hum. Genet. 1998;103:475–482. doi: 10.1007/s004390050853. [DOI] [PubMed] [Google Scholar]
- Liu YI, et al. The “etiome”: identification and clustering of human disease etiological factors. BMC Bioinformatics. 2009;10(Suppl. 2):S14. doi: 10.1186/1471-2105-10-S2-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio T, Collins F. The HapMap and genome-wide association studies in diagnosis and therapy. Annu. Rev. Med. 2009;60:16–1. doi: 10.1146/annurev.med.60.061907.093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, et al. A Bayesian hierarchical mixture model for genotype calling in a multi-cohort study. 2007 [Google Scholar]
- Meigs J, et al. Genotype score in addition to common risk factors for prediction of Type 2 Diabetes. New Engl. J. Med. 2008;359:2208. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, et al. Localization of Type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan J. Simplifying Decision Trees. AI Memo 930; 1986. [Google Scholar]
- Rhodes DR, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl Acad. Sci. USA. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torfs C, et al. Genetic interrelationship between insulin-dependent diabetes mellitus, the autoimmune thyroid diseases, and rheumatoid arthritis. Am. J. Hum. Genet. 1986;38:170. [PMC free article] [PubMed] [Google Scholar]
- Torkamani A, et al. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for Type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]