

Abstract
We have generated an antiserum to the variable domain of mouse collagen XXVII, a recently discovered novel member of the fibrillar collagen family. Collagen XXVII protein is first detectable in the mouse at embryonic day 12.5. By E14.5 days, the protein localises to cartilage, developing dermis, cornea, inner limiting membrane of the retina and major arteries of the heart. However, at E18.5 days, collagen XXVII protein is no longer apparent in most tissues and appears restricted mainly to cartilage where expression continues into adulthood. Type XXVII collagen immunolocalises to 10 nm thick non-striated fibrils that are distinct from fibrils formed by the classical fibrillar collagens. The transient nature of its expression and unusual fibrillar structure suggest that collagen XXVII plays a developmental role distinct from those of the classical fibrillar collagens.
INTRODUCTION
Fibrillar collagens are key structural components of the vertebrate skeleton and most other connective tissues. Types I, III and V collagen are expressed in tissues such as bone, skin, aorta, intestine, tendon and ligament whereas types II and XI collagen are expressed predominantly in cartilage (1). During and upon secretion from the cell, these collagens assemble into heterotypic fibrils that have characteristic banded patterns when negatively stained and viewed by transmission electron microscopy (1, 2). Mutations in each of the genes encoding these fibrillar collagen types cause diseases such as chondrodysplasias, osteoarthritis, sub-types of Ehlers-Danlos syndrome and osteogenesis imperfecta (3). On phylogenetic analysis, the classical fibrillar collagen chains described above cluster into two related clades (A and B) (4). We and others have recently discovered a new fibrillar collagen gene, (COL27A1) that encodes an alpha chain that differs in key characteristics from its classical fibrillar collagen ‘cousins’ and, together with its sibling, COL24A1, form a novel C-clade of vertebrate fibrillar collagens (5-7).
Type XXVII collagen has an unusual molecular structure in that it has two interruptions in the major collagenous domain whereas the classical fibrillar collagens have none; the major triple helix is only 997 amino acid residues long compared to 1014-20 residues for the other fibrillar collagen α chains; and the major triple helical domain fuses directly with its N-terminal non-collagenous domain and collagen XXVII therefore lacks the N-terminal telopeptide-like region and minor triple helical domain present in the classical fibrillar collagens (5, 6). Collagen XXVII also has a short chain selectivity sequence in the C-propeptide that is reminiscent of invertebrate fibrillar collagens although the origins and evolutionary significance of the length of this motif remains uncertain (5, 8-10). All of these unusual characteristics for a vertebrate fibrillar collagen are shared with its sibling gene encoding type XXIV collagen (7). Collagen XXVII is highly conserved across vertebrates and, together with type XXIV collagen, is considered to have arisen from a common progenitor, the modern day equivalent of which is found in the primitive chordate Ciona intestinalis (9-11). Although this high degree of conservation implies that collagen XXVII is playing an important role in the extracellular matrices where it is expressed, the function of this novel fibrillar collagen is yet to be determined.
During embryonic development, the collagen XXVII messenger RNA is expressed at high levels in cartilage, the developing dermis, the eye, and in some epithelial cell layers such as the epithelial linings of embryonic lung and ameloblasts in developing teeth (5, 6). We have generated an antisera against the variable domain of mouse collagen XXVII and report that the protein is first detectable in mouse at embryonic day 12.5; that by E14.5 days, the protein localises to cartilage, developing dermis, cornea, inner limiting membrane of the eye and aorta; that at E18.5 days, collagen XXVII protein is no longer apparent in most tissues and becomes restricted mainly to cartilage; that the expression continues into adulthood; and finally, that type XXVII collagen immunolocalises to 10 nm thick non-striated fibrils that are distinct from fibrils formed by the classical fibrillar collagens.
EXPERIMENTAL PROCEDURES
Antisera
The variable domain of Col27A1 was amplified by PCR from mouse genomic DNA, sequenced and ligated, in frame, into the pTrc-His bacterial expression vector (Invitrogen, UK). Primers used for the PCR were: 5′ATATAGGATCCAGTCCCCAGGTGGGAACCCT3′ and 5′ATAGAATTCTCACATCAGCATCGGGAAAGGTG3′. The construct was then transformed into BL21 E. coli cells and cultures grown with shaking at 37°C. Protein expression was induced by addition of IPTG (final concentration 1 mM). Induced bacterial cells were pelleted, resuspended in 25 ml of ice cold lysis buffer (1 × PBS, 1% (v/v) Triton X-100 and lysozyme to a final concentration of 0.25 mg/ml) containing a cocktail of protease inhibitors (Roche, cat # 1836153, 1 tablet per 50 ml lysis buffer) and incubated on ice for 30 minutes. The solution was then transferred to a Duonce homogeniser (Jencons, UK) and cells further lysed with 20-30 strokes. The lysate was then spun at 13000 rpm for 10 minutes and the pellet, containing the insoluble recombinant variable domain, washed (by resuspending and subsequently re-centrifuging) sequentially in 1 × PBS containing 1% (v/v) Triton X-100, 2 mM EDTA, followed by 1 M Tris.HCl pH 8, 1.5 M KCl, 5 mM EDTA, 1 % (v/v) Triton X-100, then 1 M Tris.HCl pH 8, 5 mM EDTA, 0.15 M NaCl. Finally, the pellet was resuspended in 1 ml 6 M urea and incubated at room temperature for 1 hour. The solubilised recombinant variable domain was electrophoresed on a 6 % polyacrylamide gel (12). The gel slices containing the correct size band were excised, homogenised in PBS and utilized for rabbit antisera production by a commercial supplier (Eurogentec).
Western blotting and coomassie staining
Western blotting of highly purified bacterial collagenase-digested 24 hr conditioned media from ATDC 5 cells grown under differentiating conditions for 6 days was performed as previously described (13).
Immunohistochemistry
Tissue samples were fixed in 5% acetic acid/95% ethanol (v/v), dehydrated and embedded in paraffin wax. Where required, bone was demineralised in 0.25M EDTA after fixing and prior to embedding. Five μm serial sections were prepared through the full thickness of each block and representative sections used for immunohistochemistry. Endogenous peroxidase activity was blocked by incubation in methanol containing 0.3% (v/v) hydrogen peroxide for 30 minutes at room temperature (RT). The sections were then treated with 0.2 % (w/v) bovine testicular hyaluronidase (Sigma, cat # H-3884, UK) for 15 minutes at 37°C in a humidified box. Non-specific binding of primary or secondary antibodies was blocked by incubation in PBS containing 1% BSA (w/v) and 0.6% (v/v) goat serum (Dako A/S, Denmark) for 1 hour at RT. Each section was overlaid with primary antiserum in dilution buffer (PBS containing 1% BSA), and incubated at RT for 1 hour. The secondary biotinylated goat - anti-rabbit antiserum (Dako A/S, Denmark) was applied at 1:400 in dilution buffer (PBS containing 1% BSA, 0.6% goat serum) for 1 hour at RT. The sections were then incubated in freshly prepared Vectastain ABC reagent (Vector Laboratories Inc, Balingame) for 30 minutes at RT. The colour was developed using DAB peroxidase substrate tablet sets (Sigma) for 1 to 5 minutes at RT. The reactions were stopped by washing the sections in water for 5 minutes, before counterstaining in methyl green (0.5 g in 100 ml 0.1 M sodium acetate, pH 4.2) for 8 minutes. Incubations replacing the primary antiserum with control immunoglobulins and/or omitting the primary antiserum were used as negative controls.
In situ hydridisation
In situ analysis of collagen XXVII expression was conducted using 35S-labelled ribroprobe exactly as described in (5).
Immuno transmission electron microscopy
The growth plates from the ribs of new born pups were dissected from freshly sacrificed mice and immediately frozen on a metal block cooled with liquid nitrogen. Frozen ribs were crushed with a hammer and the pieces were put in buffer solution, transferred to a Dounce homogenizer and 15-20 strokes were made. The resulting homogenate was put on carbon/formvar coated copper grids for 1 min (14). After a brief wash in a buffer containing 50 mM Tris.HCl, pH 7.4, 50 mM EDTA, 100 mM sucrose and 150 mM NaCl, grids were blocked with 0.5% cold fish gelatin and 1% BSA. Collagen XXVII primary antiserum was applied for 30 minutes Secondary antibodies or protein A conjugated with 10 nm gold particles, were then applied for 15 minutes After extensive washing in distilled water, grids were stained in aqueous 1% uranyl acetate for 1 minute The excess stain was removed with filter paper and the grids were left to air dry. Samples were observed in an FEI Tecnai 12 electron microscope with an accelerating voltage of 100 kV. Images were recorded on film.
RESULTS
Generation of collagen XXVII antiserum
Attempts to produce significant quantities of soluble recombinant collagen XXVII variable domain with either a His-tag or as a GST fusion were not successful (data not shown) although significant expression of insoluble protein was achieved. As an alternative approach to generate sufficient antigen, we extracted inclusion bodies, produced by expression of the His tagged variable domain, in 6 M urea and resolved the solubilised material on a preparative SDS/PAGE gel. Gel slices containing intact recombinant variable domain were excised from preparative SDS/PAGE gels, homogenised in PBS (Fig. S1) and used as an immunogen in rabbits. Western-blotting of conditioned medium from the chondrogenic ATDC5 cell line probed with the newly generated type XXVII variable domain antiserum revealed a collagenase-sensitive set of bands matching that generated by a previously characterised peptide-derived antiserum to collagen XXVII (Ref. 5 and Fig. S1).
Immunohistochemical localisation of collagen XXVII during embryonic development
Collagen XXVII is first detectable (weakly) by immunohistochemistry in the cartilaginous anlage of E12.5 day mouse embryos (Fig. S2). By E14.5 days, the signal for collagen XXVII is strong and widespread (Fig. 1 and S3). Collagen XXVII is immunolocalised to the extracellular matrix of the cartilage anlagen and is particularly concentrated around hypertrophic chondrocytes (Fig.1A & B). The developing lung does not give any discernable signal for collagen XXVII although the adjacent cartilaginous elements of developing ribs stains well (Fig. 1B). In the developing eye, immunostaining for collagen XXVII is apparent at the inner limiting membrane that lines the surface of the retina, and weak staining is apparent in the corneal stroma (Fig. 1C). Collagen XXVII localises to the stellate reticulum of developing teeth at E18.5 days (Fig. 1D).
Fig. 1.
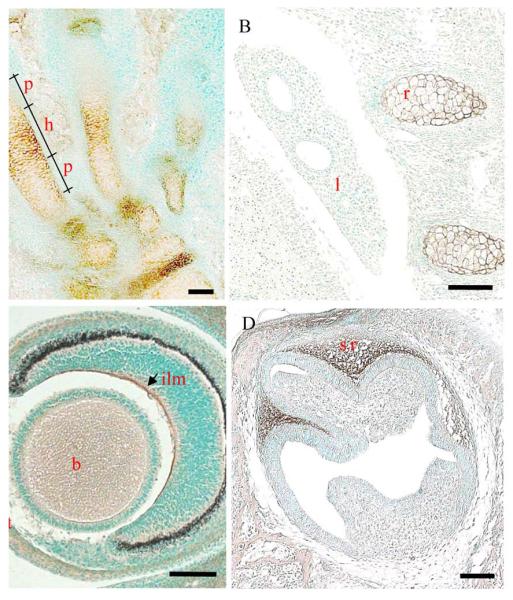
Immunolocalisation of collagen XXVII in embryonic mouse tissues.
Collagen XXVII (brown staining) localises to the cartilaginous skeletal elements and in particular, the hypertrophic regions, shown here in the developing foot (A) and ribs (B), the inner limiting membrane of the retinal and developing corneal stroma (C) at E14.5 days. (Preimmune and no chromogen controls can be seen in supplementary material - Fig. S3) Strong staining for collagen XXVII is detected in the stellate reticulum of developing teeth at E18.5 days. p, prehypertrophic; h, hypertrophic; l, lung; r, rib; st, corneal stroma; b, non-specific staining of the lens (see Fig. S3); ilm, inner limiting membrane of retina; sr, stelate reticulum. Bar = 10 μM
Collagen XXVII expression becomes restricted to cartilage during later development
In addition to those tissues described above (Fig. 1), strong expression of collagen XXVII can be seen in the walls of the major arteries serving the heart at E14.5 days by both immunolocalisation and in situ (Fig. 2A and C respectively). However, the protein is no longer apparent in the same major blood vessels at day E18.5 (Fig. 4D). Similarly, the expression of collagen XXVII is readily detectable in regions of developing dermis at E14.5 days (Fig. 2F-H) but, again, no immunolocalisation is apparent in the dermis at E18.5 days (Fig.2I). The pattern of collagen XXVII immunolocalisation in the growth plate is maintained in new born and 6 week old animals (Fig. 3A, D & G). At 6 weeks, the staining within the articular cartilage has become predominantly pericellular or ‘territorial’ (Fig. 3D & G). Collagen XXVII immunolocalisation in cartilage is distinct from both collagen II, which has a more even intensity of signal across the different cartilage growth plate compartments (Fig. 3B, E & H), and from collagen X which is localised to regions of hypertrophy (Fig. 6C, F & I).
Fig. 2.

Expression of collagen XXVII is down-regulated in some tissues between E14.5 and E18.5 days of development.
Strong collagen XXVII is detected by both immunohistochemical (A, F) and 35S in situ hybridisation (C, G) in the major arteries associated with the heart (A,C) and the dermis (F, G) of E14.5 day mouse embryos. However, at E18.5 days, immunolocalisation at these anatomical sites is no longer apparent (heart, D; dermis, I). Preimmune serum treated controls for heart at E14.5 (B) and E18.5 days (E). H is the brightfield image of G. a, aorta; d, dermis. Bar = 10 μM
Fig. 4.

Collagen XXVII immunolocalises to 10 nm thick non-striated fibrils.
Fibril extracts of new born mouse rib growth plate were absorbed to grids, treated with the collagen XXVII antiserum, washed and bound antibody detected using protein A labelled with 10 nm gold particles. A. An area showing the localisation of 10 nm particles to thin fibrils. B. An adjacent area showing no localisation to the much thicker, cross-striated fibrils containing collagen II. Further micrographs are presented in supplementary material (Fig. S4). Bar = 100 nM
Fig. 3.

Localisation of collagen XXVII in new born and 6 week old mouse tibia.
Collagen XXVII (A, D, G), collagen II (B, E, H) and collagen X (C, F, I) immunolocalisation in tibia of newborn (A, B, C) and 6 week old mice (D-I). G, H & I are enlargements of the boxed areas in the adjacent micrographs. a, articular cartilage; r, p and h refer to the reserve proliferative and hypertrophic zones of the growth plate. Bar = 10 μM
Immunogold localisation of collagen XXVII to thin fibrils
Attempts to localise type XXVII collagen in tissue sections using immunogold EM were unsuccessful, presumably because of the low levels of protein. It was therefore decided to immunogold-label fibrillar material extracted from tissues and mounted on grids. Extracts were made from growth plate cartilage of new born pups where collagen XXVII expression is known to be relatively high. The immunogold label localised consistently to narrow (~10 nm thick) non-striated fibrillar material (Fig. 4 & S4). No labelling was seen associated with the thicker, cross-striated collagen II rich fibrils on the same grids (Fig. 4 & S4).
DISCUSSION
Characterising the collagen XXVII antiserum
Our strategy for generating antibodies against collagen XXVII was to use the N-terminal variable domain since equivalent domains of most B-clade collagen chains remain part of the processed collagen and are incorporated into the fibril (15). Gel purified, intact recombinant protein with a His-tag was used to produce rabbit antiserum against the recombinant variable domain of mouse collagen XXVII.
The antisera recognised the same pattern of collagenase-sensitive bands from medium conditioned by a chondrogenic cell line (Fig. S1) as previously identified using an antiserum generated to peptides derived from the mouse variable domain (5). The latter peptide-derived antiserum, however, did not work for immunolocalisation studies (5).
Collagen XXVII expression is developmentally regulated
The deposition of collagen XXVII within the extracellular matrix can first be detected in the developing mouse at E12.5 days (Fig. S2) and has accumulated to significant levels in the anlagen, eye, dermis and major arteries of the heart by E14.5 days (Fig. 1, 2 and S3). Within cartilage, collagen XXVII immunolocalisation is most prominent in the matrix surrounding hypertrophic chondrocytes (Fig. 1A). It is of note that significant expression of collagen XXVII mRNA by epithelial cells lining developing bronchioles in the E14.5 lung has been demonstrated (5) and yet no significant immunolocalisation is apparent in this tissue despite the prominent staining of adjacent rib cartilage (Fig. 1B). It seems unlikely that the high level of expression of collagen XXVII mRNA detected in the developing lung in both mouse and man (5) could represent previously reported aberrantly spliced transcripts with premature stop codons since this type of transcript was retained in the nucleus and thought to be unstable (6). We are investigating further the nature of the collagen XXVII mRNA in the lung.
A surprising finding was that collagen XXVII is present only transiently in several locations in the developing embryo including aorta and dermis (Fig. 2). Continued expression of collagen XXVII into adulthood only seems to take place within cartilage and particularly within the growth plate - a ‘transient’ tissue in its own right that is rapidly eroded and renewed during endochondral bone growth. The transient appearance and disappearance of collagen XXVII in a narrow window of development in several tissues raises the possibility that this fibrillar collagen may not be playing a predominantly structural role typical of the classical fibrillar collagens for which protein half lifes are not counted in days but years (16). Rather, this transient expression is more reminiscent of a protein involved in signalling-type events or processes orchestrating development.
Collagen XXVII forms a fibrillar structure independent of the classical collagen fibrils
The classical fibrillar collagens are a highly interdependent family of proteins because of their propensity to form heterotrimeric collagens and to co-polymerise forming heterotypic fibrils (17). As a consequence, all of the classical fibrillar collagen alpha chains have a similar length major collagenous domain (1014-1020 amino acid residues) and they can not tolerate in-frame deletions resulting in a shortening of the collagenous domain or interruptions in the Gly-X-Y amino acid repeat which disrupt the linear nature of the collagen trimer (18). Collagen XXVII (and its sibling collagen XXIV) break all of these rules in that their collagenous domains are not only interrupted (twice) but are also ~18 amino acid residues shorter than the classical fibrillar collagen chains (5, 6). These differences between the classical fibrillar collagens and collagen XXVII are further emphasized by the immunogold labelling data that revealed collagen XXVII localising to a network of narrow fibrils that are entirely distinct from the thicker cross-striated fibrils formed by the classical fibrillar collagens (Fig. 4 and S4). It is possible that the relatively high abundance of lysine (and by implication, hydroxylysine) and bulky amino acids in the collagenous domain of collagen XXVII compared to the classical fibrillar collagens is a factor in collagen XXVII assembling to form a thin fibril (19 and refs therein).
In conclusion, we provide the first insight into the function of the collagen XXVII protein. The protein is transiently expressed in a narrow time frame during developmental processes in several tissues and localises to a fine fibrillar network distinct from classical collagen fibrils. These features suggest that the function of collagen XXVII may be somewhat different from its classical fibrillar collagen ‘cousins’ which provide the structural framework and tensile strength of most interstitial matrices. The findings reported here are a prerequisite for the analyses of mouse models expressing mutant forms of collagen XXVII which are in progress.
Supplementary Material
Acknowledgments
This work was funded by the Arthritis Research Campaign (Grants 15967 & 17243) and the NIH. DAP was the recipient of an MRC PhD studentship.
Abbreviations used
- PCR
polymerase chain reaction
- His
histidine
- RT
room temperature
- GST
glutathione S-transferase
- SDS/PAGE
sodium dodecyl sulphate polyacrylamide gel electrophoresis
- Gly
glycine
REFERENCES
- 1).Canty EG, Kadler KE. J. Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 2).Kadler KE, Holmes DF, Trotter JA, Chapman JA. Biochem. J. 1996;316:1–11. doi: 10.1042/bj3160001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Myllyharju J, Kivirikko KL. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 4).Bailey WJ, Kim J, Wagner GP, Ruddle FH. Mol. Biol. Evol. 1997;14:843–853. doi: 10.1093/oxfordjournals.molbev.a025825. [DOI] [PubMed] [Google Scholar]
- 5).Boot-Handford RP, Tuckwell D, Plumb DA, Rock C. Farrington, Poulsom R. J. Biol. Chem. 2003;278:31067–77. doi: 10.1074/jbc.M212889200. [DOI] [PubMed] [Google Scholar]
- 6).Pace JM, Corrado M, Missero C, Byers PH. Matrix Biol. 2003;22:3–14. doi: 10.1016/s0945-053x(03)00007-6. [DOI] [PubMed] [Google Scholar]
- 7).Koch M, Laub F, Zhou P, Hahn RA, Tanaka S, Burgeson RE, Gerecke DR, Ramirez F, Gordon MK. J. Biol. Chem. 2003;278:44236–44. doi: 10.1074/jbc.M302112200. [DOI] [PubMed] [Google Scholar]
- 8).Boot-Handford RP, Tuckwell DS. Bioessays. 2003;25:142–151. doi: 10.1002/bies.10230. [DOI] [PubMed] [Google Scholar]
- 9).Aouacheria A, Cluzel C, Lethias C, Gouy M, Garrone R, Exposito JY. J. Biol. Chem. 2004;279:47711–19. doi: 10.1074/jbc.M408950200. [DOI] [PubMed] [Google Scholar]
- 10).Wada H, Okuyama M, Satoh N, Zhang S. Evol. Dev. 2006;8:370–7. doi: 10.1111/j.1525-142X.2006.00109.x. [DOI] [PubMed] [Google Scholar]
- 11).Huxley-Jones J, Robertson DL, Boot-Handford RP. Matrix Biol. 2007;26:2–11. doi: 10.1016/j.matbio.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 12).Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 13).Atsumi T, Miwa Y, Kimata K, Ikawa YA. Cell Differ. Dev. 1990;30:109–16. doi: 10.1016/0922-3371(90)90079-c. [DOI] [PubMed] [Google Scholar]
- 14).Holmes DF, Kadler KE. J. Mol. Biol. 2005;345:773–784. doi: 10.1016/j.jmb.2004.10.078. [DOI] [PubMed] [Google Scholar]
- 15).Gopalakrishnan B, Wang W-M, Greenspan DS. J. Biol. Chem. 2004;279:30904–12. doi: 10.1074/jbc.M402252200. [DOI] [PubMed] [Google Scholar]
- 16).Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM. J. Biol. Chem. 2000;275:39027–31. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- 17).Boot-Handford RP, Tuckwell DS. Bioessays. 2003;25:142–151. doi: 10.1002/bies.10230. [DOI] [PubMed] [Google Scholar]
- 18).Kuivaniemi H, Tromp G, Prockop DJ. Hum. Mutation. 1997;9:300–315. doi: 10.1002/(SICI)1098-1004(1997)9:4<300::AID-HUMU2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 19).Zhang X, Boot-Handford RP, Huxley-Jones J, Forse LN, Mould AP, Robertson DL, Li L, Athiyal M, Sarras MP., Jr. J. Biol. Chem. 2007 Jan 3; doi: 10.1074/jbc.M607528200. 2007 as doi:10.1074/jbc.M607528200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.