

Abstract
The discovery of the new crosstalk between the aryl hydrocarbon receptor (AhR) and the NF-κB subunit RelB may extend our understanding of the biological functions of the AhR and at the same time raises a number of questions, which will be addressed in this review. The characteristics of this interaction differ from that of AhR with RelA in that the latter appears to be mostly negative unlike the collaborative interactions of AhR/RelB. The AhR/RelB dimer is capable of binding to DNA response elements including the dioxin response element (DRE) as well as NF-κB binding sites supporting the activation of target genes of the AhR as well as NF-κB pathway. Further studies show that AhR/RelB complexes can be found not only in lymphoid cells but also in a human hepatoma cell line (HepG2) or breast cancer cell line (MDA-MB-231). RelB has been implicated in carcinogenesis of breast cancer for instance and RelB is known to be a critical factor for the function and differentiation of dendritic cells; interestingly the participation of AhR in both processes has been suggested recently, which offers the great potential to expand the scope of the physiological roles of the AhR. There is evidence indicating that RelB may serve as a pro-survival factor, including its ability to promote “inflammation resolution” besides the association of RelB with inflammatory disorders. Based on such information, a hypothesis has been proposed in this review that AhR together with RelB functions as a coordinator of inflammatory responses.
Keywords: AhR, IDO, IL-8, NF-kappaB RelB, TCDD
1. Introduction
The AhR is a member of basic helix-loop-helix (bHLH-PAS) transcription factors including Period (Per), AhR nuclear translocator (ARNT), and single minded (SIM) regulating hypoxia, circadian rhythm, and cellular processes like differentiation and apoptosis [1]. The AhR is generally considered as a ligand-dependent transcription factor which dimerizes with ARNT to activate gene transcription through DRE located on the promoter of these target genes. Numerous exogenous compounds (e.g. polycyclic aromatic hydrocarbons, benzimidazoles and flavonoids) with various binding affinities have been shown to bind to and activate the AhR [2]. A number of endogenous ligands with varying binding affinities to the AhR have also been proposed. Especially derivates from the essential amino acid tryptophan have been identified as high-affinity ligands to the AhR and are considered as potential endogenous AhR ligands [3, 4]. The non-activated form of the AhR is complexed with heat shock protein 90 (HSP90) and X-associated protein 2 (XAP2) in the cytosol, but depending on cell type and physiological conditions the AhR is also located in the nucleus even in absence of exogenous ligand [5]. XAP2 may enhance the rate of nuclear translocation of the ligand-bound human AhR complex and modulates the sub-cellular localization of the mouse AhR [6, 7]. The mechanism of classical AhR/Arnt signaling pathway will be discussed in detail elsewhere.
Although AhR agonists are known to induce immunosuppression, there is increasing evidence that one of the significant toxic actions of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is elicitation of chronic active inflammation in different organs and sites of the body [8-11]. TCDD is a prototype of polycyclic aromatic hydrocarbons (PAH) and exogenous compounds which binds with high affinity to the AhR. One of the routes of propagation of inflammatory signaling may be induction of proinflammatory genes including Tumor necrosis factor (TNF)α, Interleukin (IL)-1β, Cyclooxygenase 2 (COX-2), and others by TCDD, which has been confirmed in numerous studies [12-15]. Recently we found that TCDD induces also IL-8 in human macrophages as well as in liver of C57BL/6 mice in an AhR-dependent manner [16-19]. IL-8 is a member of the CXC chemokine family. One of its functions is to control the process of leukocyte invasion into injured tissue which is a hallmark of inflammation [20]. The important point of showing all these examples of TCDD-induced inflammation and immunosuppression occurring in various tissues and cells is that these are the type of events, which cannot be readily explained on the basis of the existing model of the classical action mechanism of TCDD that is based on binding of AhR/ARNT to DRE.
Although tissue inflammation stimulates adaptive immunity, certain types of inflammation, particularly excessive or chronic inflammation, paradoxically suppresses adaptive immunity as well. In general, inflammation induces compensatory anti-inflammatory mechanisms that are essential to limit potentially dangerous immune responses and reduce tissue damage. Therefore it should be considered that inflammation enhances immunogenicity, but in some contexts, inflammation may also stimulate active suppression. The mechanism underlying TCDD’s immunosuppressive effect is not well understood and it is still an open question if the AhR-mediated inflammatory responses might be the first event which eventually leads to immune suppression.
This review summarizes existing and original data of the new crosstalk between the AhR and NF-κB RelB and provides a new perspective of how AhR together with RelB may participate in immuneregulatory functions.
2. TCDD and the AhR
2.1. General mechanisms of action of TCDD
Following the original elucidation of the structure of the AhR by Chris Bradfield and his colleagues [21], there has been tremendous progress made in the field of AhR biology in terms of its basic structure [22], its cytosolic association with XAP2 [23] along with HSP90 proteins, its nuclear translocation and formation of its gene transactivation complex with its dimerization partner ARNT and several nuclear coactivators [24]. This line of investigation has been very fruitful and constructive in establishing the identity of this important receptor, particularly in mediating toxic signaling of TCDD and related dioxin-type pollutants with special emphasis on induction of a number of xenobiotic metabolizing enzymes of the AhR gene battery. Besides dimerization with its well known partner ARNT, the AhR may also bind to other transcription factors to regulate gene expression [25-30]. Despite the above accomplishments, some questions remain unanswered in this field of science. The most pressing questions are: how does activation of AhR leads to toxic consequences especially inflammation as well as immunosuppression and how are developmental processes and physiological functions regulated through AhR?
2.2. Physiological roles of AhR
In this regard, the most illustrative findings indicating the role of AhR have been provided by several groups of scientists, who created strains of the AhR null mice. Among those, AhR null mice from one strain created by Frank Gonzalez and his colleagues [31] showed liver abnormality during the neonatal development which is associated with high rates of apoptosis. More recently, Chris Bradfield’s group has reported that their strain of AhR null mice develop liver abnormality which is characterized with a patent developmental structure known as the ductus venosus (DV) during the first 48 h of their postnatal life. This abnormality is caused by the failure of the DV to close (decrease the diameter of the portal and the umbilical veins [32]. These observations clearly support the physiological role of AhR in the normal development of embryos and neonates. In addition, AhR null mice exhibit high production of TGFβ3 in liver, which is accompanied with excess vitamin A [33]. Furthermore, Rodriguez-Sosa et al. [34] have reported that spleen of AhR null mice over-produces Interferon (IFN)γ and IL-12 when challenged with ovalbumin, which is in line with previous findings [35] that AhR plays an important role in the normal development and function of the immune system. AhR null mice show decreased accumulation of lymphocytes in the spleen and lymph nodes [35]. This suggests the AhR is also located in the nucleus to regulate these physiological processes in the absence of exogenous ligands. Nuclear localization and activity of the AhR during embryonic development has also been reported [36].
Interestingly, from recent studies it becomes clear now that the AhR can regulate the generation of regulatory T cells (Treg) or pro-inflammatory T cells producing IL-17 (TH17) depending on the nature of its ligand. Activation of the AhR by TCDD induces differentiation of T-cell progenitor cells into Treg whereas ligand binding of AhR by 6-formylindolo[3,2-b]carbazole (FICZ) leads to differentiation of those progenitor cells into TH17 cell which produces interleukin (IL)-22 [37, 38]. FICZ is a tryptophan-derived photoproduct that is thought to be an endogenous ligand with high affinity for the AhR [4]. It has long been recognized that derivates of tryptophan can activate AhR [3]. In contrast to FICZ, the tryptophan-derived AhR endogenous ligand 2-(1’H-indole-3’-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) suppressed autoimmune encephalomyelitis (EAE) in mice as demonstrated for TCDD [37]. Dioxin (= TCDD) is a prototype of a class of environmental contaminants with high affinity to the AhR. AhR activation by TCDD has broad effects on the immune system in humans [39] and the AhR-dependent induction of a T-cell population with characteristics of Treg has been described earlier [40, 41]. The concept emerging from recent studies on T cell differentiation regulated by AhR proposes that activation of AhR dependent on its activating ligand can induce immunity or tolerance. Our most recent findings show, that activation of AhR by TCDD induces indoleamine-2,3-dioxygenase 1 (IDO1) and indoleamine-2,3-dioxygenase-like protein (IDO2) [42]. Expression of IDO1 and IDO2 is known to generate Treg which suggest a critical role of IDO to mediate immunosuppressive effects after activation of AhR by TCDD.
Future studies are necessary to show, if these ligand-specific effects on T cell differentiation are due to the capability of the AhR to interact with different transcription factors leading to distinct different outcomes or to the different pharmacological features of the AhR activating compounds.
It must be added that, while the connection between such a function of AhR to regulate the balancing of Treg versus TH17 and that of RelB is not apparent so far, the possibility of the involvement of RelB will be elaborated in a later section.
3. The NF-κB subunit RelB
3.1 Physiological roles of the NF-κB subunit RelB
The NF-κB or Rel proteins comprise a family of structurally related eukaryotic transcription factors that play critical roles in diverse cellular processes including adaptive and innate immunity, inflammatory responses, cell differentiation, proliferation, and apoptosis. The functions of Rel/NF-κB transcription factors are conserved from the fruit fly Drosophila melanogaster to humans; more recently, Rel/NF-κB homologs have also been found to occur even in organisms as simple as Cnidarians (e.g., sea anemones and corals) and Porifera (sponges). Transcriptionally active NF-κB dimers are formed by combinatorial association of five subunits: p50, RelA (p65), p52, c-Rel, and RelB [43]. Nuclear translocation of the NF-κB subunits is controlled by two main signaling pathways. The well known classical (canonical) NF-κB pathway consists majorly of the classic inducible NF-κB heterodimer p50 and RelA (=p65) subunits, each contacting one half of the DNA binding site. All Rel members can form heterodimers or homodimers, except for RelB, which can only form heterodimers. The individual dimers have specific DNA-binding sites and slight variations in the 10 base pair NF-κB consensus sequence (5’-GGGGYNNCCY-3’) which is critical for the preference of selected Rel combinations [44] and regulation of a distinct set of genes. The activity of Rel dimers is controlled by a family of inhibitor proteins, known as IκBs, whose degradation can be induced by a variety of stimuli and involves signal responsive activation of IkB kinase (IKK).
The activation of the IKK complex in the classical NF-κB pathway is generally regulated by IKKβ. In contrast, the alternative (non-canonical) NF-κB pathway is activated by phosphorylation of p100 through an IKKα homodimer lacking the regulatory subunit IKKγ. IKKγ, however, is absolutely required for activation of the classical pathway through ligation of members of the tumor necrosis factor receptor (TNFR) superfamily or initiation of pathogen sensing receptors the toll like receptors (TLR). Activation of the non-classical or alternative NF-κB pathway has been described to be mediated by several developmental stimuli such as Lymphotoxin (LT)-α1β2 or TNFR ligands like B cell activating factor (BAFF), CD40 ligand (CD40L), or receptor activator of NF-κB ligand (RANKL). Upon binding of these ligands NF-κB-inducing kinase (NIK) and IKKα will be activated and p100 will be processed to mature p52 followed by nuclear translocation of RelB/p52 NF-κB dimers. Activation of the alternative NF-κB pathway through ligation of Lymphotoxin β receptor (LTβR) for instance regulates expression of organogenic chemokines such as SLC (CCL12), BLC (CXCL13), ELC (CCL19), or SDF-1 (CXCL12) which is needed for organization of secondary lymphoid organs like maturation of spleen and lymph nodes [45-47]. NF-κB activation through the alternative pathway in response to developmental signals is weaker and delayed compared to inflammatory signals mediated via the classical pathway, which is usually a transient signal [48].
Genetic studies revealed that lymph node development requires the activity of both NF-κB pathways the classical (RelA and IKKβ) and the alternative (RelB and IKKα) pathway. For instance, signals mediated via CD40R or BAFFR which control B cell maturation were also shown to utilize the NIK pathway for RelA activation. Thus, the NF-κB RelA/p50 dimer does not only control a critical role in immune activation but plays also a role in immune development. For instance, development and maintenance of lymph nodes requires both RelA and RelB mediated NF-κB pathways. Their results further indicate that constitutive nuclear RelA/p50 activity is required to ensure expression of RelB and thus LTβR mediated activation of the alternative RelB/p52 dimer. Recent studies performed by Alexander Hoffmann’s group support the crosstalk of the classical and alternative NF-κB pathway showing that inflammatory and developmental signaling of NF-κB dimers are not separate but highly interconnected pathways [49].
The cross-regulation of both NF-κB systems has been shown in RelA deficient mice which are defective in RelB activation. Evidence came from recent reports, showing that stimulation of the alternative pathway via LTβR implicates a first phase of RelA activity which is important for expression and de novo synthesis of RelB [50]. RelB deficient mice showed increased inflammatory infiltration in multiple organs as well as severe deficits in adaptive immunity [51]. The most impressive findings regarding the function of RelB have been reported by Burkly et al. [52]. In RelB deficient mice, they could demonstrate that the expression of RelB is required for the development of thymic medulla and dendritic cells (DC). More recent studies have shown that the expression of RelB is upregulated early during differentiation of DC and is required for proper development and function of DC [53]. DCs are considered as key regulators of immunity or tolerance and are also an important source of IDO1 to induce Treg [54]. It has been shown that the alternative NF-κB pathway is critical for the induction of IDO1 in DC through ligation of CD40 for instance [55]. Thus, it is not unlikely that the TCDD-mediated induction of IDO1 does not only require AhR as shown recently [42], but also involves RelB as in the case of IL-8 [18].
3.2 The alternative NF-κB pathway in pathophysiology and the etiology of diseases
NF-κB transcription factors are persistently active in a number of disease states, including arthritis, chronic inflammation, asthma, neurodegenerative diseases, cardiovascular disease, and cancer. Inflammation caused by deregulation of NF-κB activity has been well identified as a tumor promoter. Both the classical and the alternative NF-κB pathways were found to be activated by viral oncoproteins, in particular the HTLV1-encoded Tax protein in adult T-cell leukemia and the EBV-encoded LMP1 protein in B-cell lymphoma [56].
Several reports indicate that the alternative NF-κB pathway is activated in specific subtypes of lymphoid leukemia and lymphoma. Chromosomal translocations disrupting the p100 gene that generate truncated p100 proteins and constitutive processing of p100 to p52 were identified in cutaneous T-cell lymphoma, and, more rarely, in B-cell non-Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma [45, 56, 57]. Transgenic expression of a truncated p100 protein led to the development of B-cell lymphomas in mice, thus demonstrating the oncogenic potential of p100 mutations [58]. Recently, we demonstrated the AhR-mediated development of lymphoma in vivo which was associated with increased expression of inflammatory markers such as COX-2. In addition, the expression of Bcl-xl an anti-apoptotic gene of the Bcl-2 family was significantly increased in clearly enlarged superficial lymph nodes of TCDD treated animals [59]. Interestingly, RelB/p52 complexes have been shown to induce Bcl-2 promoter activity [60] and future studies are needed to verify the possible involvement of AhR/RelB in lymphoproliferative disorder promoted by the TCDD-activated AhR.
In other cancer types such as breast cancer NF-κB has been shown to be constitutively active and located in the nucleus in over 90% of the breast cancers [61]. This could be due to chronic stimulation of the IKK pathway or defective IkB genes. Both NF-κB pathways can affect tumor growth. More recently the overexpression of RelB has been demonstrated in mammary tumorigenesis. Therefore the alternative signaling pathway has been considered to play a critical role in controlling the activity of NF-κB which is associated with cancer cells. Beside breast cancers (ER-negative, inflammatory type) [62], RelB has also been found to be over-expressed in some of prostate cancers [63]. It has been observed that in the above types of cancers as those cancer cells progress toward more advanced states of malignancy the nuclear titer of RelA protein decreases and that of RelB protein increases. The important point is that these are the type of breast cancer cells, which also exhibit overexpression of AhR and require RelB, not RelA to survive in the hostile environment (Vogel and Matsumura, unpublished data, 2008). Such an observation of RelB contributing to cancer cell survival should include their acquisition of immune tolerance. Studies investigating the epithelial-mesenchymal transition (EMT) in mammary tumorigenesis showed overexpression and nuclear translocation of RelB contributes to the phenotype of inflammatory breast cancer which is the most aggressive breast cancer with high metastatic potential [64, 65]. Especially in estrogen receptor (ER)α negative breast tumor cell types the expression of RelB is increased which is obviously based on a negative control of RelB de novo synthesis by ERα [60]. From their studies the authors concluded an inverse relationship exists between the expression of ERα and RelB in MCF-7, transformed mammary epithelial cells. Other data suggest that estrogenic status is also important in AhR regulation and can influence the effects of xenobiotics-mediated carcinogenicity [66]. A strong link between mammary tumorigenesis and AhR overexpression derived from studies performed by David Sherr’s group [67]. Recent reports by G. Lazennec show that development of this type of breast tumor is associated with inflammation and an inappropriate production of chemokines especially IL-8 which is a critical phase in breast tumor metastasis [68-71]. Besides these recent findings little is known about the role of AhR together with RelB in human breast disease. Since we know that AhR/RelB complexes are involved in the regulation of IL-8 and that IL-8, AhR, and RelB are overexpressed and active in many breast cancer cells, we consider that it is not a mere coincidence that formation of the AhR/RelB dimer is found in those ERα-negative, transformed mammary epithelial cells. Rather the AhR/RelB dimer likely contributes to mammary tumorigenesis. This hypothesis is supported by the association of elevated AhR activity in breast tumor cells with formation of the AhR/RelB complex as found by co-immunoprecipitation in MDA-MB-231 breast cancer cells (personal communication, D. Sherr and G. Sonenshein, Boston University). Our own data confirm a distinct increase (10-fold) of AhR/RelB binding activity in EMSA with nuclear extracts of the breast epithelial cell line MCF10A after long-term treatment with estradiol for 20 weeks (P. Wong, C.F. Vogel and F. Matsumura, unpublished data). A better understanding of the overexpression of inflammatory markers such as IL-8 in breast cancer is crucial to get a better insight into the role of the cytokines/chemokines in mammary tumorigenesis.
4. AhR and RelB
4.1. Implication for the interaction of AhR and RelB
Recently it was reported from this laboratory [18] that in U937 macrophages the NF-κB subunit RelB is functionally associated with the AhR and mediates transcription of chemokines such as IL-8 via activation of AhR that is assisted by protein kinase A (PKA). RelB was found to physically interact with AhR, as attested to by their co-immnoprecipitation and their association as detected in electromobility shift assays (EMSA). We have shown that this dimer binds to a previously unrecognized RelB/AhR response element (RelBAhRE) of the IL-8 promoter linking two signaling pathways to activate gene transcription. We found a time-dependent recruitment of AhR to the RelBAhRE site of IL-8 mediated by the AhR ligand, TCDD and via activation of PKA. Furthermore, NF-κB-binding sites that are preferentially recognized by RelB/p52 are a target for RelB/AhR complexes without addition of any exogenous ligands, implicating the endogenous function of the AhR. RelB/AhR complexes are also found to bind on DRE as well as NF-κB consensus elements, and RelB drastically increases the TCDD-induced DRE-Luc reporter activity.
Since this discovery of the interaction of AhR and RelB is so recent, not many data are available from the literature showing the crosstalk of the AhR and RelB. However, some recent reports from other groups support the interaction between both transcription factors AhR and RelB [72] and may give us some more insight into the physiological and toxicological relevance of this novel heterodimer.
In a previous report we showed that TCDD dose-dependently induces the expression of IL-8 in human U937 macrophages [19]. Mechanistic studies on IL-8 activation showed that TCDD or forskolin (FSK) mediated nuclear entry of the AhR which represents a new alternative mechanism of AhR regulation which dependent at least partly on activation of PKA and the association of AhR with RelB [18]. Previous studies from other groups have shown that FSK induces IL-8 [73] and that FSK or cAMP may activate AhR through a PKA-dependent mechanism [74]. Interestingly, the PKA-dependent activation of AhR was associated with nuclear translocation, but without dimerization with ARNT. The potential dimerization partner of AhR remained unknown at that time. Recently, we found that the FSK-and TCDD-induced activation of the IL-8 involves an unidentified RelB/AhR binding element (RelBAhRE) close to the TATA box of the IL-8 gene. This RelBAhRE sequence is distinctly different from DRE or NF-κB consensus elements since it does not bind to ARNT nor to the NF-κB subunit p50, which are well known dimerization partners of AhR and RelB, respectively. Further studies in our laboratory have revealed that NF-κB binding sites, which preferentially bind RelB/p52 complex, are also bound by the AhR/RelB dimer. Besides IL-8, such RelB/AhR responsive elements have been found on genes like B-cell activating factor of the tumor necrosis factor family (BAFF), B-lymphocyte chemoattractant (BLC), CC-chemokine ligand 1 (CCL1), and the transcription factor interferon γ responsive factor (IFR3), which are known targets of the alternative NF-κB pathway [29]. Results from these studies also showed that the RelB/AhR heterodimer binds to DRE as well as NF-κB consensus elements suggesting that AhR together with RelB may also regulate classical NF-κB and AhR signaling pathways. Binding of AhR/RelB on DRE elements is not limited to hematopoetic cell lines like U937 and was also found in the human hepatoma cell line HepG2 (Fig. 1) and other cell lines like mouse Hepa1c1c7 or 3T3 L1 fibroblasts (Data not shown). This is an important point since Chris Bradfield’s group has found that DNA binding of the AhR is required to mediate liver development as well as toxic endpoints elicited by TCDD [75]. Based on these data we proposed a model of an alternative AhR/RelB pathway for the action of the non-liganded AhR in which the crosstalk between AhR and RelB regulates the expression of inflammatory genes like IL-8 [18].
Figure 1.
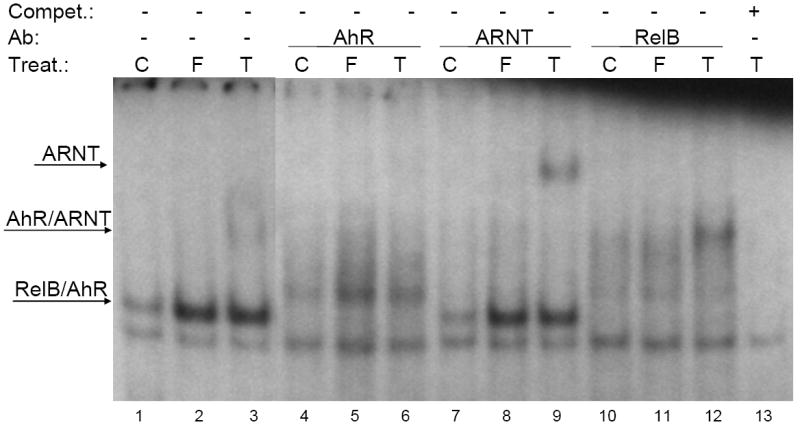
Nuclear protein extracts from control (C), FSK- (F), or TCDD-stimulated (T) HepG2 cells were incubated with 32P-labeled oligonucleotide containing a DRE consensus element of the CYP1A1 promoter. Nuclear proteins were extracted after treatment (Treat.) of HepG2 cells (ATCC, Manassas, VA) with 10 nM TCDD or 5 μM FSK for 2h. TCDD (>99% purity) was originally obtained from Dow Chemical Co. (Midland, MI) and FSK was purchased from Sigma (St. Louis, MO). A possible binding of AhR, ARNT, and RelB was identified by supershift analyses using AhR-, ARNT-, or RelB-specific antibodies (Ab). Monoclonal ARNT (Santa Cruz Biotech Inc, Santa Cruz, CA), RelB (Active Motif, Carlsbad, CA) and polyclonal AhR (Novus Biologicals, Littleton, CO) antibodies were used for supershift analysis. To confirm specificity a 100-fold excess of unlabeled DRE consensus oligonucleotide was added. A 100-fold molar excess of unlabeled oligonucleotides was added as competitor (Compet.).
The unexpected finding that the AhR also binds on NF-κB consensus sites suggests that the AhR may affect the classical NF-κB pathway as well. Previous studies have shown that TCDD-mediated activation of the AhR may suppress the effects elicited by LPS or TNFα for instance [26]. The antagonistic action of AhR with the classical NF-κB pathway will be discussed in more detail in a different chapter of this issue. However, there is no doubt that TCDD together with LPS or TNFα may also act synergistically on target genes such IL-8 or COX-2 [76]. In collaboration with Nancy Kerkvliet we asked the question if the AhR may play a role in the induction mechanism of the classical NF-κB pathway mediated by inducers like LPS, for instance. For this purpose AhR null mice were challenged in Nancy Kerkvliet’s lab with 50 μg LPS for 3 and 6 hrs. Then we analyzed the expression of NF-κB target genes like KC (=IL-8 equivalent in mice) and COX-2 in liver and lung of LPS treated AhR null mice and compared the response towards LPS in wild type C57BL/6 mice. Surprisingly, we found that the LPS-induced up-regulation of KC mRNA expression in liver as well as that of COX-2 mRNA in lung was significantly lower in AhR null mice compared to wild type C57BL/6 mice (Fig. 2A and D). In contrast, LPS-mediated induction of KC in lung and COX-2 in liver was not significantly different in AhR null mice from that what was found in the matched wild type mice (Fig. 2B and C). As expected, TCDD had no effect on the expression of KC or COX-2 in AhR null mice (Fig. 2A-D). Previous studies have shown that both KC and COX-2 are targets of the AhR activated by TCDD [17, 77], and therefore, these data indicate that even in the absence of exogenously added ligand the AhR must be helping those cells in liver and lung to maintain their responsiveness to LPS. The main implication of this finding may be that the AhR functions not only in AhR signaling but also NF-κB signaling that is activated by classical inducers like LPS. Results from another study by Thatcher et al. [72] showing the accelerated degradation of RelB in AhR null mice after LPS challenge, support the critical role of AhR and RelB in response to LPS. Thatcher et al. [72] investigated the inflammatory response in lung of LPS and cigarette smoke exposed mice. They found that mice which inhaled cigarette smoke or LPS express higher levels of TNFα and IL-6 in the bronchoalveolar lavage of AhR null mice compared to wild type mice. In contrast to our findings (Fig. 2) and a previous report [17], these authors found that both constitutive as well as LPS-induced expression of KC was expressed at a higher level in AhR null mice compared to wild type BL/6 mice. The authors concluded that this phenomenon might be due to an elevated NF-κB activity in control animals of AhR null mice which is not the case in wild type mice under normal and healthy conditions [72]. Interestingly, the increased inflammatory response in cigarette smoke or LPS exposed AhR deficient mice was associated with a rapid loss of RelB protein. In a more recent study the same group reported that the heightened inflammatory response observed in fibroblasts from AhR null mice is not the result of NF-κB (p50/RelA) activation [78] as speculated previously [72]. The authors concluded that AhR helps to maintain the expression of RelB and thus limits pro-inflammatory COX-2 and prostaglandin (PG) production induced by cigarette smoke [78]. This report is in contrast to a previous study performed by the same group showing that the AhR is required and plays an important role in cigarette smoke-mediated COX-2 and PG production in human lung fibroblasts and may contribute to tobacco-associated inflammation [79]. One possibility is that specific components of cigarette smoke such as PAHs require AhR to mediate their effects whereas other components of cigarette smoke rather activate the classical NF-κB pathway through oxidative stress and may benefit from the absence of the AhR and RelB to induce an inflammatory response.
Figure 2.

C57BL/6 wild type (wt) and AhR homozygous null mice (AhR-/-) were treated with 50 μg LPS E. coli 055:B5 (Sigma L4005, St. Louis, MO). In addition AhR-/- mice were treated with 15 μg/kg TCDD. After 6 hrs animals were sacrificed and tissues were prepared for RNA analysis by real time PCR. Values of KC and prostaglandin endoperoxide H synthase-2 (PGHS-2 or COX-2) mRNA are normalized to the expression of β-actin. C57BL/6J (BL6) and AhR null (AhR-/-) mice were originally purchased from The Jackson Laboratory (Bar Habor, ME).
* significantly different from LPS-treated wild type mice p≤0.05
Nevertheless, these studies confirm our previous findings [17] on the critical role of the AhR in regulation of the inflammatory response. More importantly these studies support our reports of the interaction of AhR complexed with RelB [18], if we interpret their data to mean that the presence of AhR helps stabilization of RelB in normal mice by extending their observation that in the absence of AhR, RelB protein is more susceptible for degradation.
Lee et al. [80] investigated the effect of TCDD on the differentiation of DCs. Activation of the AhR through environmental pollutants like dioxin is well known to induce profound suppression of immune responses, especially T cell dependent responses, which is the primary toxicity associated with the exposure to dioxin [81]. Emerging from recent studies it becomes clear that the AhR can regulate the generation of regulatory T cells (Treg) which could explain TCDD’s immune suppressive effects. Interestingly, induction of Treg after activation of AhR by an anti-inflammatory compound (VAF347) has been found to be mediated at least in part through DCs [82]. Therefore, DCs should be considered as a potential target for TCDD. DCs are the most potent antigen-presenting cells (APC) and are key regulators of the choice between immunity and tolerance [83]. One of the most potent ways in which DCs can create immunosuppression is by activating Treg cells [84]. Lee et al. [80] showed that treatment of mouse DC derived from bone marrow with TCDD significantly affected the differentiation and maturation of DCs. TCDD-treated DCs expressed significantly higher level of DC differentiation markers such as MHC class II and the co-stimulatory molecule CD86, whereas the expression of CD11c and the production of IL-10 was reduced by TCDD. These results agree with studies performed in Nancy Kerkvliet’s lab [85, 86] showing an increased expression of the DC surface markers CD86, MHC class II, and ICAM-1, whereas LFA-1 was found to be decreased in splenic DC of TCDD treated mice. In summary, the authors described an activation-like stimulus to DC after treatment with TCDD in an AhR-dependent manner which is supported by our most recent report [42]. In that we could show that activation of AhR by TCDD or the tryptophan derived photoproduct FICZ increases the expression of surface markers especially CD86 in human U937 monocyte-derived DCs, which was associated with morphological changes of DCs into more elongated and stellate cells typical for dendritic cells. In addition, we found that activation of AhR by TCDD leads to indolamine-2,3,-dioxygenase (IDO) expressing DCs and induction of IDO1 and IDO2 in spleen of BL/6 mice. The induction of IDO in spleen was associated with the increase of the Treg marker Foxp3 which was suppressed by treatment with a pharmacological inhibitor of IDO. These data indicate that induction of IDO by TCDD may play an important role in AhR-dependent regulation of T cell differentiation and immune suppression.
Lee et al. [80] reported that DCs from TCDD-treated mice showed enhanced ability to stimulate allogeneic T cell proliferation in a mixed lymphocyte reaction. Furthermore, the authors found that TCDD as well as Benzo(a)pyren downregulate the expression of RelB upon stimulation of DC maturation with LPS in an AhR-dependent manner. As mentioned earlier one critical physiological function of RelB is regulation of differentiation of DCs. Thus the TCDD-mediated and AhR-dependent effects on DC differentiation seem to implicate the function of RelB. With respect to our findings describing the role of the AhR/RelB complex in the regulation of cytokines and chemokines it is likely that the interference on DC differentiation caused by TCDD is mediated through an activated AhR complexed with RelB.
In another study Ruby et al. [87] investigated whether TCDD would alter the activation of NF-κB/Rel in DCs. The dendritic cell line DC2.4 was exposed to TCDD before treatment with TNF-α or anti-CD40, to activate both the classical and alternative NF-κB/Rel pathway. TCDD suppressed the binding of NF-κB/Rel to its cognate response element in TNF-α- and anti-CD40-treated cells and blocked translocation to the nucleus. The AhR was shown to associate with RelA, after coimmunoprecipitation, and seemed to block its binding to DNA. In contrast to the studies of Lee et al. [80] and our studies in U937 macrophages, this interaction was limited to RelA, but RelB did not seem to interact with AhR in DCs transfected with AhR. Ruby et al. [87] concluded from their results that the defects in the DCs and suppression of the immune response mediated by TCDD may be due to a shifted balance between NF-κB/Rel heterodimers and transcriptional inhibitory p50 homodimers in DCs. In contrast to TCDD, Lawrence et al. [88] reported recently that an anti-inflammatory low molecular weight compound (VAF347) activates the AhR signaling pathway resulting in anti-inflammatory effects on DC by inhibiting expression of the DC markers CD86 and MHC class II. Thus, the role of the AhR mediating activation-like or anti-inflammatory responses in DC might be due to its capacity to interact with different transcription factors in a ligand-specific manner as suggested for the differentiation of T cells into Treg or TH17. Another explanation could be that VAF347 affects additional signaling pathways independent of the AhR. The knowledge of how activation of AhR by specific ligands results in biologically different endpoints might improve our understanding of the toxicity of various AhR ligands and more importantly the physiological role of the AhR.
5. Concluding Remark
Evidence has been accumulating to indicate that there are close interactions between AhR and NF-κB family members such as RelA and RelB. Interestingly interactions appear to occur between RelB with the form of AhR either bound with its ligand or without. The consequence of RelA/AhR interactions appears to be mostly negative (i.e. antagonistic) as in the case of CYP1A1 [26] or IL-6 [89] gene activity by forming complexes with AhR which have obviously no DNA-binding capacity or transcriptional activity with the exception of the c-myc gene (Fig. 3). Kim et al. [90] reported that AhR and RelA may functionally cooperate to bind to NF-κB elements and induce c-myc gene expression. So far as we have found, the interaction of RelB/AhR appears to be rather positive by increasing DRE-reporter activity of CYP1A1 and transcription of NF-κB target genes such as IL-8 and other chemokines [17,18] through binding on specific RelBAhRE sequences (Fig. 4). However, we have to consider that the interaction of the AhR with NF-κB signaling pathways (classical or alternative) seems to be much more complex and should not exclude the possibility that enhanced formation of AhR/RelB dimers could also have suppressive action on the activity of certain target genes under certain conditions. It must be added that in both cases (i.e. RelA/AhR and RelB/AhR interactions), their joint effects are exacerbated when AhR is activated by its prototypical ligand TCDD. The main question we must raise is then why does such an opposite consequence of their interactions exist between RelA/AhR versus RelB/AhR? This difference between RelA and RelB cannot be coincidental; after all, if RelA and RelB serve for the same purpose, there would be no need for two elaborate although interacting NF-κB signaling pathways (i.e. the classical pathway for RelA and the alternative pathway for RelB). One of the possibilities is that AhR assists the function of RelB not only to mediate chronic inflammation but also to promote RelB’s function in resolution of inflammation via negative feedback mechanisms for instance [91,92], whereas AhR antagonizes the action of RelA to moderate acute cellular inflammation, particularly after stimulation by LPS or other potent inflammation inducing agents and/or protect cells from unwanted side-effects of full activation of inflammatory effects of RelA. Viewed in this way, it makes it easier to understand the reason why RelB/AhR interactions tend to produce mostly synergistic consequences of inflammation, while that of RelA/AhR could act antagonistically.
Figure 3.

Putative crosstalk between the classical pathway of NF-κB and AhR activation.
Ligand engagement of specific membrane receptors (TNFR, IL-1R or TLR) triggers phosphorylation and activation of IKKβ. Free NF-κB translocates to the nucleus and activates target genes via p50/RelA dimers. Activated free RelA may also bind to ligand-activated AhR generating transcriptionally inactive complexes that could suppress the classical activation pathways of NF-κB [89] and AhR [26] or positively regulate gene expression as shown for c-myc [90]. IL-1R, IL-1 receptor; NEMO, NF-κB essential modulator; TLR, Toll like receptor; TNFR, TNF receptor.
Figure 4.

New mechanism of cross talk between AhR and RelB.
Ligand-activated or unliganded AhR activated by PKA translocates into the nucleus and interacts with RelB and occupy RelB/AhR-response elements (RelBAhRE) of promoters as in the case of IL-8 (alternative AhR/RelB pathway). AhR agonists induce the recruitment of AhR/ARNT complexes to DRE-responsive promoters such as CYP1a1 (classical AhR/ARNT pathway). Alternative pathway of NF-κB activation through the LTβR, RANK, CD40, or BAFFR leads to the release of active NIK which phosphorylates IKK. After processing of p100, RelB/p52 heterodimers are generated and activate target genes. BAFFR, B-cell activating factor receptor; FSK, forskolin; IKK, IKB kinase; LTβR, Lymphotoxin β receptor; NIK, NF-κB-inducing kinase; NF-κB, nuclear factor-κB; PKA, Protein kinase A; RANK, receptor activator of NF-κB. From Vogel et al. [18] reproduced with permission from Molecular Endocrinology 2008.
Exacerbated activation of the alternative NF-κB RelB pathway is associated with various inflammatory disorders including cancer. Recent reports indicate that activation of RelB through IKKα (as opposed to activation of RelA, which largely depends on activation of IKKβ) may also lead to resolution (a term used to express the effect of anti-inflammatory agents such as aspirin or the negative feedback of cells to reduce the effects of inflammation) of RelA-induced inflammation [93]. Indeed, Matos and Jordan [94] consider that RelB acts essentially as the negative feedback to the inflammation pathway mediated by RelA. Furthermore, there is now enough evidence to indicate that RelB acts as a major cell survival factor such as conferring radiation resistance [95] and anti-apoptotic properties of cancer cells [96]. Since the finding of the RelB/AhR interaction is so recent, the possible assisting role of AhR in the above cases has not been addressed adequately yet. If such a view is confirmed, it would provide the important clue to one of the major functions of AhR to act as the factor promoting inflammation in cells and alternately it can act as a factor promoting resolution of inflammation to protect cells that are experiencing intense inflammation.
Certainly studies on the interaction of NF-κB proteins and signaling pathways are evolving rather fast, and we must closely keep up with future changes occurring in this fascinating research area. Nevertheless, the realization that AhR participates in NF-κB signaling pathways in regulating inflammation, immune responses, resolution, survival, and probably other functions, this aspect of the biological roles of the AhR merits close attention in the future. Furthermore, this finding also fits the model of AhR being capable of directly forming dimers with individual members of other types of nuclear transcription factors, even without its ligand, e.g. with ERα as in the case of BRCA-1 gene activation [97]. With the additional evidence of direct interactions of AhR with other transcription factors presented in this special issue, such a mode of AhR action to coordinate its activities with other major cellular regulatory pathways through direct AhR protein interactions in the nucleus should be considered as one of the major paradigm of AhR operations. Finally studies on the interaction between AhR and RelB support our previous findings that one of the cellular physiological roles of AhR includes regulation of the cellular inflammatory state.
Acknowledgments
We thank Nancy Kerkvliet and Linda Steppan for providing us with samples from LPS-treated BL/6 wildtype and AhR-null mice and Helen Woldai and Danitza Alvizar-Trujillo for preparation of animal tissues. We also thank David Sherr and Gail Sonenshein for providing us data from Co-IP studies with AhR and RelB. This work was supported by research grants R01-ES05233 from the National Institute of Environmental Health Sciences, research grant FA0703859 from Susan G. Komen for the Cure, and a Grant-in-Aid of the American Heart Association.
Abbreviations
- AhR
aryl hydrocarbon receptor
- ARNT
AhR nuclear translocator
- COX-2
Cyclooxygenase 2
- DC
dendritic cell
- DRE
dioxin response element
- FICZ
6-formylindolo[3,2-b]carbazole
- IDO
indoleamine-2,3-dioxygenase
- IL-8
Interleukin 8
- IFN
Interferon
- RelBAhRE
RelB/AhR response element
- PKA
protein kinase A
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- Treg
regulatory T cells
- XAP2
X-associated protein 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Ann Rev Pharmacol and Toxicol. 2000;40:519–61. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 2.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Ann Rev Pharmacol and Toxicol. 2003;43:309–34. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 3.Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, et al. Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. J Biol Chem. 1987;262:15422–7. [PubMed] [Google Scholar]
- 4.Fritsche E, Schafer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci USA. 2007;104:8851–6. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh SS, Hord NG, Perdew GH. Characterization of the activated form of the aryl hydrocarbon receptor in the nucleus of HeLa cells in the absence of exogenous ligand. Arch Biochem Biophys. 1996;329:47–55. doi: 10.1006/abbi.1996.0190. [DOI] [PubMed] [Google Scholar]
- 6.Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Divergent roles of hepatitis B virus X-associated protein 2 (XAP2) in human versus mouse Ah receptor complexes. Biochemistry. 2004;43:700–9. doi: 10.1021/bi035827v. [DOI] [PubMed] [Google Scholar]
- 7.Petrulis JR, Hord NG, Perdew GH. Subcellular localization of the aryl hydrocarbon receptor is modulated by the immunophilin homolog hepatitis B virus X-associated protein 2. J Biol Chem. 2000;275:37448–53. doi: 10.1074/jbc.M006873200. [DOI] [PubMed] [Google Scholar]
- 8.Matsumura F. On the significance of the role of cellular stress response reactions in the toxic actions of dioxin. Bioch Pharmacol. 2003;66:527–40. doi: 10.1016/s0006-2952(03)00157-6. [DOI] [PubMed] [Google Scholar]
- 9.National Toxicology Program (U.S.), United States. Dept. of Health and Human Services., United States. Public Health Service., National Institutes of Health (U.S.) NTP technical report on the toxicology and carcinogenesis studies of a mixture of 2,3,7,8-tetrachlorodibenzo- rho -dioxin (TCDD) (CAS no. 1746-01-6), 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) (CAS no. 57117-31-4), and 3,3’,4,4’,5-pentachlorobiphenyl (PCB 126) (CAS no. 57465-28-8) in female Harlan Sprague-Dawley rats (gavage studies) Research Triangle Park, N.C.: National Toxicology Program National Institutes of Health Public Health Service U.S. Dept. of Health and Human Services; 2006. [Google Scholar]
- 10.Nyska A, Jokinen MP, Brix AE, Sells DM, Wyde ME, Orzech D, et al. Exocrine pancreatic pathology in female Harlan Sprague-Dawley rats after chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin and dioxin-like compounds. Environ Health Perspect. 2004;112:903–9. doi: 10.1289/ehp.6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pande K, Moran SM, Bradfield CA. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol Pharmacol. 2005;67:1393–8. doi: 10.1124/mol.105.010983. [DOI] [PubMed] [Google Scholar]
- 12.Sutter TR, Guzman K, Dold KM, Greenlee WF. Targets for dioxin: genes for plasminogen activator inhibitor-2 and interleukin-1 beta. Science. 1991;254:415–8. doi: 10.1126/science.1925598. [DOI] [PubMed] [Google Scholar]
- 13.Taylor MJ, Lucier GW, Mahler JF, Thompson M, Lockhart AC, Clark GC. Inhibition of acute TCDD toxicity by treatment with anti-tumor necrosis factor antibody or dexamethasone. Toxicol Appl Pharmacol. 1992;117:126–32. doi: 10.1016/0041-008x(92)90227-j. [DOI] [PubMed] [Google Scholar]
- 14.Fan F, Yan B, Wood G, Viluksela M, Rozman KK. Cytokines (IL-1beta and TNFalpha) in relation to biochemical and immunological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology. 1997;116:9–16. doi: 10.1016/s0300-483x(96)03514-7. [DOI] [PubMed] [Google Scholar]
- 15.Vogel C, Schuhmacher US, Degen GH, Bolt HM, Pineau T, Abel J. Modulation of prostaglandin H synthase-2 mRNA expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice. Arch Biochem Biophys. 1998;351:265–71. doi: 10.1006/abbi.1997.0555. [DOI] [PubMed] [Google Scholar]
- 16.Matsumura F, Vogel CF. Evidence supporting the hypothesis that one of the main functions of the aryl hydrocarbon receptor is mediation of cell stress responses. Biol Chem. 2006;387:1189–94. doi: 10.1515/BC.2006.146. [DOI] [PubMed] [Google Scholar]
- 17.Vogel CF, Nishimura N, Sciullo E, Wong P, Li W, Matsumura F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys. 2007;461:169–75. doi: 10.1016/j.abb.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21:2941–55. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogel CF, Sciullo E, Wong P, Kuzmicky P, Kado N, Matsumura F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Env Health Perspec. 2005;113:1536–41. doi: 10.1289/ehp.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250:91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- 21.Swanson HI, Bradfield CA. The AH-receptor: genetics, structure and function. Pharmacogenetics. 1993;3:213–30. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Poellinger L. Mechanistic aspects--the dioxin (aryl hydrocarbon) receptor. Food Add Contam. 2000;17:261–6. doi: 10.1080/026520300283333. [DOI] [PubMed] [Google Scholar]
- 23.Carlson DB, Perdew GH. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J Biochem Mol Toxicol. 2002;16:317–25. doi: 10.1002/jbt.10051. [DOI] [PubMed] [Google Scholar]
- 24.Hankinson O. Role of coactivators in transcriptional activation by the aryl hydrocarbon receptor. Arch Biochem Biophys. 2005;433:379–86. doi: 10.1016/j.abb.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 25.Ge NL, Elferink CJ. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle. J Biol Chem. 1998;273:22708–13. doi: 10.1074/jbc.273.35.22708. [DOI] [PubMed] [Google Scholar]
- 26.Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274:510–5. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- 27.Klinge CM, Kaur K, Swanson HI. The aryl hydrocarbon receptor interacts with estrogen receptor alpha and orphan receptors COUP-TFI and ERRalpha1. AArch Biochem Biophys. 2000;373:163–74. doi: 10.1006/abbi.1999.1552. [DOI] [PubMed] [Google Scholar]
- 28.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545–50. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 29.Vogel CF, Sciullo E, Matsumura F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem Biophys Res Commun. 2007;363:722–6. doi: 10.1016/j.bbrc.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marlowe JL, Fan Y, Chang X, Peng L, Knudsen ES, Xia Y, et al. The Aryl Hydrocarbon Receptor Binds to E2F1 and Inhibits E2F1-induced Apoptosis. Mol Biol Cell. 2008;19:3263–71. doi: 10.1091/mbc.E08-04-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez FJ, Fernandez-Salguero P. The aryl hydrocarbon receptor: studies using the AHR-null mice. Drug Metabol Dispos. 1998;26:1194–8. [PubMed] [Google Scholar]
- 32.Lahvis GP, Pyzalski RW, Glover E, Pitot HC, McElwee MK, Bradfield CA. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol Pharmacol. 2005;67:714–20. doi: 10.1124/mol.104.008888. [DOI] [PubMed] [Google Scholar]
- 33.Andreola F, Hayhurst GP, Luo G, Ferguson SS, Gonzalez FJ, Goldstein JA, et al. Mouse liver CYP2C39 is a novel retinoic acid 4-hydroxylase. Its down-regulation offers a molecular basis for liver retinoid accumulation and fibrosis in aryl hydrocarbon receptor-null mice. J Biol Cchem. 2004;279:3434–8. doi: 10.1074/jbc.M305832200. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Sosa M, Elizondo G, Lopez-Duran RM, Rivera I, Gonzalez FJ, Vega L. Overproduction of IFN-gamma and IL-12 in AhR-null mice. FEBS letters. 2005;579:6403–10. doi: 10.1016/j.febslet.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–6. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 36.Walisser JA, Bunger MK, Glover E, Harstad EB, Bradfield CA. Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. T J Biol Cchem. 2004;279:16326–31. doi: 10.1074/jbc.M400784200. [DOI] [PubMed] [Google Scholar]
- 37.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 38.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 39.Baccarelli A, Mocarelli P, Patterson DG, Jr, Bonzini M, Pesatori AC, Caporaso N, et al. Immunologic effects of dioxin: new results from Seveso and comparison with other studies. Environ Health Perspect. 2002;110:1169–73. doi: 10.1289/ehp.021101169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Funatake CJ, Marshall NB, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation of alloreactive CD8+ T cells toward a regulatory T cell phenotype by a mechanism that is dependent on aryl hydrocarbon receptor in CD4+ T cells. J Immunotoxicol. 2008;5:81–91. doi: 10.1080/15476910802019037. [DOI] [PubMed] [Google Scholar]
- 41.Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J Immunol. 2005;175:4184–8. doi: 10.4049/jimmunol.175.7.4184. [DOI] [PubMed] [Google Scholar]
- 42.Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.07.156. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–3. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 44.Kunsch C, Ruben SM, Rosen CA. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–21. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161–79. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–35. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 47.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO. 2004;23:4202–10. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 49.Basak S, Hoffmann A. Crosstalk via the NF-kappaB signaling system. Cytokine & Growth Factor Rev. 2008;19:187–97. doi: 10.1016/j.cytogfr.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–81. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–40. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 52.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–6. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 53.Cejas PJ, Carlson LM, Kolonias D, Zhang J, Lindner I, Billadeau DD, et al. Regulation of RelB expression during the initiation of dendritic cell differentiation. Mol Cell Biol. 2005;25:7900–16. doi: 10.1128/MCB.25.17.7900-7916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–70. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- 55.Tas SW, Vervoordeldonk MJ, Hajji N, Schuitemaker JH, van der Sluijs KF, May MJ, et al. Noncanonical NF-kappaB signaling in dendritic cells is required for indoleamine 2,3-dioxygenase (IDO) induction and immune regulation. Blood. 2007;110:1540–9. doi: 10.1182/blood-2006-11-056010. [DOI] [PubMed] [Google Scholar]
- 56.Jost PJ, Ruland J. Aberrant NF-kappaB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109:2700–7. doi: 10.1182/blood-2006-07-025809. [DOI] [PubMed] [Google Scholar]
- 57.Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine & Growth Factor Rev. 2006;17:281–93. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 58.Zhang B, Wang Z, Li T, Tsitsikov EN, Ding HF. NF-kappaB2 mutation targets TRAF1 to induce lymphomagenesis. Blood. 2007;110:743–51. doi: 10.1182/blood-2006-11-058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, et al. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol. 2007;171:1538–48. doi: 10.2353/ajpath.2007.070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nature Cell Biol. 2007;9:470–8. doi: 10.1038/ncb1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, et al. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–60. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Laere SJ, Van der Auwera I, Van den Eynden GG, van Dam P, Van Marck EA, Vermeulen PB, et al. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 overexpression and MAPK hyperactivation. British J Cancer. 2007;97:659–69. doi: 10.1038/sj.bjc.6603906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lessard L, Begin LR, Gleave ME, Mes-Masson AM, Saad F. Nuclear localisation of nuclear factor-kappaB transcription factors in prostate cancer: an immunohistochemical study. British J Cancer. 2005;93:1019–23. doi: 10.1038/sj.bjc.6602796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Laere SJ, Van der Auwera I, Van den Eynden GG, Elst HJ, Weyler J, Harris AL, et al. Nuclear factor-kappaB signature of inflammatory breast cancer by cDNA microarray validated by quantitative real-time reverse transcription-PCR, immunohistochemistry, and nuclear factor-kappaB DNA-binding. Clin Cancer Res. 2006;12:3249–56. doi: 10.1158/1078-0432.CCR-05-2800. [DOI] [PubMed] [Google Scholar]
- 65.Zhang J, Warren MA, Shoemaker SF, Ip MM. NFkappaB1/p50 is not required for tumor necrosis factor-stimulated growth of primary mammary epithelial cells: implications for NFkappaB2/p52 and RelB. Endocrinol. 2007;148:268–78. doi: 10.1210/en.2006-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singhal R, Shankar K, Badger TM, Ronis MJ. Estrogenic status modulates aryl hydrocarbon receptor--mediated hepatic gene expression and carcinogenicity. Carcinogenesis. 2008;29:227–36. doi: 10.1093/carcin/bgm288. [DOI] [PubMed] [Google Scholar]
- 67.Schlezinger JJ, Liu D, Farago M, Seldin DC, Belguise K, Sonenshein GE, et al. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol Chem. 2006;387:1175–87. doi: 10.1515/BC.2006.145. [DOI] [PubMed] [Google Scholar]
- 68.Ali S, Lazennec G. Chemokines: novel targets for breast cancer metastasis. Cancer Metastasis Rev. 2007;26:401–20. doi: 10.1007/s10555-007-9073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Freund A, Jolivel V, Durand S, Kersual N, Chalbos D, Chavey C, et al. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene. 2004;23:6105–14. doi: 10.1038/sj.onc.1207815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bieche I, Chavey C, Andrieu C, Busson M, Vacher S, Le Corre L, et al. CXC chemokines located in the 4q21 region are up-regulated in breast cancer. Endocr Relat Cancer. 2007;14:1039–52. doi: 10.1677/erc.1.01301. [DOI] [PubMed] [Google Scholar]
- 71.Wu K, Katiyar S, Li A, Liu M, Ju X, Popov VM, et al. Dachshund inhibits oncogene-induced breast cancer cellular migration and invasion through suppression of interleukin-8. Proc Natl Acad Sci USA. 2008;105:6924–9. doi: 10.1073/pnas.0802085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, et al. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170:855–64. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA. 2003;100:12147–52. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oesch-Bartlomowicz B, Huelster A, Wiss O, Antoniou-Lipfert P, Dietrich C, Arand M, et al. Aryl hydrocarbon receptor activation by cAMP vs. dioxin: divergent signaling pathways. Proc Natl Acad Sci USA. 2005;102:9218–23. doi: 10.1073/pnas.0503488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bunger MK, Glover E, Moran SM, Walisser JA, Lahvis GP, Hsu EL, et al. Abnormal Liver Development and Resistance to 2,3,7,8-Tetrachlorodibenzo-p-dioxin Toxicity in Mice Carrying a Mutation in the DNA Binding Domain of the Aryl Hydrocarbon Receptor. Toxicol Sci. 2008 doi: 10.1093/toxsci/kfn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li W, Vogel CF, Fujiyoshi P, Matsumura F. Development of a human adipocyte model derived from human mesenchymal stem cells (hMSC) as a tool for toxicological studies on the action of TCDD. Biol Chem. 2008;389:169–77. doi: 10.1515/BC.2008.015. [DOI] [PubMed] [Google Scholar]
- 77.Vogel C, Boerboom AM, Baechle C, El-Bahay C, Kahl R, Degen GH, et al. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway. Carcinogenesis. 2000;21:2267–74. doi: 10.1093/carcin/21.12.2267. [DOI] [PubMed] [Google Scholar]
- 78.Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappa B family member RELB. J Biol Chem. 2008 Aug 12; doi: 10.1074/jbc.M800685200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, Phipps RP. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005 Sep;289(3):L391–9. doi: 10.1152/ajplung.00062.2005. [DOI] [PubMed] [Google Scholar]
- 80.Lee JA, Hwang JA, Sung HN, Jeon CH, Gill BC, Youn HJ, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin modulates functional differentiation of mouse bone marrow-derived dendritic cells Downregulation of RelB by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Letters. 2007;173:31–40. doi: 10.1016/j.toxlet.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 81.Kerkvliet NI. Recent advances in understanding the mechanisms of TCDD immunotoxicity. International Immunopharmacol. 2002;2:277–91. doi: 10.1016/s1567-5769(01)00179-5. [DOI] [PubMed] [Google Scholar]
- 82.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschlager M, et al. Activation of the aryl hydrocarbon receptor promotes allograft specific tolerance through direct-and DC-mediated effects on regulatory T cells. Blood. 2008 doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 83.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci USA. 2002;99:351–8. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mahnke K, Ring S, Johnson TS, Schallenberg S, Schonfeld K, Storn V, et al. Induction of immunosuppressive functions of dendritic cells in vivo by CD4+CD25+ regulatory T cells: role of B7-H3 expression and antigen presentation. European J Immunol. 2007;37:2117–26. doi: 10.1002/eji.200636841. [DOI] [PubMed] [Google Scholar]
- 85.Vorderstrasse BA, Dearstyne EA, Kerkvliet NI. Influence of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the antigen-presenting activity of dendritic cells. Toxicol Sci. 2003;72:103–12. doi: 10.1093/toxsci/kfg012. [DOI] [PubMed] [Google Scholar]
- 86.Vorderstrasse BA, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin affects the number and function of murine splenic dendritic cells and their expression of accessory molecules. Toxicol Appl Pharmacol. 2001;171:117–25. doi: 10.1006/taap.2000.9119. [DOI] [PubMed] [Google Scholar]
- 87.Ruby CE, Leid M, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses tumor necrosis factor-alpha and anti-CD40-induced activation of NF-kappaB/Rel in dendritic cells: p50 homodimer activation is not affected. Mol Pharmacol. 2002;62:722–8. doi: 10.1124/mol.62.3.722. [DOI] [PubMed] [Google Scholar]
- 88.Lawrence BP, Denison MS, Novak H, Vorderstrasse BA, Harrer N, Neruda W, Reichel C, Woisetschläger M. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low-molecular-weight compound. Blood. 2008 Aug 15;112(4):1158–65. doi: 10.1182/blood-2007-08-109645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study. Environ Health. 2003 Dec 16;2(1):16. doi: 10.1186/1476-069X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene. 2000;48:5498–506. doi: 10.1038/sj.onc.1203945. [DOI] [PubMed] [Google Scholar]
- 91.Xia Y, Pauza ME, Feng L, Lo D. RelB regulation of chemokine expression modulates local inflammation. Am J Pathol. 1997;151:375–87. [PMC free article] [PubMed] [Google Scholar]
- 92.Pereira SG, Oakley F. Nuclear factor-kappaB1: regulation and function. Int J Biochem Cell Biol. 2008;40:1425–30. doi: 10.1016/j.biocel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 93.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–43. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 94.Matos P, Jordan P. Rac1, but not Rac1B, stimulates RelB-mediated gene transcription in colorectal cancer cells. J Biol Chem. 2006;281:13724–32. doi: 10.1074/jbc.M513243200. [DOI] [PubMed] [Google Scholar]
- 95.Josson S, Xu Y, Fang F, Dhar SK, St Clair DK, St Clair WH. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene. 2006;25:1554–9. doi: 10.1038/sj.onc.1209186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lwin T, Hazlehurst LA, Li Z, Dessureault S, Sotomayor E, Moscinski LC, et al. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-kappaB (RelB/p52) in non-Hodgkin’s lymphoma cells. Leukemia. 2007;21:1521–31. doi: 10.1038/sj.leu.2404723. [DOI] [PubMed] [Google Scholar]
- 97.Hockings JK, Thorne PA, Kemp MQ, Morgan SS, Selmin O, Romagnolo DF. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res. 2006;66:2224–32. doi: 10.1158/0008-5472.CAN-05-1619. [DOI] [PubMed] [Google Scholar]