

Abstract
Altered folate homeostasis is associated with many clinical and pathological manifestations in the CNS. Notably, folate-mediated one-carbon metabolism is essential for methyltransferase-dependent cellular methylation reactions. Biogenesis of protein phosphatase 2A (PP2A) holoenzyme containing the regulatory Bα subunit, a major brain tau phosphatase, is controlled by methylation. Here, we show that folate deprivation in neuroblastoma cells induces downregulation of PP2A leucine carboxyl methyltransferase-1 (LCMT-1) expression, resulting in progressive accumulation of newly synthesized demethylated PP2A pools, concomitant loss of Bα, and ultimately cell death. These effects are further accentuated by overexpression of PP2A methylesterase (PME-1) but cannot be rescued by PME-1 knockdown. Overexpression of either LCMT-1 or Bα is sufficient to protect cells against the accumulation of demethylated PP2A, increased tau phosphorylation, and cell death induced by folate starvation. Conversely, knockdown of either protein accelerates folate deficiency-evoked cell toxicity. Significantly, mice maintained for 2 months on low-folate or folate-deficient diets have brain-region-specific alterations in metabolites of the methylation pathway. Those are associated with downregulation of LCMT-1, methylated PP2A, and Bα expression and enhanced tau phosphorylation in susceptible brain regions. Our studies provide novel mechanistic insights into the regulation of PP2A methylation and tau. They establish LCMT-1- and Bα-containing PP2A holoenzymes as key mediators of the role of folate in the brain. Our results suggest that counteracting the neuronal loss of LCMT-1 and Bα could be beneficial for all tauopathies and folate-dependent disorders of the CNS.
Keywords: folate, methylation, methylesterase, methyltransferase, PP2A, tau
Introduction
Folate is a necessary cofactor for enzymatic reactions mediating conversion of homocysteine (Hcy) to methionine in one-carbon metabolism (Friso and Choi, 2005). Dietary folate deficiency and/or folate-associated gene polymorphisms are risk factors for many developmental, vascular, neurological, and psychiatric disorders in the CNS, including Alzheimer's disease (AD) (Reynolds, 2006). Filamentous lesions containing phosphorylated tau accumulate in AD and other tauopathies (Goedert and Spillantini, 2006). Lower folate levels correlate with higher phosphorylated tau levels in CSF from patients with neurodegenerative disorders (Obeid et al., 2007a). By impairing one-carbon metabolism, folate deficiency severely disturbs normal neuronal homeostasis. It promotes oxidative damage and mitochondrial dysfunction, compromises DNA repair, and ultimately leads to cell death (Duan et al., 2002; Kruman et al., 2002; Ho et al., 2003; Tjiattas et al., 2004). Mechanisms implicated in folate deficiency-evoked neurotoxicity include the following: (1) decreased methionine and S-adenosylmethionine (SAM) synthesis and increased levels of S-adenosylhomocysteine (SAH), resulting in altered SAM/SAH ratio and inhibition of protein, phospholipid, and DNA methylation reactions; and (2) elevation of neurotoxic Hcy and homocysteic acid, causing activation of NMDA and glutamate receptors, increased intracellular calcium, and oxidative stress (Obeid and Herrmann, 2006; Boldyrev and Johnson, 2007).
Decreased SAM/SAH ratio leads to downregulation of protein phosphatase 2A (PP2A) methylation in N2a cells and in mouse brain tissue (Sontag et al., 2007). PP2A catalytic C subunit is methylated on Leu-309 by SAM-dependent leucine carboxyl methyltransferase-1 (LCMT-1) [or PP2A methyltransferase-1 (PPMT1)] (De Baere et al., 1999; Leulliot et al., 2004) and demethylated by protein phosphatase methylesterase (PME-1) (Lee et al., 1996; Ogris et al., 1999). This process modulates the recruitment of specific regulatory B subunits to the (AC) core enzyme, thereby contributing to regulation of PP2A biogenesis and substrate specificity (Janssens et al., 2008). Methylation is required for generation of mammalian ABαC holoenzymes containing the Bα (PPP2R2A or PR55α) subunit (Ogris et al., 1997; Bryant et al., 1999; Tolstykh et al., 2000; Yu et al., 2001; Lee and Pallas, 2007; Longin et al., 2007; Nunbhakdi-Craig et al., 2007), which bind to and dephosphorylate tau (Sontag et al., 1996). Downregulation of LCMT-1 and PP2A methylation occurs in AD (Sontag et al., 2004b) and in hyperhomocysteinemic mice (Sontag et al., 2007) and correlates with enhanced tau phosphorylation. Injection of homocysteine (Zhang et al., 2008) or incubation of neurons with folate antagonists (Yoon et al., 2007) also promote PP2A demethylation and tau phosphorylation. However, underlying molecular mechanisms remain unclear. Here, we investigated how folate deficiency alone impacts the regulation of PP2A methylation in cultured cells and in vivo. We show that LCMT-1 or Bα overexpression, but not PME-1 knockdown, protects cells against folate deprivation-induced accumulation of demethylated PP2A, enhanced tau phosphorylation, and cell death. Dietary folate deficiency in wild-type mice induces brain-region-dependent changes in SAM and SAH levels that are associated with alterations in LCMT-1, Bα, and phosphorylated tau levels. Our results provide new mechanistic insights into the regulation of PP2A methylation and folate-dependent pathways that are critical for neuronal homeostasis.
Materials and Methods
Cell culture and transfection.
Mouse Neuro-2a (N2a) (American Type Culture Collection) were propagated in DMEM (Invitrogen) containing 10% fetal bovine serum (FBS) (HyClone). When indicated, cells were transfected using Metafectene Pro reagent following the instructions of the manufacturer (Biontex Laboratories). N2a cells stably overexpressing hemagglutinin (HA)-tagged LCMT-1 and Myc-tagged PME-1 have been characterized in previous studies (Sontag et al., 2007). N2a overexpressing Bα were obtained after transfection with pcDNA3.1 vector encoding HA-tagged Bα (Nunbhakdi-Craig et al., 2002). Specific Bα knockdown was performed in N2a cells using short hairpin-Bα (sh-Bα) as described previously (Nunbhakdi-Craig et al., 2007). The small interfering RNA (siRNA) targets for mouse PME-1 (GenBank accession number NM_028292) and mouse LCMT-1 (GenBank accession number NM_025304) were obtained from Applied Biosystems/Ambion (Silencer Pre-designed siRNA) and Qiagen (HP Genomewide siRNA) (supplemental Table 1, available at www.jneurosci.org as supplemental material). si2-PME-1 and si4-LCMT-1 were the most efficient of the siRNAs tested in these experiments and were later referred to as “si-PME-1” and “si-LCMT-1” in all following studies. Mismatch siRNAs for si2-PME-1 and si4-LCMT-1 were designed based on published guidelines and synthesized (Qiagen). No significant homology was found with any gene when sequences of both mismatch controls were submitted to Washington University BLAST (basic local alignment search tool). In addition to mismatch siRNAs for si2-PME-1 and si4-LCMT-1, a nonspecific negative control from Ambion (Silencer Negative Control #1) was used as control in each of the silencing experiments performed throughout this study. For LCMT-1 and PME-1 silencing experiments, duplicate sets of N2a cells were plated in regular cell culture medium (50,000 cells per well; 24-multiwell plates) and transfected the next day with the indicated siRNA. Preliminary experiments showed that, under our experimental conditions, maximal and specific protein knockdown was obtained with 20 nm siRNA 3–4 d after transfection and persisted for up to 6 d. Thus, folate starvation studies were started in cells 3–4 d after transfection. Similar results were obtained when cells were either mock transfected or transfected with any of the siRNA controls. Efficiency of protein knockdown was systematically verified by Western blotting at the end of each experiment performed in this study. In some experiments, cells were transfected with corresponding cyanine 3-labeled siRNAs and analyzed by fluorescence-activated cell sorting (FACS) to verify transfection efficiency and assess cell death.
Cell treatment.
For folate starvation studies, exponentially growing cells were trypsinized and plated in regular cell culture medium. Once attached (after 4–6 h), cells were washed in folate-free RPMI 1640 medium (Invitrogen) and immediately incubated for the indicated time in RPMI 1640 medium containing either 100% folate (∼1 mg/L folate; normal folate “NF” medium), 1% folate (low folate “LF” medium), or 0% folate (folate-deficient “FD” medium) (Invitrogen). Unless otherwise indicated, all incubation media were supplemented with 10% FBS. Note that the amount of folate contributed by the presence of FBS (∼25 μg/L folate) in these media is negligible. To block protein synthesis, cells were first preincubated for 1 h with 10 μg/ml cycloheximide (Sigma) in NF medium and then switched for the indicated time to FD medium containing the same amount of this antibiotic.
Analysis of cell death.
Cells (1 × 106 cells per well) were plated in triplicate in NF medium in multiwell dishes. Once attached, cells were washed in folate-free RPMI 1640 medium and immediately incubated for 24 h in either NF or FD media. To assess cell viability, the cell culture medium was collected and combined with the remaining adherent cells that had been harvested by trypsinization. After centrifugation for 5 min at 500 × g, the cell pellet was resuspended in 300 μl of Dulbecco's PBS (Invitrogen). Cells were stained with 2 μl of propidium iodide (250 μg/ml stock; Immunochemistry Technologies), placed on ice, and immediately analyzed for viability by flow cytometry on a Becton Dickinson FAC Scan.
Analysis of cell morphology.
Cells were plated on poly-l-lysine-coated six-well dishes in NF medium at a density of 30,000 cells per well. Once attached, cells were rinsed with folate-free RPMI 1640 medium and then incubated for 48 h in either NF or LF medium supplemented with 0.1% FBS to promote cell differentiation. Note that we chose to use LF medium because cell death was too significant 48 h after incubation in FD medium plus 0.1% FBS. The cell medium was changed to phenol-free medium just before observation, which resulted in the removal of dead cells and debris. Cells were examined by phase contrast (15× magnification) on a Nikon Eclipse TE2000-U microscope equipped with a digital camera. Images were directly acquired using the MetaMorph software (Molecular Devices) and transferred to Adobe Photoshop CS2 (Adobe Systems) for printing. Western blot analyses of total cell extracts were performed in parallel to verify the efficiency of protein overexpression or silencing.
Animals.
All experiments involving animals were approved by the Institutional Animal Care and Use Committee at Baylor Research Institute. Male C57BL/6J mice (4 weeks of age) were obtained from The Jackson Laboratory and housed in separate cages with a 12 h light/dark cycle. Animals were allowed access to food and water ad libitum. Mice were assigned to one of three dietary treatment groups (six mice in each). These were specially prepared by Harland Teklad based on an amino acid defined composition containing either a normal folate (6.7 mg/kg, NF), low folate (0.2 mg/kg, LF), or folate-deficient (0.0 mg/kg, FD) content. All diets contained succinylsulfathiazole (10 mg/kg) to inhibit the growth of cecal bacteria and prevent absorption of folate from this source. Mice were maintained on the diets for 8 weeks, after which they were killed by asphyxiation with carbon dioxide. Blood was obtained by cardiac puncture, and brain tissue was removed for regional dissection of left and right hemispheres. Tissues were stored at −80°C until time of analysis. Brain regions from the left side were used for SAM and SAH analysis, and brain regions from the right side were processed for Western blot analysis as described previously (Sontag et al., 2007). Total plasma Hcy, plasma folate, and regional brain tissue SAM and SAH levels were determined according to previously published procedures (Sontag et al., 2007).
Gel electrophoresis and Western blotting.
PP2A methylation, tau phosphorylation, and LCMT-1, PME-1, and Bα protein expression levels were determined in cell and tissue extracts as described previously (Sontag et al., 2007). Equivalent amounts of proteins (30 μg) from cellular extracts or equivalent volumes (5–10 μl) of brain homogenates were analyzed in duplicate on 4–12% Bis–Tris gels using the NU-PAGE system (Invitrogen) followed by Western blotting. Blots were first probed with mouse anti-LCMT-1 (1:500) and anti-Bα (1:200) antibodies (Sontag et al., 2004b), washed, and reprobed with rabbit anti-PME-1 (1:5000) (Sontag et al., 2007). Blots were stripped with BlotFresh Western Blot Stripping Reagent (SignaGen Laboratories) and reprobed with either anti-tubulin (1:4000; Sigma) and/or rabbit anti-actin (1:10,000; Cytoskeleton) to verify and normalize for protein loading. To analyze endogenous tau phosphorylation, blots were first probed with either rabbit anti-phospho-tau pSer422 antibody (1:500; BioSource International) or monoclonal “PHF-1” (for paired helical filament) recognizing tau phosphorylated at the Ser396/Ser404 epitope (1:2500; a gift from Dr. Peter Davies, Albert Einstein College of Medicine, New York, NY). Blots were then stripped and reprobed with rabbit anti-tau (1:10,000; rPeptide) to normalize for total tau expression levels. To analyze endogenous PP2A methylation levels, blots were probed with either monoclonal anti-methyl-C or anti-demethyl-C antibodies (Upstate Biotechnology), stripped, and reprobed with anti-C antibodies (1:1000; BD Biosciences) to assess total C expression and with anti-tubulin antibodies for normalization. Immunoreactivity of mouse brain extracts with anti-methyl-C antibodies was quite poor because of low amounts of protein analyzed. As a result, PP2A steady-state methylation levels were quantified using monoclonal anti-demethyl C (1:10,000) antibodies and alkaline treatment following published protocols (Yu et al., 2001; Sontag et al., 2004b). In all cases, protein expression levels were quantified after scanning blots by densitometry using Kodak Image software (Eastman Kodak).
Statistical analysis.
Data were analyzed using the Student's t test. Differences with p values <0.05 were considered statistically significant.
Results
Downregulation of LCMT-1 in folate-starved N2a cells correlates with accumulation of demethylated PP2A, loss of Bα, and enhanced tau phosphorylation
Switching N2a neuroblastoma cells from NF to FD medium was associated with a time-dependent increase in PP2A demethylation (Fig. 1A). Pretreatment of cells with the protein synthesis inhibitor cycloheximide induced a rapid loss of demethylated C (Fig. 1B), indicating the existence of a fast intracellular turnover of demethylated PP2A pools under normal conditions. Most importantly, it abolished folate deprivation-induced increase in demethylated C. Thus, the accumulation of demethylated C in response to folate deficiency results from the buildup of de novo synthesized C subunits in an unmethylated state rather than from cumulative demethylation of preexisting PP2A enzymes. The accumulation of demethylated C also correlated with a loss of Bα in folate-starved cells (Fig. 1C,D), in agreement with previous studies showing that decreasing PP2A methylation below a certain threshold prevents formation of ABαC holoenzymes and eventually leads to Bα degradation (Lee and Pallas, 2007; Longin et al., 2007; Sontag et al., 2007). Interestingly, folate deficiency also induced a time-dependent loss of LCMT-1 without significantly affecting PME-1 expression levels.
Figure 1.

Accumulation of demethylated PP2A, downregulation of LCMT-1 and Bα, and enhanced tau phosphorylation in folate-starved N2a cells. A, Incubation of N2a cells in FD medium induced a progressive accumulation of demethylated C. B, This effect was blocked when cells were treated with the protein synthesis inhibitor cycloheximide. Data in A and B are mean ± SD; n = 4. C, Folate starvation induced time-dependent changes in LCMT-1 and Bα expression without significantly affecting PME-1 levels. D, Compared with control cells cultured in NF medium, there was a downregulation of LCMT-1 and Bα expression in cells incubated for 4 h in FD medium that correlated with increased levels of phosphorylated tau (P-tau) at the PHF-1 and Ser422 epitopes. Note that endogenous tau isoforms migrate as several bands.
Folate deficiency rapidly promotes oxidative and metabolic stress and DNA damage, resulting in neurodegeneration (Kruman et al., 2002; Ho et al., 2003). Indeed, signs of neurite swelling and retraction were visible 8 h after incubation of N2a cells in FD medium and were followed by progressive cell rounding and death. Because incubation of cells for 4 h in FD medium was sufficient to induce a clear loss of LCMT-1 expression and near-maximal PP2A demethylation and minimize folate deficiency-induced toxicity, additional comparative analyses of protein expression and tau phosphorylation levels were preferentially performed in cells that were incubated for this duration in either NF or FD medium (Fig. 1D). We have reported that enhanced demethylation of PP2A in N2a cells correlates with increased steady-state phosphorylation of tau at many epitopes (Sontag et al., 2007). Not surprisingly, folate deprivation-induced PP2A demethylation was associated with enhanced phosphorylation of endogenous tau at two selected epitopes: (1) the phospho-Ser396/Ser404 epitope recognized by the monoclonal PHF-1 antibody (Greenberg et al., 1992), which is considered a “late marker” of tau pathology because it reacts with mature, phosphorylated tau proteins primarily found in late-stage neurofibrillary tangles in AD; and (2) the phospho-Ser422 epitope, considered an “earlier marker” of tau pathology because anti-phospho-Ser422 antibodies label phosphorylated tau before tangle formation in AD and non-AD tauopathies (Guillozet-Bongaarts et al., 2007).
Overexpression of either LCMT-1 or Bα protects N2a cells against folate deficiency-induced tau phosphorylation and cell death
To gain additional mechanistic insights into the contribution of LCMT-1, PME-1, and ABαC enzymes in neuronal homeostasis and folate metabolic pathways, we next investigated the effects of overexpressing or knocking down LCMT-1, PME-1, and Bα in our experimental cell model. We have shown previously that endogenous PP2A methylation and Bα expression levels are increased by ∼30–44% in N2a cells stably overexpressing LCMT-1 (N2a–LCMT-1) (Sontag et al., 2007), probably as a result of the stabilization of methylated ABαC heterotrimers by the methyltransferase (Tolstykh et al., 2000). Overexpression of LCMT-1 protected N2a cells against folate deprivation-induced downregulation of both LCMT-1 and Bα (Fig. 2A,B). At the same time, relative to control cells, basal PME-1 expression levels were significantly reduced in N2a–LCMT-1 cultured in NF medium and further decreased after cells were subjected to folate starvation. PME-1 downregulation closely correlated with the loss of demethylated PP2A pools in N2a–LCMT-1 cells. Overexpression of LCMT-1 also prevented folate deprivation-induced tau phosphorylation (Fig. 2C). As reported previously in HeLa cells (Lee and Pallas, 2007; Longin et al., 2007), LCMT-1 knockdown induced significant downregulation of PP2A methylation and Bα expression in N2a or N2a–LCMT-1 cells (Fig. 2D) (supplemental Fig. S1, available at www.jneurosci.org as sup-plemental material). Although LCMT-1 knockdown had no major effect on PME-1 expression in cells cultured in NF medium, it promoted the accumulation of PME-1 in folate-starved cells. Next, we investigated how manipulating LCMT-1 expression affects folate deficiency-induced cell toxicity. Potential effects on cell death were assessed 24 h after incubation in FD medium using FACS analysis (Fig. 2E). The morphology of cells was also compared after incubation for 48 h in either NF or LF medium containing low serum concentrations (0.1% FBS) (Fig. 2F,G). These experimental conditions allowed the development of neurites in N2a cells cultured in NF medium (Fig. 2F). As reported previously in HeLa cells (Lee and Pallas, 2007; Longin et al., 2007), persistent LCMT-1 knockdown led to cell death in N2a cells cultured in NF medium. In addition, it accelerated the neurite retraction, cell rounding, and cell death induced by folate starvation in N2a cells. Conversely, overexpression of LCMT-1 dramatically prevented the morphological changes and cell death induced by folate deprivation, and these effects could be abolished by LCMT-1 knockdown.
Figure 2.
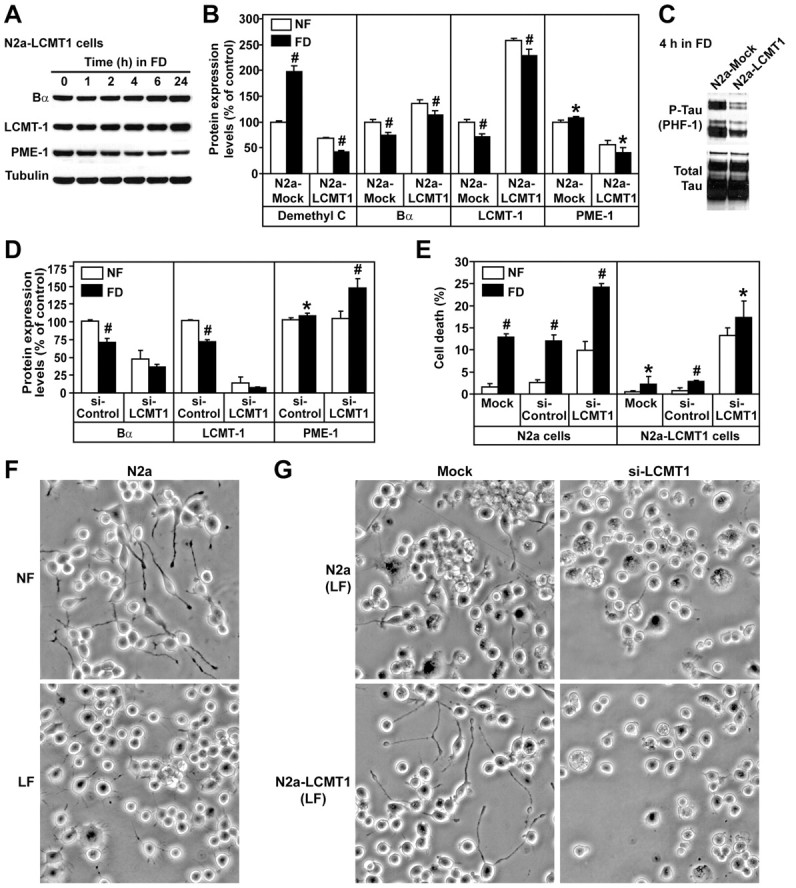
Overexpression of LCMT-1 protects N2a cells against folate deficiency-induced tau phosphorylation and cell death. A, Relative expression levels of Bα, LCMT-1, and PME-1 in N2a cells stably expressing HA-tagged LCMT-1 (N2a–LCMT-1) that were incubated for 0–24 h in FD medium. Note that both endogenous and ectopic LCMT-1 proteins migrated together under the electrophoretic conditions used. B, The amounts of demethylated C, Bα, LCMT-1, and PME-1 were compared in mock-transfected N2a (N2a-Mock) and N2a–LCMT-1 cells 4 h after incubation in NF (white bars) or FD (black bars) medium. Data are expressed as percentage of protein expression levels in control N2a cells incubated in NF medium (mean ± SD; n = 4; FD vs NF, #p < 0.001, *p < 0.05). C, N2a mock-transfected and N2a–LCMT-1 cells were incubated for 4 h in FD medium and analyzed by Western blotting for phosphorylated tau at the PHF-1 epitope. D, N2a cells were transfected with siRNA targeted for LCMT-1 (si-LCMT1) or control siRNA (si-Control). The relative amounts of Bα, LCMT-1, and PME-1 were quantified by Western blotting and densitometry 4 h after incubation of cells in NF or FD medium. Data are expressed as percentage of protein expression levels in control N2a cells incubated in NF medium (mean ± SD; n = 5; FD vs NF, #p < 0.001, *p < 0.05). E, FACS analysis was used to comparatively assess the effect of LCMT-1 overexpression and knockdown on cell survival 24 h after incubation of cells in NF or FD medium (mean ± SD; n = 4; FD vs NF, #p < 0.001, *p < 0.05). F, N2a cell morphology was analyzed by phase contrast 48 h after incubation in NF or LF medium containing 0.1% FBS. G, The effects of LCMT-1 overexpression and knockdown on cell morphology were analyzed by phase contrast 48 h after incubation of cells in LF medium containing 0.1% FBS. The morphology of mock-transfected cells or cells transfected with several distinct control siRNAs was similar to the morphology of untransfected cells (data not shown). Note that equivalent amounts of cells were plated in each condition. Nonviable cells that became detached from the substratum were removed when the culture medium was replaced before cell viewing. For F and G, representative images are shown. Similar results were obtained in at least five separate experiments.
Because changes in LCMT-1 and Bα expression levels were closely correlated in our experiments, we next tested the hypothesis that ABαC holoenzymes play a key role in folate metabolism. As observed in N2a–LCMT-1 cells, overexpression of Bα in N2a cells was associated with a similar downregulation of basal demethylated C and PME-1 expression levels (Fig. 3A–C). Remarkably, it also prevented the downregulation of LCMT-1 but induced an additional loss of PME-1 and demethylated PP2A pools during the course of folate starvation. Bα overexpression promoted tau dephosphorylation (Fig. 3A), whereas Bα knockdown induced tau phosphorylation (Fig. 3D) in N2a cells, in agreement with the finding that ABαC holoenzymes are major tau phosphatases (Sontag et al., 1996). Interestingly, Bα knockdown also caused a downregulation of LCMT-1 and an increase in PME-1 expression in N2a cells cultured in NF medium, and these effects were further intensified in folate-starved cells (Fig. 3E). Bα knockdown promoted cell death in N2a cells cultured in NF medium as reported previously in HeLa cells (Lee and Pallas, 2007) and significantly accelerated cell death in response to folate starvation (Fig. 3F,G). Conversely, as observed in N2a–LCMT-1 cells, cells overexpressing Bα were more resistant to folate deficiency-induced toxic effects.
Figure 3.
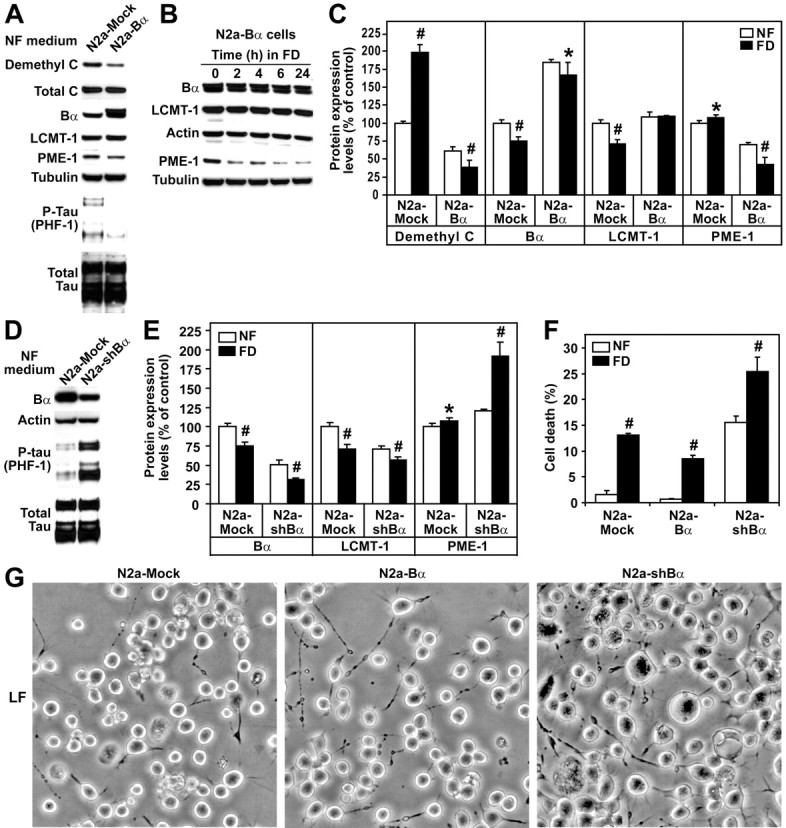
Overexpression of Bα protects N2a cells against folate deficiency-induced tau phosphorylation and cell death. A, N2a mock-transfected (N2A-Mock) and N2a cells expressing HA-tagged Bα (N2a-Bα) cultured in NF medium were analyzed by Western blotting for demethylated C, LCMT-1, PME-1, and PHF-1-phosphorylated tau levels. Note that HA-tagged Bα migrated slower than endogenous Bα. B, Western blot analysis of total cell extracts from N2a–Bα cells incubated for 0–24 h in FD medium. C, Demethylated C, Bα, LCMT-1, and PME-1 expression levels were compared in N2a mock-transfected and N2a–Bα cells 4 h after incubation in NF or FD medium (mean ± SD; n = 4; FD vs NF, #p < 0.001, *p < 0.05). D, N2a cells were mock transfected (N2a-Mock) or transfected with sh-Bα (N2a-shBα) to induce selective Bα knockdown (Nunbhakdi-Craig et al., 2007). Representative immunoblots show that Bα knockdown was associated with increased tau phosphorylation in cells cultured in NF medium. E, Bα, LCMT-1, and PME-1 protein expression levels were quantified in N2a cells mock transfected and N2a cells transfected with sh-Bα 4 h after incubation in NF or FD medium (mean ± SD; n = 4; FD vs NF, #p < 0.001, *p < 0.05). F, Effects of Bα overexpression or knockdown on cell death in N2a cells incubated for 24 h in NF or FD medium (mean ± SD; n = 4; FD vs NF, #p < 0.001). G, Representative images showing the effects of Bα overexpression or knockdown on the morphology of N2a cells incubated for 48 h in LF medium containing 0.1% FBS.
PME-1 knockdown cannot rescue the effects of folate deficiency
Because overexpressed LCMT-1 and Bα both promoted a decrease of PME-1 levels in NF and FD medium, we tested the hypothesis that decreasing PME-1 expression is a key protective factor under conditions of folate deprivation. We have shown that PP2A methylation levels are reduced by ∼47–64% in N2a cells overexpressing PME-1 (N2a–PME-1 cells) (Sontag et al., 2007). Notably, overexpression of PME-1 not only induced the accumulation of demethylated C but also protected folate-starved cells against the dwindling of demethylated C pools induced by cycloheximide (Fig. 4A), in agreement with the finding that PME-1 interacts with and stabilizes inactive PP2A (Ogris et al., 1999; Longin et al., 2008; Xing et al., 2008). Relative to control cells, the accumulation of demethylated C coincided with a reduction in LCMT-1 and Bα amounts in N2a–PME-1 cells cultured in NF medium. These effects were further emphasized after incubation of N2a–PME-1 cells in FD medium (Fig. 4B,C). Bα downregulation likely mediated the increase in tau phosphorylation observed in N2a–PME-1 cells cultured in either NF (Sontag et al., 2007) or FD (Fig. 4D) medium. The high level of necrotic (Fig. 4F), enlarged, and vacuolized (Fig. 4G) cells in folate-starved N2a–PME-1 cells was reminiscent of the phenotype of N2a cells after Bα silencing (Fig. 3G). However, compared with controls, knockdown of PME-1 in either N2a or N2a–PME-1 cells cultured in NF medium increased basal methylated C levels without significantly altering Bα or LCMT-1 expression levels (Fig. 4E) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) or cell survival (Fig. 4F). Above all, it was unable to prevent the loss of Bα and LCMT-1 (Fig. 4E) and rescue N2a or N2a–PME-1 cells from death (Fig. 4F,G) in response to folate starvation.
Figure 4.

Effects of PME-1 overexpression and knockdown in N2a cells cultured in NF and FD medium. A, Compared with control N2a cells (Fig. 1B), demethylated C levels remain constant in N2a–PME-1 cells after pretreatment with cycloheximide and incubation in FD medium. B, Western blot analysis of total cell extracts from N2a–PME-1 cells incubated for 0–24 h in FD medium. Note that myc-tagged PME-1 migrated slower than endogenous PME-1. C, Demethylated C, Bα, LCMT-1, and PME-1 expression levels were compared in N2a mock-transfected and N2a–PME-1 cells 4 h after incubation in NF or FD medium (mean ± SD; n = 5; FD vs NF, #p < 0.001, *p < 0.05). D, Representative immunoblot showing that overexpressed PME-1 further enhances tau phosphorylation induced by incubation of cells for 4 h in FD medium. E, Relative expression levels of Bα, LCMT-1, and PME-1 in N2a cells transfected with siRNA targeted for PME-1 (si-PME1) or control siRNA (si-Control), 4 h after incubation in NF or FD medium (mean ± SD; n = 5; FD vs NF, #p < 0.001). F, Effects of PME-1 overexpression or knockdown on cell death 24 h after incubation in NF or FD medium (mean ± SD; n = 4; FD vs NF, #p < 0.001). G, Representative images illustrating the effects of PME-1 overexpression and knockdown on cell morphology 48 h after incubation of cells in LF medium containing 0.1% FBS.
Altogether, results from our cellular studies suggest that, by stabilizing Bα-containing PP2A holoenzymes, LCMT-1 plays a critical role in folate-dependent pathways that regulate tau phosphorylation and survival.
Experimental dietary folate deficiency in mice induces brain-region-specific downregulation of the SAM/SAH ratio, LCMT-1, and Bα
To further test our hypothesis in vivo, we analyzed brain homogenates from 1-month-old wild-type mice that had been fed for 8 weeks with either an NF, LF, or FD diet. As expected, serum folate levels were significantly decreased, whereas total plasma Hcy levels were increased in mice on the LF and FD diets (Fig. 5A). SAM levels were not significantly affected by folate deprivation in all the brain regions examined, except for the striatum, in which they were decreased (Fig. 5B). In contrast, SAH levels were increased in response to the FD diet in all regions, except in the striatum. Relative to mice fed on the control NF diet, the SAM/SAH ratio was decreased in the frontal cortex, cortex, midbrain, and cerebellum of mice fed on the LF and FD diets, but the extent of this decrease was brain region specific. In stark contrast to other regions, the basal SAM/SAH ratio was very low in the striatum and essentially remained unaffected by dietary folate fluctuations. Reductions in SAM/SAH ratio were associated with a concomitant decrease in steady-state methylated PP2A levels in susceptible brain regions (Fig. 6A). Relative to control mice on the NF diet, PP2A demethylation was especially pronounced in the cortex and cerebellum from mice fed on the LF and FD diets. Dietary folate deficiency did not affect much PP2A methylation in the striatum compared with other regions. As observed in cultured cells, acute PP2A demethylation was accompanied by Bα downregulation (Fig. 6B,C). Moreover, marked decrease in the SAM/SAH ratio promoted LCMT-1 downregulation. Slight variations in PME-1 expression levels were observed in all brain region homogenates from mice fed on the LF or FD diets relative to control mice. Notably, the extent of the drop in the LCMT-1/PME-1 ratio correlated best with the extent of Bα downregulation in each specific brain region. Last, statistically significant increases in regional brain tau phosphorylation at the PHF-1 and Ser422 epitopes (Fig. 7) were only observed under conditions in which there was a prominent decrease in Bα expression levels.
Figure 5.

Effects of dietary folate deficiency on plasma and regional brain metabolites in wild-type mice. A, B, Groups of 1-month-old C57BL/6J wild-type male mice were fed for 8 weeks on amino acid-defined diets that contain either normal folate levels (NF; 6.7 mg/kg folic acid; white bars) or low folate levels (LF; 0.2 mg/kg folic acid; gray bars) or were folate deficient (FD; 0 mg/kg folic acid; black bars). Data are mean ± SD, n = 6 mice per group; #p < 0.001; *p < 0.05. A, Total plasma Hcy and plasma folate. B, Regional brain tissue SAM and SAH levels and SAM/SAH ratio. “Cortex” is defined as the cortical region minus the frontal cortex.
Figure 6.

Dietary folate deficiency differentially affects PP2A methylation, LCMT-1, PME-1, and Bα expression levels in mouse brain regions. A, B, Equivalent aliquots of total homogenates from the striatum (ST), frontal cortex (FC), cortex (CX), midbrain (MB), and cerebellum (CB) from the same mice described in Figure 5 were analyzed by gel electrophoresis and Western blotting followed by densitometry. In all experiments, brain homogenates were analyzed simultaneously to allow accurate comparison of protein expression levels across mouse groups. All values were determined from three separate experiments. Data are mean ± SD; n = 6 mice per group; #p < 0.001; *p < 0.05. A, Methylated C levels. B, Representative blots. Note that the panels were assembled from the same exposed autoradiograph. C, Bα, LCMT-1, and PME-1 protein expression levels and LCMT-1/PME-1 protein ratio.
Figure 7.

Brain-region-specific effects of dietary folate deficiency on tau phosphorylation. A, B, The same mouse brain homogenates described in Figure 6 were analyzed for tau phosphorylated at the Ser422 and PHF-1 epitopes after normalization for total tau levels. A, Representative immunoblots for the striatum (ST) and cerebellum (CB). B, Relative phosphorylated tau expression levels in each brain region. Data are mean ± SD; n = 6 mice per group; #p < 0.001; *p < 0.05.
These in vivo data underscore the importance of a vital link between brain-region-sensitive, folate-dependent LCMT-1-mediated methylation pathways that critically regulate the expression of Bα-containing PP2A holoenzymes and tau phosphorylation.
Discussion
Pathological conditions associated with abnormal folate status range from genetic to acquired disorders, highlighting the importance of this vitamin in key physiological processes in the CNS (Djukic, 2007; Obeid et al., 2007b). Because regulation of folate metabolism is highly complex, CNS folate deficiency or impaired availability can occur in the settings of normal or decreased systemic folate levels. Both cause altered methyltransferase-catalyzed reactions, leading to defects in amino acid metabolism, phospholipid and neurotransmitter biosynthesis, DNA repair, and gene expression. In cultured cells, folate deficiency inhibits phosphatase activity (Chan et al., 2008), and folate antagonists induce PP2A demethylation (Yoon et al., 2007). Methylation differentially modulates the affinity of PP2A core enzyme for specific regulatory subunits and is essential for ABαC formation (Janssens et al., 2008). However, the regulatory mechanisms underlying the interplay between LCMT-1, PME-1, and PP2A and their physiological significance for neuronal homeostasis remain essentially unknown. Using cultured neuroblastoma cells, we show that the major pathway by which folate deficiency induces tau phosphorylation and cell death involves downregulation of LCMT-1 and subsequent loss of ABαC. Our experiments indicate that folate deficiency does not demethylate preexisting PP2A holoenzymes, in agreement with previous in vitro studies suggesting that binding of B subunits to the methylated core enzyme prevents demethylation by PME-1 (Tolstykh et al., 2000). Rather, folate deprivation induced the de novo accumulation of PP2A enzymes in an unmethylated state. This is in line with previous studies of PP2A biogenesis proposing that ABαC holoenzyme assembly requires preactivation of inactive PP2A by PP2A phosphatase activator (PTPA) and sequential methylation by LCMT-1 (Fellner et al., 2003; Hombauer et al., 2007). Our data suggest that folate starvation precludes the methylation of newly synthesized PP2A enzymes by (1) inhibiting LCMT-1 activity toward PP2A as a result of decreased SAM/SAH ratio and (2) inducing LCMT-1 downregulation through mechanisms that remain to be elucidated. As dwindling pools of methylated C became available for Bα binding, Bα subunits that are unstable as monomers become targeted for degradation (Janssens et al., 2008). Accordingly, the sizeable accumulation of demethylated C was associated with Bα downregulation in cellular and brain tissue extracts. Ultimately, Bα downregulation led to N2a cell death, as reported in HeLa cells (Lee and Pallas, 2007). Knockdown of LCMT-1 in N2a cells substantially reduced PP2A methylation, leading to Bα downregulation and cell death, as reported in HeLa and COS7 cells (Lee and Pallas, 2007; Longin et al., 2007). Conversely, methylation likely promotes ABαC stabilization (Tolstykh et al., 2000), and methylated C and Bα expression levels increased in N2a–LCMT-1 cells. Downregulation of demethylated C pools was observed in both N2a–LCMT-1 and N2a–Bα cells. Thus, shifting intracellular PP2A composition toward preferential enrichment in methylated ABαC isoforms protects PP2A against demethylation.
Remarkably, overexpression of either LCMT-1 or Bα was sufficient to protect cells against folate deficiency-mediated tau phosphorylation and cell death. In this context, it is noteworthy that, in yeast, overexpression of the Bα homolog cdc55 reverses at least one phenotype induced by deletion of the LCMT-1 homolog PPM1 (Wu et al., 2000). It is well established that LCMT-1-mediated methylation regulates ABαC formation. Our data provide surprising new evidence for the influence of Bα bioavailability on LCMT-1 regulation. Bα overexpression prevented the loss of LCMT-1 induced by folate starvation, and Bα knockdown promoted LCMT-1 downregulation. Thus, ABαC holoenzymes likely form a close complex with or stabilize LCMT-1 enzymes. Indeed, whereas demethylated C was enriched with PME-1 in the nucleus as observed previously in HeLa cells (Longin et al., 2008), methylated ABαC and LCMT-1 colocalized in the cytosol and other subcellular structures of N2a cells (data not shown). The differential spatial regulation of PME-1/demethylated and LCMT-1/methylated PP2A pools may explain why silencing PME-1 is not equivalent to overexpressing LCMT-1 in our cellular assays.
Significantly, overexpression of either LCMT-1 or Bα induced the downregulation of demethylated C and PME-1 in N2a cells cultured in NF medium, an effect that was further accentuated in FD medium. Conversely, PME-1 accumulated in folate-starved cells after LCMT-1 or Bα knockdown. PME-1 induces PP2A inactivation and demethylation (Xing et al., 2008). It forms a stable complex with and sequesters inactive PP2A (Ogris et al., 1999; Longin et al., 2004; Hombauer et al., 2007). Our data further point to a role for PME-1 in regulating PP2A turnover and protecting demethylated C pools against degradation. They also suggest that endogenous PME-1 expression levels become readjusted in response to detrimental fluctuations in demethylated C concentrations.
There is increasing evidence linking alterations in one-carbon metabolism with PP2A deregulation and tau phosphorylation. Downregulation of LCMT-1 and PP2A methylation correlate with enhanced tau phosphorylation in hyperhomocysteinemic mice (Sontag et al., 2007). Injection of Hcy into rat cerebral ventricles induces C subunit downregulation and tau phosphorylation (Luo et al., 2007). Vena caudalis Hcy injection for 2 weeks inactivates PP2A and increases PME-1 expression and tau phosphorylation in the hippocampus without affecting LCMT-1 expression (Zhang et al., 2008). In cultured cells, folate deficiency promotes tau phosphorylation by increasing Hcy levels and inhibiting Ser/Thr phosphatase activity (Chan et al., 2008). We observed that folate deficiency-mediated tau phosphorylation and toxicity were associated with LCMT-1 and Bα downregulation, but these effects could not be rescued by decreasing intracellular PME-1 levels. Although we cannot exclude the possibility that PME-1 expression and activity are regulated by Hcy, our results strongly suggest that folate deficiency regulates PP2A methylation and tau phosphorylation via a pathway primarily involving SAH-induced LCMT-1 inhibition and/or downregulation. Because Hcy likely becomes rapidly oxidized into neurotoxic homocysteic acid after injection, additional mechanisms such as oxidative stress may contribute to the deregulation of PME-1, PP2A, and tau in the studies by Zhang et al. (2008).
Tau phosphorylation increases in mice fed a diet combining folate and vitamin E deficiency with iron supplementation (Chan and Shea, 2006), but the contribution of vitamin deprivation and oxidative stress in tau changes cannot be distinguished. Here, we demonstrate that dietary folate deficiency alone affects the regulation of methylation metabolites, PP2A, and tau in mouse brain, in a region-specific manner. Under our experimental conditions, the extent of PP2A demethylation in response to dietary changes was the most significant in the cortex and cerebellum. It was associated with a significant decrease of LCMT-1 and Bα expression. In agreement with the observation that ABαC is a predominant tau phosphatase (Sontag et al., 1996), enhanced tau phosphorylation only occurred under conditions in which Bα expression levels decreased below a certain threshold relative to basal levels. Compared with other regions examined, folate deprivation did not significantly affect the SAM/SAH ratio and PP2A methylation in the striatum. The high demand for methylation reactions by catecholamine-O-methyltransferase in dopaminergic terminals probably differentially affects striatal SAM metabolism (Zhu, 2002). Compared with other brain areas, the striatum is also highly enriched in Bα mRNA and proteins (Strack et al., 1998), which may protect neurons against folate starvation-induced PP2A demethylation. Although LF and FD diets decreased the SAM/SAH ratio and PP2A methylation levels in the midbrain, these effects were relatively modest compared with the cortical and cerebellar regions. In response to the FD diet, there was no marked loss of LCMT-1 and Bα and little changes in tau phosphorylation in the midbrain. Basal methylated PP2A levels were lower in the midbrain than in other regions, which may render PP2A pools in this region less sensitive to SAH fluctuations. It is likely that compensatory or other mechanisms, e.g., increase in LCMT-1 and/or Bα expression and activation of signaling pathways leading to inhibition of tau kinases, could be triggered in response to folate deficiency and protect selective neurons against the loss of ABαC and/or tau phosphorylation. For instance, tau phosphorylation is controlled by the balance between PP2A and glycogen-synthase kinase 3β activities (Meske et al., 2008). These protective mechanisms could operate in a time-dependent manner, and specific brain regions and neuronal cell populations may be able to cope better than others with low folate-induced toxicity.
In conclusion, our studies suggest that LCMT-1 is a critical intermediate of the role of folate in the CNS. Notably, the extent of the downregulation of LCMT-1 and Bα correlates with the severity of phosphorylated tau pathology in AD (Sontag et al., 2004a,b). Our cellular and mouse data reinforce the strong connection between LCMT-1-dependent ABαC holoenzyme formation/stabilization and tau regulation. Thus, offsetting the neuronal loss of LCMT-1 or Bα could be a valuable therapeutic approach for tauopathies and folate-dependent CNS disorders.
Footnotes
This work was supported by National Institutes of Health (NIH)/National Institute on Aging Grant AG18883 (E.S.) and NIH/National Center for Complementary and Alternative Medicine Grant AT002311 (T.B.). We thank Dr. Peter Davies (Albert Einstein College of Medicine, New York, NY) for providing PHF-1 antibody and Dr. Egon Ogris (Medical University of Vienna, Vienna, Austria) for providing antibodies against Bα and PME-1.
References
- Boldyrev AA, Johnson P. Homocysteine and its derivatives as possible modulators of neuronal and non-neuronal cell glutamate receptors in Alzheimer's disease. J Alzheimers Dis. 2007;11:219–228. doi: 10.3233/jad-2007-11209. [DOI] [PubMed] [Google Scholar]
- Bryant JC, Westphal RS, Wadzinski BE. Methylated C-terminal leucine residue of PP2A catalytic subunit is important for binding of regulatory Balpha subunit. Biochem J. 1999;339:241–246. [PMC free article] [PubMed] [Google Scholar]
- Chan A, Shea TB. Dietary and genetically-induced oxidative stress alter tau phosphorylation: influence of folate and apolipoprotein E deficiency. J Alzheimers Dis. 2006;9:399–405. doi: 10.3233/jad-2006-9405. [DOI] [PubMed] [Google Scholar]
- Chan AY, Alsaraby A, Shea TB. Folate deprivation increases tau phosphorylation by homocysteine-induced calcium influx and by inhibition of phosphatase activity: alleviation by S-adenosyl methionine. Brain Res. 2008;1199:133–137. doi: 10.1016/j.brainres.2008.01.008. [DOI] [PubMed] [Google Scholar]
- De Baere I, Derua R, Janssens V, Van Hoof C, Waelkens E, Merlevede W, Goris J. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry. 1999;38:16539–16547. doi: 10.1021/bi991646a. [DOI] [PubMed] [Google Scholar]
- Djukic A. Folate-responsive neurologic diseases. Pediatr Neurol. 2007;37:387–397. doi: 10.1016/j.pediatrneurol.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Duan W, Ladenheim B, Cutler RG, Kruman II, Cadet JL, Mattson MP. Dietary folate deficiency and elevated homocysteine levels endanger dopaminergic neurons in models of Parkinson's disease. J Neurochem. 2002;80:101–110. doi: 10.1046/j.0022-3042.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- Fellner T, Lackner DH, Hombauer H, Piribauer P, Mudrak I, Zaragoza K, Juno C, Ogris E. A novel and essential mechanism determining specificity and activity of protein phosphatase 2A (PP2A) in vivo. Genes Dev. 2003;17:2138–2150. doi: 10.1101/gad.259903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friso S, Choi SW. Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005;6:37–46. doi: 10.2174/1389200052997339. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- Guillozet-Bongaarts AL, Glajch KE, Libson EG, Cahill ME, Bigio E, Berry RW, Binder LI. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol. 2007;113:513–520. doi: 10.1007/s00401-007-0209-6. [DOI] [PubMed] [Google Scholar]
- Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, Rogers E. Folate deprivation induces neurodegeneration: roles of oxidative stress and increased homocysteine. Neurobiol Dis. 2003;14:32–42. doi: 10.1016/s0969-9961(03)00070-6. [DOI] [PubMed] [Google Scholar]
- Hombauer H, Weismann D, Mudrak I, Stanzel C, Fellner T, Lackner DH, Ogris E. Generation of active protein phosphatase 2A is coupled to holoenzyme assembly. PLoS Biol. 2007;5:e155. doi: 10.1371/journal.pbio.0050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends Biochem Sci. 2008;33:113–121. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. J Neurosci. 2002;22:1752–1762. doi: 10.1523/JNEUROSCI.22-05-01752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Chen Y, Tolstykh T, Stock J. A specific protein carboxyl methylesterase that demethylates phosphoprotein phosphatase 2A in bovine brain. Proc Natl Acad Sci U S A. 1996;93:6043–6047. doi: 10.1073/pnas.93.12.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Pallas DC. Leucine carboxyl methyltransferase-1 is necessary for normal progression through mitosis in mammalian cells. J Biol Chem. 2007;282:30974–30984. doi: 10.1074/jbc.M704861200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot N, Quevillon-Cheruel S, Sorel I, de La Sierra-Gallay IL, Collinet B, Graille M, Blondeau K, Bettache N, Poupon A, Janin J, van Tilbeurgh H. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J Biol Chem. 2004;279:8351–8358. doi: 10.1074/jbc.M311484200. [DOI] [PubMed] [Google Scholar]
- Longin S, Jordens J, Martens E, Stevens I, Janssens V, Rondelez E, De Baere I, Derua R, Waelkens E, Goris J, Van Hoof C. An inactive protein phosphatase 2A population is associated with methylesterase and can be re-activated by the phosphotyrosyl phosphatase activator. Biochem J. 2004;380:111–119. doi: 10.1042/BJ20031643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longin S, Zwaenepoel K, Louis JV, Dilworth S, Goris J, Janssens V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic subunit. J Biol Chem. 2007;282:26971–26980. doi: 10.1074/jbc.M704059200. [DOI] [PubMed] [Google Scholar]
- Longin S, Zwaenepoel K, Martens E, Louis JV, Rondelez E, Goris J, Janssens V. Spatial control of protein phosphatase 2A (de)methylation. Exp Cell Res. 2008;314:68–81. doi: 10.1016/j.yexcr.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Luo Y, Zhou X, Yang X, Wang J. Homocysteine induces tau hyperphosphorylation in rats. Neuroreport. 2007;18:2005–2008. doi: 10.1097/WNR.0b013e3282f29100. [DOI] [PubMed] [Google Scholar]
- Meske V, Albert F, Ohm TG. Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3-dependent phosphorylation of Tau. J Biol Chem. 2008;283:100–109. doi: 10.1074/jbc.M704292200. [DOI] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Machleidt T, Ogris E, Bellotto D, White CL, 3rd, Sontag E. Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction complex. J Cell Biol. 2002;158:967–978. doi: 10.1083/jcb.200206114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunbhakdi-Craig V, Schuechner S, Sontag JM, Montgomery L, Pallas DC, Juno C, Mudrak I, Ogris E, Sontag E. Expression of protein phosphatase 2A mutants and silencing of the regulatory Balpha subunit induce a selective loss of acetylated and detyrosinated microtubules. J Neurochem. 2007;101:959–971. doi: 10.1111/j.1471-4159.2007.04503.x. [DOI] [PubMed] [Google Scholar]
- Obeid R, Herrmann W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006;580:2994–3005. doi: 10.1016/j.febslet.2006.04.088. [DOI] [PubMed] [Google Scholar]
- Obeid R, Kasoha M, Knapp JP, Kostopoulos P, Becker G, Fassbender K, Herrmann W. Folate and methylation status in relation to phosphorylated tau protein(181P) and β-amyloid(1–42) in cerebrospinal fluid. Clin Chem. 2007a;53:1129–1136. doi: 10.1373/clinchem.2006.085241. [DOI] [PubMed] [Google Scholar]
- Obeid R, McCaddon A, Herrmann W. The role of hyperhomocysteinemia and B-vitamin deficiency in neurological and psychiatric diseases. Clin Chem Lab Med. 2007b;45:1590–1606. doi: 10.1515/CCLM.2007.356. [DOI] [PubMed] [Google Scholar]
- Ogris E, Gibson DM, Pallas DC. Protein phosphatase 2A subunit assembly: the catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene. 1997;15:911–917. doi: 10.1038/sj.onc.1201259. [DOI] [PubMed] [Google Scholar]
- Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem. 1999;274:14382–14391. doi: 10.1074/jbc.274.20.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006;5:949–960. doi: 10.1016/S1474-4422(06)70598-1. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–1207. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL., 3rd Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004a;63:287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL., 3rd Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004b;63:1080–1091. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, Dayal S, Lentz SR, Arning E, Bottiglieri T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci. 2007;27:2751–2759. doi: 10.1523/JNEUROSCI.3316-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Zaucha JA, Ebner FF, Colbran RJ, Wadzinski BE. Brain protein phosphatase 2A: developmental regulation and distinct cellular and subcellular localization by B subunits. J Comp Neurol. 1998;392:515–527. [PubMed] [Google Scholar]
- Tjiattas L, Ortiz DO, Dhivant S, Mitton K, Rogers E, Shea TB. Folate deficiency and homocysteine induce toxicity in cultured dorsal root ganglion neurons via cytosolic calcium accumulation. Aging Cell. 2004;3:71–76. doi: 10.1111/j.1474-9728.2004.00086.x. [DOI] [PubMed] [Google Scholar]
- Tolstykh T, Lee J, Vafai S, Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000;19:5682–5691. doi: 10.1093/emboj/19.21.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Tolstykh T, Lee J, Boyd K, Stock JB, Broach JR. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J. 2000;19:5672–5681. doi: 10.1093/emboj/19.21.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133:154–163. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Choi HI, Choi JE, Sul CA, Choi JM, Kim DH. Methotrexate decreases PP2A methylation and increases tau phosphorylation in neuron. Biochem Biophys Res Commun. 2007;363:811–816. doi: 10.1016/j.bbrc.2007.09.060. [DOI] [PubMed] [Google Scholar]
- Yu XX, Du X, Moreno CS, Green RE, Ogris E, Feng Q, Chou L, McQuoid MJ, Pallas DC. Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol Biol Cell. 2001;12:185–199. doi: 10.1091/mbc.12.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CE, Tian Q, Wei W, Peng JH, Liu GP, Zhou XW, Wang Q, Wang DW, Wang JZ. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus. Neurobiol Aging. 2008;29:1654–1665. doi: 10.1016/j.neurobiolaging.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Zhu BT. Catechol-O-methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr Drug Metab. 2002;3:321–349. doi: 10.2174/1389200023337586. [DOI] [PubMed] [Google Scholar]