

The crystal structure of an ATP-dependent DNA ligase from A. fulgidus has been determined. It resembles, but shows some variations from, the closed conformation of DNA ligase from P. furiosus.
Keywords: ATP-dependent DNA ligases, Archaeoglobus fulgidus, Af_0623
Abstract
DNA ligases join the breaks in double-stranded DNA by catalyzing the formation of a phosphodiester bond between adjacent 3′-hydroxyl and 5′-phosphate termini. They fall into two classes that require either ATP or NAD+ as the source of an AMP group that is covalently attached to a strictly conserved lysine. Conformational flexibility is essential for the function of multi-domain DNA ligases because they must undergo large conformational changes involving domain rearrangements during the course of the reaction. In the absence of the nicked DNA substrate, both open and closed conformations have been observed for the ATP-dependent DNA ligases from Sulfolobus solfataricus and Pyrococcus furiosus. Here, the crystal structure of an ATP-dependent DNA ligase from Archaeoglobus fulgidus has been determined in the DNA-unbound unadenylated state. It resembles the closed conformation of P. furiosus DNA ligase but was even more closed, thus enhancing our understanding of the conformational variability of these enzymes.
1. Introduction
DNA ligases play an essential role in joining the breaks in double-stranded DNA, a crucial step in the replication and repair of DNA and in genetic recombination, by catalyzing the formation of a phosphodiester bond between adjacent 3′-hydroxyl and 5′-phosphate termini (Lehman, 1974 ▶; Lindahl & Barnes, 1992 ▶; Tomkinson & Levin, 1997 ▶). They fall into two groups depending on the cofactor specificity. One group of DNA ligases depend on ATP and are found in eukarya, viruses, archaea, bacteria and bacteriophages. The other group are NAD+-dependent and are present in bacteria and in some eukaryotic viruses. Apart from the difference in cofactor requirement, the reactions catalyzed by these two classes of DNA ligase are identical (Lehman, 1974 ▶; Tomkinson & Levin, 1997 ▶). In the first step, an AMP moiety derived from the cofactor is covalently attached to a conserved lysine residue within the KxDG motif. In the second step, the AMP moiety is transferred from the adenylated enzyme intermediate to the free 5′-phosphoryl group at a nicked site of duplex DNA. In the final step, the AMP group is released from the adenylated DNA intermediate as the phosphodiester bond is formed.
The crystal structures of two ATP-dependent archaeal DNA ligases from Pyrococcus furiosus (Nishida et al., 2006 ▶) and Sulfolobus solfataricus (Pascal et al., 2006 ▶) revealed that they consist of three domains: a DNA-binding domain, an adenylation domain and an OB-fold domain. Despite the structural similarity of each domain, these archaeal DNA ligases, both in the absence of DNA, showed strikingly different domain arrangements. P. furiosus DNA ligase adopted a closed conformation (Nishida et al., 2006 ▶), whereas S. solfataricus DNA ligase showed an extended highly open conformation (Pascal et al., 2006 ▶). A partially open conformation was also observed in the structure of N-terminally truncated human DNA ligase I, the three domains of which completely encircle the bound DNA substrate (Pascal et al., 2004 ▶). These results demonstrate a high degree of conformational flexibility of these multi-domain DNA ligases. These structures support the model involving large conformational changes associated with domain rearrangements which has been proposed for the action of a multi-domain NAD+-dependent DNA ligase (Lee et al., 2000 ▶). In order to provide further structural information on the conformational variability of archaeal DNA ligases, we have overexpressed, crystallized and determined the crystal structure of an ATP-dependent DNA ligase from Archaeoglobus fulgidus, which shows 49% and 37% sequence identity to the DNA ligases from P. furiosus and S. solfataricus, respectively. There is also a significant level (26%) of sequence identity between A. fulgidus DNA ligase and the corresponding fragment of human DNA ligase I. Our structure of A. fulgidus DNA ligase reveals that it comprises three domains like other archeal DNA ligases and adopts a more closed conformation than that of P. furiosus DNA ligase. Unexpectedly, the arrangement of the two subdomains of the DNA-binding domain in A. fulgidus DNA ligase is more similar to that of the DNA-bound human DNA ligase I in the partially open conformation than to the DNA-unbound P. furiosus DNA ligase in the closed conformation.
2. Materials and methods
2.1. Cloning, protein expression and purification
The gene encoding ATP-dependent DNA ligase from A. fulgidus was cloned into the expression vector pET-28b(+) (Novagen), adding a hexahistidine-containing 20-residue fusion tag to the N-terminus of the recombinant enzyme. The recombinant protein was overexpressed in Escherichia coli Rosetta2(DE3)pLysS cells using Terrific Broth culture medium. Protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside and the cells were incubated for an additional 18 h at 298 K following growth to mid-log phase at 310 K. The cells were lysed by sonication in lysis buffer [50 mM Tris–HCl pH 7.9, 500 mM sodium chloride and 10%(v/v) glycerol] containing 50 mM imidazole. The crude lysate was centrifuged at 3800g for 60 min. The supernatant was applied onto a nickel–nitrilotriacetic acid–agarose affinity chromatography column (Qiagen). The protein was eluted with lysis buffer containing 500 mM imidazole and the eluted sample was diluted fivefold with buffer A [10 mM Tris–HCl pH 8.5, 5%(v/v) glycerol and 10 mM β-mercaptoethanol]. The diluted sample was applied onto a Source 15Q ion-exchange column (GE Healthcare Bioscience) which was previously equilibrated with buffer A. The protein was eluted with a linear gradient of 0–1.0 M sodium chloride in buffer A. The next step was size-exclusion chromatography on a HiLoad 16/60 Superdex-200 prep-grade column (GE Healthcare Bioscience), employing an elution buffer consisting of 20 mM Tris–HCl pH 7.2 and 200 mM sodium chloride. Fractions containing A. fulgidus DNA ligase were concentrated to 18 mg ml−1 using an Amicon Ultra-15 centrifugal filter unit (Millipore).
2.2. Crystallization and X-ray data collection
Crystallization was performed at 297 K using the hanging-drop vapour-diffusion method. Each hanging drop was prepared by mixing 2 µl each of protein solution (18 mg ml−1 in a buffer consisting of 20 mM Tris–HCl pH 7.2, 200 mM sodium chloride and 10 mM β-mercaptoethanol) and reservoir solution (100 mM Tris–HCl pH 9.0, 0.4 M sodium dihydrogen phosphate, 1.2 M dipotassium hydrogen phosphate and 10 mM magnesium chloride). Bipyramidal crystals grew to approximate dimensions of 0.1 × 0.1 × 0.2 mm within a few days.
For cryoprotection, 10 µl of the drop solution containing A. fulgidus DNA ligase crystals was dried in air for 10 min prior to flash-cooling in a nitrogen cryostream at 100 K. X-ray diffraction data were collected at 100 K using a Quantum 210 charge-coupled device area-detector system (Area Detector Systems Corporation, Poway, California, USA) at the BL-4A experimental station of the Pohang Light Source, Pohang, Korea. The crystal was rotated through a total of 150° with a 1.0° oscillation range per frame. The raw data were processed and scaled using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶). The diffraction intensity was consistent with space groups P41212 or P43212, with unit-cell parameters a = b = 94.36, c = 197.42 Å. A monomeric molecule of A. fulgidus DNA ligase was present in each asymmetric unit, giving a solvent fraction of 64.6%. The correct space group was determined to be P41212, since the structure could only be solved in this space group.
2.3. Structure determination and refinement
The structure was solved by molecular replacement, using the AMP-bound structure of DNA ligase from P. furiosus (PDB code 2cfm; Nishida et al., 2006 ▶) as a search model. A cross-rotational search followed by a translational search was performed utilizing the program Phaser (Storoni et al., 2004 ▶). Subsequent manual model building was performed using the program Coot (Emsley & Cowtan, 2004 ▶). The model was refined with the program REFMAC (Murshudov et al., 1997 ▶), including bulk-solvent correction. 10% of the data were randomly set aside as test data for the calculation of R free (Brünger, 1992 ▶).
3. Results and discussion
3.1. Model quality and the domain structure
We have determined the crystal structure of A. fulgidus DNA ligase at 2.30 Å resolution (Table 1 ▶). The refined model contains 551 residues in a molecule of the recombinant A. fulgidus DNA ligase, four phosphate ions and 489 water molecules in the asymmetric unit. The missing residues (residues 320–321 and 551–555 of A. fulgidus DNA ligase, together with the first 17 residues of the 20-residue fusion tag at the N-terminus of the recombinant enzyme) were not defined in the electron density. As evaluated by the program PROCHECK (Laskowski et al., 1993 ▶), the refined model has excellent stereochemistry (Table 1 ▶), with the exception of Glu111, the main-chain conformation of which deviates slightly from the allowed regions of the Ramachandran plot. The reason for this is not clear; however, the mean B factor (39.8 Å2) of Glu111 is not much higher than the average over the protein atoms, as its side chain is involved in a salt bridge with the side chain of Arg520 of a neighbouring molecule (with a distance of 2.86 Å between the closest atoms). A. fulgidus DNA ligase consists of three domains (Fig. 1 ▶), a DNA-binding domain (DBD; residues 1–221), an adenylation domain (AdD; residues 222–419) and an OB-fold domain (OBD; residues 420–550), like other archaeal ATP-dependent DNA ligases from P. furiosus and S. solfataricus (Nishida et al., 2006 ▶; Pascal et al., 2006 ▶).
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray wavelength (Å) | 1.0000 |
| Space group | P41212 |
| Unit-cell parameters (Å) | a = b = 94.36, c = 197.42 |
| Resolution range (Å) | 20.0–2.30 (2.38–2.30) |
| Total/unique reflections | 400106/40500 |
| Completeness (%) | 100 (100) |
| I/σ(I) | 36.0 (5.1) |
| Rmerge† | 0.089 (0.44) |
| Model refinement | |
| Rwork/Rfree‡ | 0.215 / 0.277 |
| No. of non-H atoms/average B factor (Å2) | |
| Protein | 4423/36.0 |
| Water | 489/45.9 |
| Phosphate ions | 20/52.4 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.31 |
| Ramachandran plot | |
| Most favourable (%) | 92.3 |
| Allowed (%) | 7.1 |
| Generously allowed (%) | 0.4 |
| Disallowed (%) | 0.2 |
R
merge = 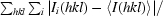
 , where I(hkl) is the intensity of reflection hkl,
, where I(hkl) is the intensity of reflection hkl,  is the sum over all reflections and
is the sum over all reflections and 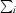 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
R = 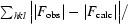
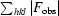 , where R
free is calculated for a randomly chosen 10% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
, where R
free is calculated for a randomly chosen 10% of reflections which were not used for structure refinement and R
work is calculated for the remaining reflections.
Figure 1.

Overall structure of A. fulgidus DNA ligase. Three phosphate ions bound in the adenine nucleotide-binding pocket of AdD are shown as stick models. All figures except Fig. 6 ▶ were drawn with PyMOL (DeLano, 2002 ▶).
3.2. Structural comparisons and the structural variation in the closed conformation of A. fulgidus DNA ligase
According to the DALI structural similarity search (Holm & Sander, 1993 ▶), A. fulgidus DNA ligase shows the highest structural similarity to P. furiosus DNA ligase (PDB code 2cfm; Nishida et al., 2006 ▶), with a root-mean-square (r.m.s.) deviation of 3.6 Å for 548 equivalent Cα positions, a Z score of 43.9 and a sequence identity of 49%. The next highest Z score is obtained with S. solfataricus DNA ligase (PDB code 2hiv; Pascal et al., 2006 ▶), with an r.m.s. deviation of 7.5 Å for 424 equivalent Cα positions, a Z score of 33.4 and a sequence identity of 37%. The r.m.s. deviation between the A. fulgidus and P. furiosus DNA ligases is considerably smaller than that between the A. fulgidus and S. solfataricus DNA ligases. This is a consequence of the conformational variability of these ligases.
The A. fulgidus and P. furiosus DNA ligases show closed conformations with no room for DNA binding, whereas S. solfataricus DNA ligase adopts the fully extended highly open conformation, which is also not suitable to enclose DNA (Fig. 2 ▶). Our study reveals that the domain arrangements of ATP-dependent DNA ligases in the closed conformation can display structural variations. Compared with P. furiosus DNA ligase, the two terminal domains of A. fulgidus DNA ligase make tighter contacts with each other (Fig. 2 ▶). That is, the DBD and OBD of A. fulgidus DNA ligase interact with each other via water-mediated hydrogen bonds (Fig. 3 ▶), whereas those of P. furiosus DNA ligase have no such interactions. A. fulgidus DNA ligase also has a moderate level of sequence and structural similarity to the DNA-bound structure of human DNA ligase I fragment (PDB code 1x9n; Pascal et al., 2004 ▶; an r.m.s. deviation of 3.7 Å for 438 equivalent Cα positions, a Z score of 27.9 and a sequence identity of 26%). While the DNA-unbound structure of A. fulgidus DNA ligase adopts a completely closed conformation, the DNA-bound structure of human DNA ligase I fragment adopts a partially open conformation in which the adenylated dsDNA is encircled by the enzyme (Fig. 2 ▶).
Figure 2.

Different conformations of archaeal and human ATP-dependent DNA ligases. Ribbon diagrams of A. fulgidus DNA ligase, P. furiosus DNA ligase, S. solfataricus DNA ligase and human DNA ligase I fragment (with bound DNA) are drawn in green, cyan, pink and blue, respectively. A. fulgidus DNA ligase shows the most closed conformation, with water-mediated hydrogen-bond interactions between the two terminal domains DBD and OBD.
Figure 3.

A stereoview of the water-mediated hydrogen-bond interactions between DBD and OBD of A. fulgidus DNA ligase. Interface residues and water molecules are shown as sticks and red spheres, respectively. No such interactions exist in the closed conformation of P. furiosus DNA ligase.
When we make structural comparisons of the individual domains between the A. fulgidus and P. furiosus DNA ligases, both of which are in closed conformations, the r.m.s. deviation for the DBD (1.60 Å for 206 Cα positions) is significantly larger than those for the AdD (0.70 Å for 175 Cα positions) or OBD (0.83 Å for 123 Cα positions). This can be explained by the slightly different arrangements of the two lobes of the DBD in these ligases. The two parts (the terminal part and the middle part) of the DBD of ATP-dependent DNA ligases are joined by two hinge loops between them, allowing the DBD to undergo a conformational change through a hinge-bending motion upon binding the DNA substrate (Nishida et al., 2006 ▶). When we overlapped the middle parts of the DBD of the archaeal DNA ligases and human DNA ligase, the overall arrangement of the terminal part and the middle part of the DBD in A. fulgidus DNA ligase is more similar to that of the human DNA ligase I than to those of the P. furiosus and S. solfataricus DNA ligases (Fig. 4 ▶). However, the r.m.s. deviation between the DBDs of A. fulgidus DNA ligase and human DNA ligase (1.84 Å for 181 Cα positions) is slightly larger than that between the A. fulgidus and P. furiosus DNA ligases owing to the much higher sequence divergence between the former pair. Therefore, the previously suggested correlation between the overall conformation of these DNA ligases and the subdomain arrangement of their DBDs (Nishida et al., 2006 ▶) may not be generally valid.
Figure 4.

Superposition of the middle part of the DBD in four DNA ligases in stereo. The DBD of A. fulgidus DNA ligase, human DNA ligase I fragment (with bound DNA), P. furiosus DNA ligase and S. solfataricus DNA ligase are coloured green, blue, cyan and pink, respectively. The superposition shows that the conformation of DBD in A. fulgidus DNA ligase is more similar to that of the human DNA ligase I fragment than to those of the P. furiosus and S. solfataricus DNA ligases.
3.3. Phosphate ions in the nucleotide-binding pocket of A. fulgidus DNA ligase
The refined model of A. fulgidus DNA ligase did not show electron density for a covalently attached adenylate group at the conserved Lys249 in the AdD, indicating that the purified recombinant enzyme was in the unadenylated state. When we cocrystallized the enzyme in the presence of either ATP or AMP and collected X-ray diffraction data, these ligands did not bind in the adenine nucleotide-binding pocket of the AdD. This is likely to be a consequence of the high concentration (1.6 M) of phosphate ions in the crystallization reservoir condition. Instead, we could identify four phosphate ions bound to the enzyme in the present structure. One of the phosphate ions was bound in a positively charged cleft between the AdD and OBD, well separated from the adenine nucleotide-binding pocket of the AdD. It interacts directly with Arg315 and Arg526, both of which are not well conserved.
Interestingly, however, three other phosphate ions were bound as a cluster in the adenine nucleotide-binding pocket of the AdD (Fig. 5 ▶). They are surrounded by the side chains of Lys249, Arg269, Arg411, Lys417, Lys419, Arg528 and Lys531. All of the equivalent residues are strictly conserved between archaeal DNA ligases and human DNA ligase I (Fig. 6 ▶) and are known to be involved in the binding of ATP (Nishida et al., 2006 ▶; Pascal et al., 2006 ▶) or the nicked DNA substrate (Pascal et al., 2004 ▶). A water molecule is centrally located and interacts with O atoms of the three bound phosphates, as well as with two other water molecules (Fig. 5 ▶). This coordination geometry resembles that of a magnesium ion. However, the interatomic distances (ranging between 2.25 and 3.17 Å, with an average of 2.71 Å) are too long to justify the assignment of the central atom as a magnesium ion. The bound phosphate ions 1 and 2 mimic the phosphate moieties of ATP. When we overlap the AdD of our structure with that of the ATP-bound structure of S. solfataricus DNA ligase (Pascal et al., 2006 ▶), the phosphate ions 1 and 2 bound to A. fulgidus DNA ligase occupy equivalent positions to the γ- and α-phosphates of ATP bound to S. solfataricus DNA ligase, respectively (Fig. 5 ▶). The bound phosphate ion 3 does not have a counterpart in other ATP-dependent DNA ligase structures. It interacts directly with the conserved Arg528 and Lys531 of motif VI in the OBD. As motif VI is known to facilitate the formation of the enzyme–AMP intermediate (Sriskanda & Shuman, 1998 ▶), phosphate ion 3 may mimic part of the pyrophosphate ion that is released from ATP upon adenylation of the conserved lysine. Motif VI of A. fulgidus DNA ligase is similarly located at the interface between the OBD and DBD as in P. furiosus DNA ligase, mimicking the incoming substrate DNA (Nishida et al., 2006 ▶).
Figure 5.

A stereoview of 2F o − F c (coloured in blue, contoured at 1.5σ) and F o − F c (coloured in red, contoured at 5σ) electron-density maps of the three phosphate ions (shown as stick models) and water molecules (shown as red spheres) bound in the adenine nucleotide-binding pocket.
Figure 6.

Sequence alignment of three archaeal DNA ligases and the human DNA ligase I fragment. Conserved residues are enclosed in blue boxes. η1, η2, η3, and η4 are 310-helices and TTs are tight β-turns. Residues in the A. fulgidus DNA ligase that interact with the bound phosphates are indicated by blue triangles. The alignment figure was made using the program ClustalX (Thompson et al., 1997 ▶) and ESPript (Gouet et al., 1999 ▶).
Supplementary Material
PDB reference: A. fulgidus DNA ligase, 3gde, r3gdesf
Acknowledgments
We thank the staff at beamlines BL-4A and BL-6C of Pohang Light Source, Korea for assistance during X-ray data-collection experiments. DJK, OK and HWK are recipients of the BK21 fellowship. This work was supported by the Basic Research Promotion Grant (grant No. KRF-2006-311-C00409) of Korea Research Foundation funded by the Korean Government (MOEST).
References
- Brünger, A. T. (1992). Nature (London), 355, 472–474. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. http://www.pymol.org/.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Holm, L. & Sander, C. (1993). J. Mol. Biol.233, 123–138. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lee, J. Y., Chang, C., Song, H. K., Moon, J., Yang, J. K., Kim, H. K., Kwon, S. T. & Suh, S. W. (2000). EMBO J.19, 1119–1129. [DOI] [PMC free article] [PubMed]
- Lehman, I. R. (1974). Science, 186, 790–797. [DOI] [PubMed]
- Lindahl, T. & Barnes, D. E. (1992). Annu. Rev. Biochem.61, 251–281. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nishida, H., Kiyonari, S., Ishino, Y. & Morikawa, K. (2006). J. Mol. Biol.360, 956–967. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pascal, J. M., O’Brien, P. J., Tomkinson, A. E. & Ellenberger, T. A. (2004). Nature (London), 432, 473–478. [DOI] [PubMed]
- Pascal, J. M., Tsodikov, O. V., Hura, G. L., Song, W., Cotner, E. A., Classen, S., Tomkinson, A. E., Tainer, J. A. & Ellenberger, T. A. (2006). Mol. Cell, 24, 279–291. [DOI] [PubMed]
- Sriskanda, V. & Shuman, S. (1998). Nucleic Acids Res.26, 4618–4625. [DOI] [PMC free article] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. (1997). Nucleic Acids Res.25, 4876–4882. [DOI] [PMC free article] [PubMed]
- Tomkinson, A. E. & Levin, D. S. (1997). Bioessays, 19, 893–901. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: A. fulgidus DNA ligase, 3gde, r3gdesf
PDB reference: A. fulgidus DNA ligase, 3gde, r3gdesf