

The modified N-domain of the Wilson disease protein has been crystallized both in its native form and as a selenomethionine derivative. X-ray diffraction data were collected from the native crystal to 1.7 Å resolution, while the selenomethionine derivative diffracted to 2.7 Å resolution.
Keywords: N-domain, Wilson’s disease, ATP7B
Abstract
Wilson disease associated protein (ATP7B) is essential for copper transport in human cells. Mutations that affect ATP7B function result in Wilson’s disease, a chronic copper toxicosis. Disease-causing mutations within the N-domain of ATP7B (WND) are known to disrupt ATP binding, but a high-resolution X-ray structure of the ATP-binding site has not been reported. The N-domain was modified to delete the disordered loop comprising residues His1115–Asp1138 (WNDΔ1115–1138). Unlike the wild-type N-domain, WNDΔ1115–1138 formed good-quality crystals. Synchrotron diffraction data have been collected from WNDΔ1115–1138 at the Canadian Light Source. A native WNDΔ1115–1138 crystal diffracted to 1.7 Å resolution and belonged to space group P42212, with unit-cell parameters a = 39.2, b = 39.2, c = 168.9 Å. MAD data were collected to 2.7 Å resolution from a SeMet-derivative crystal with unit-cell parameters a = 38.4, b = 38.4, c = 166.7 Å. The WNDΔ1115–1138 structure is likely to be solved by phasing from multiwavelength anomalous diffraction (MAD) experiments.
1. Introduction
Wilson’s disease is an autosomal recessive disorder that results from mutations in the ATP7B gene. Reduced copper efflux out of cells is the primary defect (Culotta & Gitlin, 2001 ▶). The associated protein ATP7B is a P-type ATPase that consists of a single polypeptide chain of 1465 amino-acid residues (Cox & Moore, 2002 ▶; Lutsenko et al., 2007 ▶; Pedersen & Carafoli, 1987 ▶). P1B-type ATPases such as ATP7B maintain metal homeostasis in bacteria and eukaryotic cells (Rosen, 2002 ▶). ATP7B is composed of eight transmembrane helices and four cytosolic domains (the N-terminal, A-, P- and N-domains; Arguello et al., 2007 ▶). The isolated N-domain of ATP7B (residues 1032–1196) has no ATPase activity, but does bind ATP, ADP and AMP specifically (Morgan et al., 2004 ▶). Early attempts to crystallize the complete N-domain were unsuccessful and the high-resolution structure was determined by NMR (Dmitriev et al., 2006 ▶). The NMR structure of the N-domain (Fig. 1 ▶) revealed a folded core consisting of a six-stranded β-sheet and two flanking α-helical hairpins and a flexible loop comprising residues Ala1114–Gln1142 (the residue numbering corresponds to that of the full-length ATP7B protein; Dmitriev et al., 2006 ▶). Several invariant residues in the N-domain of P1B-ATPases, Glu1064, His1069, Gly1099 and Gly1101, and the highly conserved Gly1149 were found to be located in the ATP-binding site. The exact nature of the ATP–protein interactions in the binding site could not be determined from the NMR data. A high-resolution X-ray structure of the complex of ATP bound to the N-domain would reveal these interactions at the atomic level and provide a structural basis for the design of highly specific modulators of Wilson disease ATPase.
Figure 1.

The previously reported NMR structure of the N-domain (PDB code 2arf) modified to show the deletion of the His1115–Asp1138 loop. The invariant residues involved in ATP binding are shown in magenta (Dmitriev et al., 2006 ▶).
The long loop comprising residues 1114–1142 is not involved in ATP binding (Dmitriev et al., 2006 ▶) and there are no known Wilson’s disease mutations in this region (Hsi & Cox, 2004 ▶), which implies that this region is not critical for enzyme function and that the core N-domain structure would not be altered by deletion of this loop. At the same time, the disordered state of this loop may have prevented successful crystallization of the full-length N-domain. To improve the chances of crystal formation, we deleted a fragment of the ATP7B gene corresponding to residues His1115–Asp1138. In the resulting protein WNDΔ1115–1138, the length of the loop connecting the short α-helix Val1109–Leu1113 to the β-strand Thr1143–Gly1149 is reduced from 29 to five amino-acid residues (Fig. 1 ▶). The preparation and crystallization of WNDΔ1115–1138 is described in the present communication.
2. Materials and methods
2.1. Protein modification, expression and purification
To generate the His1115–Asp1138 deletion, the pTYB12-NABD plasmid (Morgan et al., 2004 ▶) used for the expression of the full-length N-domain was amplified by the around-the-plasmid PCR. The primers GCA GTC CCC CAG ACC TTC TCT (forward) and GGC CAG GAT GCC TTC CAC GTT (reverse) were designed to anneal immediately outside the region intended for deletion. Both primers were 5′-phosphorylated. The amplified DNA product produced using these primers corresponded to the complete sequence of the plasmid with the exception of the deletion region. A total of 30 PCR cycles were performed with annealing at 328 K and elongation at 345 K for 150 s using the Phusion DNA polymerase kit (New England Biolabs). The 7.9 kb PCR product was purified by agarose gel electrophoresis and ligated overnight at 289 K with T4 ligase (New England Biolabs). DH5α cells were transformed with the ligation mixture and the transformants were selected on LB plates supplemented with 100 µg ml−1 ampicillin. Plasmid DNA isolated from the selected clones was checked for the correct restriction pattern (7.1 and 0.8 kb fragments) with NheI and KpnI enzymes, sequenced and transformed into Escherichia coli BL21 (DE3) for protein expression. Expression and purification of the modified N-domain was performed essentially as described previously (Dmitriev et al., 2006 ▶; Morgan et al., 2004 ▶). Briefly, the N-domain was expressed as a fusion protein with a chitin-binding domain and an intein. The fusion protein was bound to chitin beads (New England Biolabs) and contaminating proteins were removed by washing the column. Intein cleavage was then induced by adding dithiothreitol. Pure N-domain was eluted from the column, whereas the fragment containing the chitin-binding domain and the intein remained bound to the beads. To produce SeMet-substituted protein, the M63 medium used for protein expression was supplemented with 50 mg l−1 l-selenomethionine (Guerrero et al., 2001 ▶). For protein crystallization, samples of the modified N-domain were additionally purified by size-exclusion chromatography on a Superdex-75 HK 16/60 column (Pharmacia) in 50 mM sodium phosphate buffer pH 6.0 containing 100 mM NaCl and 5 mM DTT. The resulting fractions containing modified N-domain protein were pooled and concentrated to 15 mg ml−1 before being stored at 277 K.
2.2. NMR spectroscopy
The 1H–15N HSQC spectra of WNDΔ1115–1138 were recorded at 300 K on a Bruker 600 MHz spectrometer equipped with a Cryoprobe and z-axis gradients. The sample contained 0.5 mM protein, 50 mM sodium phosphate buffer pH 6.0, 5%(v/v) D2O, 5 mM DTT, 0.05 mM NaN3 and 0.5 mM 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) for chemical shift referencing.
2.3. Protein crystallization
Initial crystallization conditions for the WNDΔ1115–1138 protein were identified by high-throughput screening at the Hauptman–Woodward Medical Research Institute using the microbatch-under-oil method (Luft et al., 2003 ▶). Subsequent optimization of the crystallization conditions resulted in WNDΔ1115–1138 crystals being obtained from a drop consisting of equal volumes (1 µl) of protein solution (15 mg ml−1 protein, 50 mM NaH2PO4 pH 6.0, 100 mM NaCl, 0.05 mM NaN3 and 5 mM DTT) and precipitant solution (30% PEG 8000, 200 mM sodium citrate pH 5.0 and 200 mM NH4SCN) which had been covered in 100% mineral oil at room temperature. Octahedral shaped crystals appeared within 2 d and continued to grow over the next few days to approximately 0.43 mm in the largest dimension (Fig. 2 ▶). The crystals were harvested into cryosolution [220 mM glucose, 525 mM sucrose, 16%(v/v) glycerol and 16%(v/v) ethylene glycol] prior to flash-cooling in liquid nitrogen. For the selenomethionine-derivative crystals, drops were formed by mixing equal volumes (1 µl) of protein solution (12.5 mg ml−1 protein, 50 mM NaH2PO4 pH 6.0, 100 mM NaCl, 0.05 mM NaN3 and 5 mM DTT) and precipitant solution [37% PEG 8000, 200 mM Na3C3H5O(COO)3 pH 5.0 and 200 mM NH4SCN] and were then covered with 100% mineral oil at room temperature. Octahedral shaped crystals appeared after 3 d and continued to grow over the next few days to approximately 0.38 mm in the largest dimension. The crystals were subsequently flash-cooled without the need for additional cryoprotectant owing to the high concentration of PEG in the solution.
Figure 2.

Crystals of WNDΔ1115–1138. The largest dimension is approximately 0.43 mm.
2.4. Data collection and processing
Diffraction of the WNDΔ1115–1138 protein crystal took place on the Canadian Macromolecular Crystallography Facility (CMCF-1) beamline (08ID-1) at the Canadian Light Source (Saskatoon, Saskatchewan, Canada). The native data set consisted of 180 images, each of which was collected with 1 s exposure over a 1° oscillation range at a crystal-to-detector distance of 160 mm. The intensity data were indexed, integrated and scaled using XDS (Kabsch, 1993 ▶). For the selenomethionine-derivative crystal, multiwavelength anomalous diffraction (MAD) data were collected based on wavelengths selected from analysis of the selenium absorption spectrum. Each of these data sets consisted of 180 images with 1 s exposure and 1° oscillation at a crystal-to-detector distance of 180 mm. The intensity data were indexed, integrated and scaled with the HKL-2000 suite of programs including DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). A summary of the crystallographic statistics for both the native and SeMet-derivative data sets is given in Table 1 ▶.
Table 1. Data-collection statistics for native and SeMet-derivative crystals.
Values in parentheses are for the highest resolution shell.
| SeMet derivative | ||||
|---|---|---|---|---|
| Native | Inflection | Peak | Remote | |
| Space group | P42212 | |||
| Unit-cell parameters (Å) | ||||
| a | 39.2 | 38.5 | 38.4 | 38.5 |
| b | 39.2 | 38.5 | 38.4 | 38.5 |
| c | 168.9 | 166.8 | 166.7 | 166.9 |
| Wavelength (Å) | 1.00000 | 0.98118 | 0.98101 | 0.97626 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Matthews coefficient (Å3 Da−1) | 2.04 | 1.93 | 1.93 | 1.93 |
| Solvent content (%) | 40 | 36 | 36 | 36 |
| Unit-cell volume (Å3) | 259538 | 247239 | 245809 | 247387 |
| Molecular weight (Da) | 14796 | 15030 | 15030 | 15030 |
| Molecules per ASU | 1 | 1 | 1 | 1 |
| Measured reflections | 102065 | 46620 | 45772 | 45172 |
| Unique reflections | 15556 | 4819 | 4812 | 4732 |
| Resolution range (Å) | 100–1.7 (1.73–1.7) | 50–2.7 (2.82–2.7) | 50–2.7 (2.82–2.7) | 50–2.7 (2.82–2.7) |
| Completeness (%) | 98.3 (87.5) | 98.1 (97.6) | 99.5 (98.0) | 97.6 (96.7) |
| Redundancy | 6.5 (4.6) | 9.7 (9.0) | 9.5 (8.7) | 9.5 (7.5) |
| Rmerge† | 0.073 (0.340) | 0.132 (0.508) | 0.146 (0.501) | 0.166 (0.584) |
| 〈I/σ(I)〉 | 16.5 (3.77) | 33.1 (6.08) | 31.2 (6.4) | 31.0 (4.7) |
R
merge = 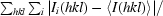
 , where 〈I(hkl)〉 is the average intensity over symmetry-related reflections and Ii(hkl) is the measured intensity.
, where 〈I(hkl)〉 is the average intensity over symmetry-related reflections and Ii(hkl) is the measured intensity.
3. Results and discussion
A photomicrograph of a crystal of WNDΔ1115–1138 protein is shown in Fig. 2 ▶ and one image of the diffraction pattern is illustrated in Fig. 3 ▶. A fingerprint 1H–15N HSQC spectrum of WNDΔ1115–1138 (Fig. 4 ▶) showed excellent chemical shift dispersion characteristic of a well folded protein with a high content of β-sheet structure. The spectrum was very similar to the spectrum of the wild-type N-domain, with the exception of the signals from residues His1115–Asp1138, which were absent, and the signals of the amino-acid residues in the immediate proximity of the deletion site, which were shifted. No significant chemical shift changes were observed for the invariant residues of the P1B-type ATPases, Glu1064, Glu1068, Gly1099 and Gly1101, which are involved in ATP binding. These data indicate that the three-dimensional structure of the folded core is very similar in the wild-type N-domain and in the WNDΔ1115–1138 variant of the protein. Consistent with this conclusion, the ATP-binding affinity of the N-domain was unaffected by the loop deletion. The dissociation constants for ATP measured by chemical shift perturbation assay using Gly1101 as a reporter group (Dmitriev et al., 2006 ▶) were found to be in the range 50–70 µM for both the wild-type N-domain and the WNDΔ1115–1138 variant. This suggested WNDΔ1115–1138 as a suitable model protein for the high-resolution structure determination of the ATP-binding site in the Wilson disease associated protein.
Figure 3.

Diffraction image of a WNDΔ1115–1138 crystal.
Figure 4.

1H–15N HSQC spectra of wild-type N-domain (red) and the loop-deletion variant WNDΔ1115–1138 (black) recorded in the presence of 5 mM ATP. The signals of the backbone amides of the invariant amino-acid residues are circled. The signal of His1069 at 8.45 and 125.4 p.p.m. (wild type) is located in a crowded area of the spectrum and is not shown.
Unfortunately, initial attempts to solve the native WNDΔ1115–1138 structure via Phaser (Storoni et al., 2004 ▶) by molecular replacement using the NMR model of WND (PDB code 2arf) or with the N-domain of the Archaeoglobus fulgidus Cu+-ATPase (Sazinsky et al., 2006 ▶), which shares 44% sequence homology with WNDΔ1115–1138, were both unsuccessful. In an effort to solve the structure through MAD phasing experiments, selenomethionine-derivatized protein was produced, purified and crystallized. Once the SeMet-derivative structure has been solved, the resulting model will be employed to solve the WNDΔ1115–1138 structure by molecular replacement. Currently, efforts are also under way to produce additional heavy-atom derivatives to aid in solving the WNDΔ1115–1138 structure.
Acknowledgments
LTJD is a Tier 1 Canada Research Chair in Structural Biochemistry. SAD is the recipient of an NSERC Postdoctoral Fellowship. This research was funded by a Saskatchewan–CIHR Regional Partnership Operating Grant to OYD and a Saskatchewan–CIHR Regional Partnership Operating Grant as well as an NSERC Discovery Grant to LTJD and in part by a Saskatchewan Health Research Foundation Team Grant to the Molecular Design Research Group at the University of Saskatchewan. The authors would like to thank the team at the CMCF-1 beamline for technical assistance at the CLS, which is supported by NSERC, NRC, CIHR and the University of Saskatchewan, and both Eva-Maria Uhlemann and Yvonne Leduc for excellent technical support.
References
- Arguello, J. M., Eren, E. & Gonzalez-Guerrero, M. (2007). Biometals, 20, 233–248. [DOI] [PubMed]
- Cox, D. W. & Moore, S. D. (2002). J. Bioenerg. Biomembr.34, 333–338. [DOI] [PubMed]
- Culotta, V. C. & Gitlin, J. D. (2001). The Molecular Basis of Inherited Disease, 8th ed., edited by C. R. Scriver, A. L. Beaudet, W. S. Sly & D. Valle, pp. 3205–3126. New York: McGraw–Hill.
- Dmitriev, O., Tsivkovskii, R., Abildgaard, F., Morgan, C. T., Markley, J. L. & Lutsenko, S. (2006). Proc. Natl Acad. Sci. USA, 103, 5302–5307. [DOI] [PMC free article] [PubMed]
- Guerrero, S. A., Hecht, H.-J., Hofmann, B., Biebl, H. & Singh, M. (2001). Appl. Microbiol. Biotechnol.56, 718–723. [DOI] [PubMed]
- Hsi, G. & Cox, D. W. (2004). Hum. Genet.114, 165–172. [DOI] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K. & DeTitta, G. T. (2003). J. Struct. Biol.142, 170–179. [DOI] [PubMed]
- Lutsenko, S., Barnes, N. L., Bartee, M. Y. & Dmitriev, O. Y. (2007). Physiol. Rev.87, 1011–1046. [DOI] [PubMed]
- Morgan, C. T., Tsivkovskii, R., Kosinsky, Y. A., Efremov, R. G. & Lutsenko, S. (2004). J. Biol. Chem.279, 36363–36371. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pedersen, P. L. & Carafoli, W. (1987). Trends Biochem. Sci.12, 146–150.
- Rosen, B. P. (2002). Comput. Biochem. Physiol. A Mol. Integr. Physiol.133, 689–693. [DOI] [PubMed]
- Sazinsky, M. H., Mandal, H. K., Argüello, J. M. & Rosenzweig, A. C. (2006). J. Biol. Chem.281, 11161–11166. [DOI] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]