

Abstract
Small molecules designed to mimic the binding of Smac protein to X-linked inhibitor of apoptosis protein (XIAP) are being pursued as a promising new class of anticancer drugs. Herein, we report the design, synthesis, and comprehensive structure-activity relationship studies of a series of conformationally constrained bicyclic Smac mimetics. Our studies led to the discovery of a number of highly potent and cell-permeable Smac mimetics and yielded important new insights into their structure-activity relationship for their binding to XIAP and for their activity in inhibition of cancer cell growth. Determination of the crystal structure of one potent Smac mimetic, compound 21, in complex with XIAP BIR3 provides the structural basis for its high-affinity binding to XIAP and for the design of highly potent Smac mimetics.
Introduction
Apoptosis is a critical cell process in normal development and homeostasis of multicellular organisms to eliminate unwanted or damaged cells. Inappropriate regulation of apoptosis plays a major role in many human diseases, including cancer.1–4 Defects in the apoptosis machinery confers apoptosis resistance on cancer cells to therapeutic agents, makes current anticancer therapies less effective and leads ultimately to their failure in the clinic.2–4 Accordingly, targeting critical apoptosis regulators aimed at overcoming apoptosis resistance of cancer cells is a promising cancer therapeutic strategy.
The X-linked inhibitor of apoptosis protein (XIAP) has been identified as a key apoptosis inhibitor, although its role in cells may not be limited to the regulation of apoptosis.5–10 XIAP inhibits apoptosis through direct binding to and inhibition of three cysteine proteases, an initiator caspase-9 and the two effectors caspase-3 and -7.5–10 XIAP contains three Baculoviral IAP Repeats (BIR) domains. While the third BIR domain (BIR3) of XIAP selectively targets caspase-9, the BIR2 domain, together with the immediate preceding linker, inhibits both caspase-3 and caspase-7. Since these caspases play a critical role in the execution of apoptosis, XIAP functions as an efficient inhibitor of apoptosis. Consistent with its potent apoptosis-suppressing function, XIAP is found to be highly expressed in many human tumor cell lines and tumor samples from patients11 and plays an important role in conferring resistance on cancer cells to a variety of anticancer drugs.8,9 Because XIAP blocks apoptosis at the down-stream effector phase, a point where multiple signaling pathways converge, it represents a particularly attractive molecular target for the design of new classes of anticancer drugs aimed at overcoming the apoptosis resistance of cancer cells.8,9,12
The anti-apoptotic function of XIAP is antagonized by Smac/DIABLO (second mitochondria-derived activator of caspases or direct IAP binding protein with low pI), a protein released from mitochondria into the cytosol in response to apoptotic stimuli.13,14 Crystal and NMR structures15,16 show that Smac, through its N-terminal AVPI (Ala1-Val2-Pro3-Ile4) motif, interacts with the XIAP BIR3 domain at the same site where caspase-9 binds and removes the inhibition of XIAP to caspase-9 by direct competition.17,18 The mechanism by which Smac removes the inhibition of XIAP to caspase-3/-7 is not entirely clear. It has been proposed that Smac protein removes the inhibition of XIAP to caspase-3/-7 by binding to the XIAP BIR2 domain through its AVPI motif.19 Hence, the AVPI binding motif in Smac plays a critical role for the interaction of Smac protein with XIAP and its functional antagonism against XIAP.
Earlier studies using Smac-based peptides containing the AVPI binding motif, tethered to a carrier peptide for intra-cellular delivery, have demonstrated that such compounds can enhance the antitumor activity of chemotherapeutic agents and of TNF-related apoptosis inducing ligand in vitro and in vivo.20–22 Although Smac-based peptides have served as useful tools for important proof-of-concept studies, they are not suitable drug candidates due to their very limited cellular activity and expected poor in vivo stability. To overcome the limitations associated with peptide-based Smac mimetics, a number of laboratories, including ours, have pursued the design of peptidic and non-peptidic small-molecule Smac mimetics with a goal to obtain more druglike compounds, which may be developed as a new class of anticancer drugs.23–30
Using a structure-based approach, our laboratory has reported the design of a number of conformationally constrained, bicyclic Smac mimetics.23,24,26,30 Our previous studies showed that these designed Smac mimetics can achieve high binding affinities to XIAP and are effective in inhibition of cell growth and induction of apoptosis in cancer cells. For example, SM-131, which contains a [7,5] bicyclic core structure, binds to XIAP BIR3 protein with a Ki of 61 nM in a competitive binding assay and directly antagonizes the XIAP inhibition of caspase-9 activity in a cell-free functional assay.26 This compound also potently inhibits cancer cell growth and induces apoptosis in cancer cells as a single agent.26
Although our previous studies23,24,26,30 have led to the discovery of potent and cell-permeable Smac mimetics, our understanding on their structure-activity relationship is still limited. Furthermore, although molecular modeling was employed to predict the binding models of our designed Smac mimetics to XIAP BIR3 protein in our previous studies, the predicted binding models have not been experimentally confirmed.
To gain a more in-depth understanding of the structure-activity relationship for our designed conformationally constrained Smac mimetics for their binding to XIAP and for their cellular activity, we have designed, synthesized and evaluated a series of new Smac mimetics. To obtain a solid structural basis for the interaction of our designed Smac mimetics with XIAP BIR3, we have determined a high-resolution crystal structure of a potent Smac mimetic (compound 21) in complex with XIAP BIR3. We report herein structure-based design, synthesis, biochemical and biological evaluation and crystallographic studies of conformationally constrained Smac mimetics as antagonists of XIAP.
Results and discussion
Structure-based design of conformationally constrained Smac mimetics and their structure-activity relationship
We have employed a structure-based strategy for the design of conformationally constrained, bicyclic Smac mimetics (Figure 1).23,24
Figure 1.

Design and examples of conformationally constrained bicyclic Smac peptide mimetics.
In the crystal structure of Smac protein in complex with XIAP BIR3, the hydrophobic side chain of Ile4 in the Smac AVPI peptide inserts into a hydrophobic pocket in XIAP BIR3 and the carbonyl group of Ile4 does not have specific interactions with the protein.15 We thus replaced the Ile4 residue with a simple benzylamine group, which led to compound 1.23,24 Compound 1 binds to XIAP BIR3 protein with a Ki of 0.29 μM in our fluorescence-polarization (FP) binding assay32 and is 2 times more potent than the natural Smac AVPI peptide (Ki = 0.58 μM) under the same assay conditions. Compound 1 was thus used as a new lead in our subsequent design.
The isopropyl side chain in Val2 has no specific interactions with the protein in the crystal structure, while the 5-membered ring of Pro3 has van der Waals contacts with the side chain of Trp323 in the protein. Modeling suggested that the isopropyl side chain in Val2 may be fused with the 5-membered ring of Pro3 to form a bicyclic lactam structure without causing steric clashes with the protein.23,24 Cyclization of the side chain of Val2 and the 5-membered ring of Pro3 generates a new chiral center. To probe the stereo-specificity of this new chiral center for binding to XIAP, we designed and synthesized stereo-isomers 2 and 3, which contain a [6,5] bicyclic lactam core structure.24 Compound 2 binds to XIAP BIR3 with a Ki value of 4.47 μM, but compound 3 shows no appreciable binding at concentrations up to 100 μM. Hence, the binding data for compounds 2 and 3 established that the R-configuration of the chiral center in the bicyclic Smac mimetics is highly preferred for their binding to XIAP BIR3. Accordingly, in our subsequent design and synthesis, we have focused on compounds with the R-configuration for the chiral center.
Although compound 2 binds to XIAP BIR3 with a good affinity, it is 8 times less potent than the Smac AVPI peptide and 15 times less potent than compound 1. Modeling showed that although compound 2 largely mimics the bound conformation of the AVPI peptide to XIAP BIR3, its rigid [6,5] bicyclic structure causes some deviations (Figure 2A), which may account for its lower binding affinity to XIAP BIR3. Therefore, we designed and synthesized compounds 423 and 5, in which the six-member ring in 2 was expanded to a seven- or eight-member ring, respectively. Modeling predicted that that these two compounds can nicely mimic the bound conformation of the Smac AVPI peptide to XIAP BIR3 protein (Figure 2B and 2C)) and suggested that both compounds should bind to XIAP BIR3 with a higher affinity than compound 2. Indeed, compounds 4 and 5 achieved Ki values of 0.15 and 0.10 μM, respectively, and are 30 or 45 times more potent than compound 2, confirming the modeling prediction.
Figure 2.

Predicted binding models of compounds 2, 4, 5, 20 and 21 to XIAP BIR3 protein in comparison with that for Smac AVPI peptide in the crystal structure. Carbon atoms in compounds 2, 4, 5, 20 and 21 and AVPI are shown in stick. The carbon atoms are colored in yellow in 2, 4, 5, 20 and 21 and in green in the AVPI peptide. The carbon atoms in several key residues in XIAP are colored in grey. Nitrogen and oxygen atoms are colored in blue and red, respectively.
We carried out further modifications in both N-terminal (region 1) and C-terminal (region 2) regions (Figure 1) in compounds 2, 4 or 5 to further explore the structure-activity relationships in these two regions. The results are summarized in Table 1.
Table 1.
Chemical structures of conformationally constrained bicyclic Smac mimetics and their binding affinities to XIAP BIR3.
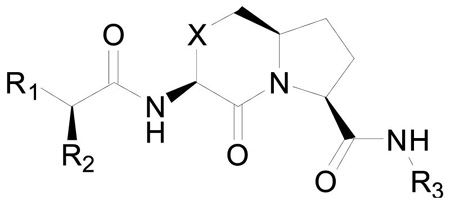 | |||||
|---|---|---|---|---|---|
| Compounds | X | R1 | R2 | R3 | XIAP BIR3 Ki (μM) |
| AVPI | 0.5823 | ||||
| 1 | 0.2923 | ||||
| 2 | CH2 | Me | NH2 | Bn | 4.4724 |
| 3 | >10024 | ||||
| 4 | (CH2)2 | Me | NH2 | Bn | 0.1523 |
| 5 | (CH2)3 | Et | NH2 | Bn | 0.10 |
| 6 | CH2 | Et | NH2 | Bn | 1.41 ± 0.16 |
| 7 | CH2 | n-Pr | NH2 | Bn | >100 |
| 8 | CH2 | i-Pr | NH2 | Bn | 43.11 ± 1.51 |
| 9 | CH2 | Me | NH2 |
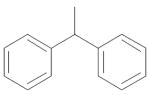
|
2.33 ± 0.68 |
| 10 | CH2 | Et | NH2 |
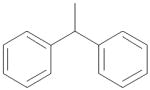
|
0.35 ± 0.01 |
| 11 | (CH2)2 | Me | NH2 |
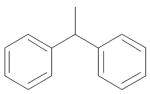
|
0.06 ± 0.02 |
| 12 | (CH2)2 | Et | NH2 |

|
0.025 ± 0.004 |
| 13 | (CH2)2 | Et | MeNH |
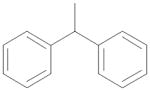
|
0.061 ± 0.006 |
| 14 | (CH2)2 | Et | Me2N |
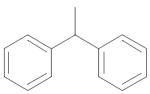
|
14.4 ± 0.6 |
| 15 | (CH2)2 | Et | OH |
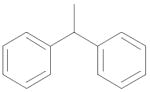
|
29.0 ± 1.4 |
| 16 | (CH2)3 | Me | NH2 |
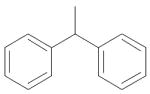
|
0.014 ± 0.003 |
| 17 | (CH2)3 | Et | NH2 |
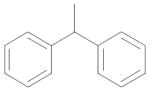
|
0.039 ± 0.004 |
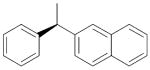
| 18 | (CH2)3 | Et | NH2 |
|
0.759 ± 0.16 |
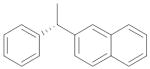
| 19 | (CH2)3 | Et | NH2 |
|
0.047 ± 0.009 |
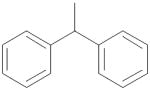
| 20 | (CH2)3 | Me | MeNH |
|
0.026 ± 0.005 |
| 21 | (CH2)3 | Et | MeNH |
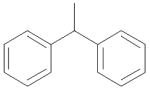
|
0.067 ± 0.018 |
| 22 | (CH2)3 | Et |

|
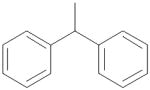
|
0.856 ± 0.075 |
| 23 | (CH2)3 | Et |
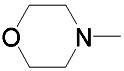
|

|
32.87 ± 4.18 |
| 24 | (CH2)3 | Me | OH |

|
12.97 ± 2.02 |
| 25 | (CH2)3 | Me | MeNH |
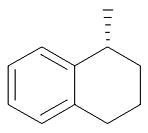
|
0.014 ± 0.005 |
| 26 | (CH2)3 | Me | MeNH |
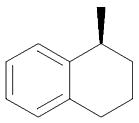
|
0.274 ± 0.037 |
| 27 | (CH2)3 | Me | MeNH |
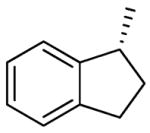
|
0.015 ± 0.008 |
In the crystal structure of Smac protein in complex with XIAP BIR3, the methyl group of Ala1 inserts into a small hydrophobic pocket. Compounds 6, 7 and 8 were designed and synthesized, in which the methyl group in 2 was replaced with an ethyl, n-propyl or isopropyl group, respectively. While compound 6 with an ethyl group is 3 times more potent than 2, compound 8 with an isopropyl group is 10-times less potent than 2 and compound 7 with an n-propyl group shows no appreciable binding up to 100 μM to XIAP BIR3. These data demonstrated that the small hydrophobic pocket in XIAP BIR3 occupied by the methyl group in Ala1 in the Smac AVPI peptide can not accommodate a group larger than an ethyl group for effective binding. Therefore, only the methyl or ethyl group was employed in this position in our subsequent design and synthesis.
The positively charged primary amino group in Ala1 in the Smac AVPI has strong charge-charge interactions with the negatively charged carboxylic group in the side chain of Glu314 of XIAP BIR3 and is critical for binding of Smac-based peptides to XIAP BIR3. However, we found that Smac mimetics with high binding affinities to XIAP BIR3 but containing this primary amino group have very weak cellular activity.23,24,26 Therefore, we replaced this primary amine with a secondary or tertiary amine or a hydroxyl group, which can also form one or more hydrogen bonds with the protein under physiological conditions.26 Their binding data indicated that replacement of the amino group with an N-methyl amino group in Smac mimetics containing either a [7,5] or [8,5] bicyclic lactam leads to reduction of the binding affinity by 2–3 times (13 vs 12, 20 vs 16 and 21 vs 17). However, replacement of the primary amino group with an N,N-dimethyl amino (14) or a cyclic tertiary amino (22 and 23) or a hydroxyl group (15 and 24) decreases the binding affinities by 20–800 times. These data show that the primary and secondary amino groups are highly preferred at this site for Smac mimetics to achieve high binding affinities to XIAP BIR3.
The crystal structure of Smac in complex with XIAP BIR3 showed that the carbonyl group of Ile4 is exposed to solvent and has no specific interaction with the protein.15 However, we hypothesized that this carbonyl group may contribute to the binding affinity of Smac to XIAP by controlling the orientation of the hydrophobic side chain of Ile4, thus reducing the entropic loss upon binding. To test this hypothesis, we replaced the benzyl group in 2, 4 and 5 with a diphenyl methyl group, which led to compounds 9, 11 and 16, respectively. These three compounds (9, 11 and 16) were found to be three times more potent than their corresponding analogues 2, 4 and 5, supporting our hypothesis.
Our modeling predicted that the pro R phenyl group enters the hydrophobic pocket and the pro S phenyl group is exposed to solvent. To test this modeling prediction, we have designed compounds 18 and 19 by replacing either the pro R or pro S phenyl group in 17 with a naphthyl group. It was found while the S form isomer 18 is as potent as 17, the R form isomer 19 is 35 times less potent than 17, thus confirming our modeling prediction.
We next tested if other structures, which can control and restrict the orientation of the phenyl group, can also improve the binding affinity. To this end, we have designed and synthesized compounds 25–27, in which the conformation of the phenyl group was further constrained by a fused second ring and was previously employed for the design of potent, Smac peptidomimetics.25 Compound 25, in which the chiral amine has the R configuration, has a Ki value of 14 nM, whereas its stereoisomer 26 has a Ki value of 207 nM. Thus, compound 25 is 15 times more potent than its isomer 26, indicating that the R form is the desirable configuration for the chiral center. Compound 27, in which the (R)-1,2,3,4-tetrahydro-naphthalenyl group in 25 was replaced by a (R)-2,3-dihydro-1H-indene group, also binds to XIAP BIR3 with a high affinity and has a Ki value of 15 nM.
Functional antagonism of Smac mimetics against XIAP BIR3 protein in a cell-free functional assay
Based upon their expected mode of action, Smac mimetics should antagonize the inhibition of XIAP BIR3 to caspase-9 by binding to the same site where caspase-9 binds. We thus established a cell-free functional assay using recombinant XIAP BIR3 protein and evaluated the ability of our designed Smac mimetics to antagonize the XIAP BIR3 protein.26 In this assay, addition of dATP and cytochrome c into cell lysates activates caspase-9, and recombinant XIAP BIR3 protein inhibits the activity of caspase-9 in a dose-dependent manner. XIAP BIR3 protein at 0.5 μM is capable of completely inhibiting the activity of caspase-9 under the assay conditions (Figure 3).
Figure 3.

Functional antagonism of compounds 20, 24, 25, and the AVPI peptide against XIAP BIR3 to promote the activity of caspase-9 in a cell-free functional assay.
We evaluated two potent Smac mimetics, compounds 20 and 25, and the Smac AVPI peptide in this cell-free functional assay for their ability to directly overcome the inhibitory effect of XIAP BIR3 protein to caspase-9 activity (Figure 3). Compound 24, which is >500-times less potent than compounds 20 and 25 in its binding affinity to XIAP BIR3, was included as a negative control. It was found that compounds 20, 25 and the AVPI peptide all effectively and dose-dependently restore the activity of caspase-9, whereas compound 24 has no significant effect at concentrations as high as 100 μM. Based upon the kinetic data, the calculated EC50 values for compounds 20, 25 and the AVPI peptide in restoration of the caspase-9 activity at 60 minutes are 0.78, 0.63 and 4.8 μM, respectively, consistent with their binding data to XIAP BIR3. Hence, our functional assay established that compounds 20 and 25 are potent antagonists of XIAP BIR3 and both of them are also more potent than the Smac AVPI peptide. Consistent with its weak binding affinity, 24 is inactive at concentrations as high as 100 μM in the functional assay.
Evaluation of the activity of Smac mimetics in cell growth inhibition and induction of cell death and apoptosis in human cancer cell lines
One major motivation for the design and synthesis of non-peptidic Smac mimetics is to dramatically improve the cellular activity over Smac-based peptides. Although Smac mimetics were initially thought to be primarily useful for sensitizing cancer cells to other anticancer drugs20,21,23,24,29, a number of studies have shown that properly designed Smac mimetics are capable of inhibiting cancer cell growth and inducing apoptosis in a subset of cancer cell lines,25,30 such as the MDA-MB-231 human breast cancer cell line and SK-OV-3 human ovarian carcinoma cell line.
We have therefore tested compounds 20, 21 and 25 for their activity in inhibition of cell growth and induction of cell death and apoptosis as single agents in the MDA-MB-231 and SK-OV-3 cancer cell lines. In our previous study,26 we showed that methylation of the primary amine group in compound 12, which resulted in compound 13, did not significantly alter the binding affinity to XIAP BIR3 but led to dramatic improvement in the cellular activity. To further confirm this observation in our Smac mimetics containing the [8,5] bicyclic ring, we have included compound 17, which contains a primary amine group and differs from compound 21 by the methylation of the amine group. Compound 24 was used as another control in these cellular assays to demonstrate the specificity of compounds 20, 21 and 25. The results are shown in Figure 4.
Figure 4.

Inhibition of cell growth by Smac mimetics in the MDA-MB-231 breast cancer and SK-OV-3 ovarian cancer cell lines. Cells were treated for 4 days and cell growth was determined using WST-based assay.
Compounds 20, 21 and 25 potently inhibit cell growth in the MDA-MB-231 breast cancer cell line in a dose-dependent manner with IC50 values of 0.41, 0.41 and 0.1 μM, respectively. Similarly, compounds 20, 21 and 25 also potently inhibit cell growth in the SK-OV-3 ovarian cancer cell line with IC50 values of 0.9, 1.0 and 0.18 μM, respectively. Consistent with its weak binding affinity to XIAP BIR3, compound 24 has IC50 values of 7.0 μM and >10 μM, respectively, being >30-times less potent than compound 25 in both cancer cell lines. Consistent with our previous observation, compound 17 has a weak cellular activity in both cell lines, indicating that the methylation on the primary amine group in 17 results in dramatic improvement of the cellular activity.
We next evaluated if our designed Smac mimetics can effectively induce apoptosis in the MDA-MB-231 cell line and the results are shown in Figure 5. As expected, compounds 20, 21 and 25 effectively induce apoptosis in a dose-dependent manner. While all three compounds induce robust apoptosis at 100 nM, compound 25 is the most potent among them, consistent with their potency in inhibition of cell growth. Similar data were obtained using the SK-OV-3 ovarian cancer cell line (data not shown). Our data thus indicate that potent Smac mimetics not only potently inhibit cell growth but also effectively induce apoptosis in cancer cells.
Figure 5.

Induction of apoptosis by Smac mimetics in the human breast cancer MDA-MB-231 cell line. Cells were treated for 48 hours and apoptosis was determined using Annexin V-propidium iodide(P.I.) staining by flow cytometry. Annexin V (+) and P.I. (−) cells are labeled as early apoptotic cells, whereas Annexin V (+) and P.I. (+) cells are labeled as late apoptotic cells. Percentage of early and late apoptotic cells are shown.
Since XIAP potently inhibits caspase activity, we next determined if compounds 20 and 25 induces apoptosis and cell death in a caspase dependent manner. Using a trypan blue exclusion assay, it was shown that both compounds effectively kills the MDA-MB-231 cancer cells in a dose-dependent manner but 25 is more potent (Figure 6). Treatment with 25 at 10, 100 and 1000 nM for 24-h kills 9%, 23% and 65% of the MDA-MB-231 cells, respectively. The cell killing by both 20 and 25 can be effectively attenuated by Z-DEVD-FMK, a cell-permeable caspase-3/7 inhibitor, indicating that cell death induction by these Smac mimetics is caspase-3/-7 dependent (Figure 6).
Figure 6.

Induction of cell death by compounds 20 and 25 in the human breast cancer MDA-MB-231 cell line and dependence on caspase-3/7. Cells were treated with different concentrations of Smac mimetic for 48 hours and cell viability was determined using trypan blue exclusion assay. To determine the dependence of cell death induction by Smac mimetics on caspase-3/-7, a cell-permeable caspase-3/-7 inhibitor, Z-DEVD-FMK, was employed.
Determination of the crystal structure of compound 21 in complex with XIAP BIR3 domain
To firmly establish the structural basis of the binding of our designed Smac mimetics to XIAP BIR3, we have determined the crystal structure of compound 21 in complex with XIAP BIR3 to 2.8 Å resolution (Figure 7).
Figure 7.

X-ray crystal structure of compound 21 in complex with human XIAP BIR3, determined to 2.8 Å resolution. (A). Overall representation of compound 21 in complex with XIAP BIR3. Compound 21 is shown in green ball-n-stick. (B). Detailed interactions between compound 21 and XIAP BIR3 residues shown as a 90° rotation of the yellow domain in A. Compound 21 is shown in yellow ball-n-stick. (C). Superposition of modeled and crystal structures of compound 21 in complex with XIAP BIR3.
The experimentally determined structure of compound 21 in complex with one XIAP BIR3 molecule is in excellent agreement with that predicted by computational modeling. The root-mean-square-deviation (RMSD) between the non-hydrogen atoms of compounds 21 in the crystal structure and the modeled structure is 0.81 Å.
The interactions of compound 21 with XIAP BIR3 closely mimic those between the Smac AVPI peptide and the protein, although some differences are observed. The methyl group in Ala1 side chain in the Smac AVPI peptide is replaced with an ethyl group in compound 21. This ethyl group makes more extensive van der waals interactions with Trp 310 and Leu 307 than the methyl group in Ala1 in Smac, which, in turn, produces a minor displacement of the positively charged N-terminal amino group. In Smac, this positively charged amino group in Ala1 has three hydrogen bonds to the side chains of Gln319 and Glu314. In compound 21, the N-methylated amino group loses contact with Gln319, but gains an additional hydrogen bonding interaction with the backbone carbonyl group of Asp309.
Similar to the Smac AVPI peptide, the first carbonyl group in compound 21 forms a less than optimal hydrogen bond to the indole NH group in Trp323. The second amino group in 21 has an optimal hydrogen bond with the backbone carbonyl group of Thr308 and the carbonyl group in the [8,5] lactam ring forms an optimal hydrogen bond with the backbone amino group of Thr308. Another strong hydrogen bond is observed between the third amino group and the backbone carbonyl group of Gly306. For hydrophobic contacts, the ethyl group in 21 inserts into a small hydrophobic pocket formed by the hydrophobic side chains of Leu307 and Trp310, as well as the hydrophobic portion of the side chain in Gln319, mimicking the methyl group in alanine residues in the Smac AVPI peptide. The 8-membered ring in 21 has extensive contacts with the side chain of Trp323. While the five-membered ring in 21 still has contacts with the side chain of Trp323, similar to that in Smac, but the interaction appears to be weaker than that in Smac. However, this five-membered ring in 21 gains additional new contacts with Tyr324. As predicted by modeling, the pro R phenyl group inserts into a hydrophobic pocket formed by the hydrophobic side chains of Leu292 and the hydrophobic portion of the side chains in Lys297 and Lys299.
Interestingly, the crystal structure also revealed a remarkable dimer, which consists of two molecules of the protein and compound 21 (Figure 7A). The dimeric interface created between the two compound 21:XIAP BIR3 complexes in this crystal structure is a unique dimer configuration when compared to other BIR3 three-dimensional structures.33,34 This dimeric interface is mainly stabilized by the presence of compound 21. The pro S phenyl group in both molecules of compound 21 makes multiple hydrophobic interactions with Trp323 and Tyr324 in the other protein molecule in the symmetric dimer. In addition, the [8,5] bicyclic lactam ring from each molecule of 21 stacks on the top of each other and has nice hydrophobic contacts. Other protein-protein interactions that stabilize this interface come from the N-terminal Phe250 from each molecule. The hydrophobic side chain of each Phe250 residue in the dimeric structure has extensive contacts with the diphenyl groups in compound 21 from the opposite side. This dimeric structure formed between 2 molecules of compound 21 and two XIAP BIR3 molecules provides a structural basis for the design of bivalent Smac mimetics to either induce dimerization for two BIR domains or concurrent targeting of two different BIR domains in the same IAP molecule.
Synthesis of designed Smac mimetics
The synthesis for Smac mimetics containing the [6,5] and [7,5] bicyclic lactam systems have been reported previously by our group.23,24 The general synthetic method for Smac mimetics containing the [8,5] bicyclic lactam system is shown in Scheme 1.
Scheme 1. General synthesis of conformationally constrained [8,5] bicyclic Smac mimetics.

Reagents and conditions: (a) i. Amine, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, overnight; ii. 4 N HCl in 1,4-dioxane, MeOH; iii. N-Boc-L-aminoacid, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2; iv. 4 N HCl in 1,4-dioxane, MeOH; (b) 1,4-dibromobutane or 2-bromoethyl ether, N,N-diisopropylethylamine, MeOH, reflux, 67% for 22 and 54% for 23; (c) i. aminodiphenylmethane, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, overnight; ii. 4 N HCl in 1,4-dioxane, MeOH; iii. L-lactic acid, EDC, HOBt, N,N-diisopropylethylamine, CH2Cl2, 71% over three steps.
The key Intermediate 28 was prepared according to a previously reported method.30,30 Condensation of 28 with corresponding amines gave a series of amides. After removal of the Boc protecting group in these amides, the resulted ammonium salts were condensed with corresponding N-Boc amino acids, followed by deprotection of the Boc protecting group to yield compounds 16–21 and 25–27. Refluxing of 17 with 1,4-dibromobutane or 2-bromoethyl ether under the existence of N,N-diisopropylethylamine in methanol furnished compounds 22 and 23. Condensation of 28 and aminodiphenylmethane furnished an amide. Removal of the Boc protecting group in this amide followed by condensation of the resulted ammonium salt with L-lactic acid yielded compound 24.
Summary
Based upon the crystal structure of Smac in complex with XIAP BIR3 domain, we have designed and synthesized a series of conformationally constrained, bicyclic Smac mimetics. Our studies have generated important structure-activity relationships for their binding affinities to XIAP BIR3 and for their activity in inhibition of cell growth in human cancer cells. Our efforts have led to the design of a number of potent and cell-permeable Smac mimetics such as 20 (SM-122), 21 (SM-130) and 25 (SM-230). These compounds bind to XIAP BIR3 with high affinities and antagonize XIAP BIR3 in cell-free functional assays. Importantly, they are potent and effective in inhibition of cell growth and in induction of apoptosis in cancer cells. Our determination of the crystal structure of compound 21 in complex with XIAP BIR3 provides the structural basis for its high-affinity interaction with XIAP BIR3 and for further design and optimization. These potent and cell-permeable Smac mimetics serve as powerful pharmacological tools to further elucidate the role XIAP and other IAP proteins in apoptosis regulation and promising lead compounds for the development of a new class of anticancer drugs.
Experimental Section
I. Chemistry
General Methods
NMR spectra were acquired at a proton frequency of 300 MHz. 1H chemical shifts are reported with TMS (0.00 ppm) or DHO (4.70 ppm) as internal standards. 13C chemical shifts are reported with CDCl3 (77.00 ppm) or 1, 4-dioxane (67.16 ppm) as internal standards. The final products were purified by HPLC on Waters Sunfire C18 reverse phase semi-preparative HPLC column (19×150 mm) using solvent A (0.1% of TFA in water) and solvent B (0.1% of TFA in CH3CN) as eluents with a flow rate of 10 mL/min.
General synthetic method for [8,5] ring contained Smac mimetics
To a solution of compound 28 (1 mmol) in 10 mL of CH2Cl2 was added corresponding amine (1.2 mmol), EDC (1.2 mmol), HOBt (1.2 mmol) and N,N-diisopropylethylamine (3 mmol) at 0 °C. The mixture was stirred at room temperature overnight. After condensation, the residue was purified by chromatography to give an amide. To a solution of this amide in 5 mL of methanol was added 2 mL of HCl solution (4 N in 1,4-dioxane). The solution was stirred at room temperature overnight and then concentrated to give an ammonium salt. To a mixture of this salt in 10 mL of CH2Cl2 was added a desired N-Boc-L-amino acid (1.2 mmol), EDC (1.2 mmol), HOBt (1.2 mmol) and N,N-diisopropylethylamine (3 mmol) at 0 °C. The mixture was stirred at room temperature overnight and then concentrated. The residue was purified by chromatography to give an amide. To a solution of this amide in 5 mL of methanol was added 1 mL of HCl solution (4 N in 1,4-dioxane). The solution was stirred at room temperature overnight and then concentrated. The residue was purified by C18 reverse phase semi-preparative HPLC to give the expected Smac mimetic.
(3S,6S,11S)-6-((S)-2-Amino-propionylamino)-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (16)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1H NMR (300 M Hz, D2O) δ 7.30-7.18 (m, 10H), 5.95 (s, 1H), 4.73 (m, 1H), 4.35 (m, 1H), 4.22 (m, 1H), 3.96 (m, 1H), 2.16 (m, 1H), 2.05 (m, 1H), 1.93-1.32 (m, 10 H), 1.39 (d, J = 7.5 Hz, 3H); 13C NMR (75 MHz, D2O) δ 173.67, 172.65, 169.93, 141.64, 141.45, 129.57, 129.45, 128.34, 128.09, 127.77, 62.50, 61.40, 58.16, 57.55, 51.42, 36.16, 33.30, 32.64, 28.07, 25.36, 22.14, 15.95; ESI MS: m/z 463.3 (M+H)+; HR ESI MS for C27H35N4O3 required: 463.2709, found: 463.2699; Anal. (C27H34N4O3·1.0HCl·1.0CF3COOH) C, H, N.
(3S,6S,11S)-6-((S)-2-Amino-butyrylamino)-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (17)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1H NMR (300 M Hz, D2O) δ 7.30-7.18 (m, 10H), 5.95 (s, 1H), 4.71 (m, 1H), 4.34 (m, 1H), 4.22 (m, 1H), 3.83 (m, 1H), 2.14 (m, 1H), 2.03 (m, 1H), 1.93-1.20 (m, 12 H), 0.88 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, D2O) δ 173.67, 172.60, 169.96, 141.64, 141.34, 129.54, 129.45, 128.37, 128.05, 127.78, 62.41, 61.39, 58.18, 54.65, 51.41, 36.22, 33.43, 32.66, 28.09, 25.34, 25.04, 22.16, 9.07; ESI MS: m/z 477.3 (M+H)+; HR ESI MS for C28H37N4O3 required: 477.2866, found: 477.2858; Anal. (C28H36N4O3·1.0HCl·2.0H2O) C, H, N.
(3S,6S,11S)-6-((S)-2-Amino-butyrylamino)-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid ((R)-naphthalen-1-yl-phenyl-methyl)-amide (18)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1HNMR (300 MHz, D2O) δ 7.75 (m, 1H), 7.65 (m, 1H), 7.58 (m, 1H), 7.32-7.10 (m, 4H), 7.10-6.92 (m, 5H), 6.58 (s, 1H), 4.63 (m, 1H), 4.27 (m, 1H), 3.80 (m, 1H), 2.12-1.65 (m, 6H), 1.65-1.20 (m, 8H), 0.86 (t, J = 7.5 Hz, 3H); 13CNMR (75 MHz, D2O) δ 173.19, 172.59, 169.83, 141.15, 136.53, 134.23, 131.35, 131.11, 129.40, 128.32, 128.04, 127.37, 126.66, 126.08, 124.02, 62.20, 61.18, 54.96, 54.60, 51.33, 36.32, 33.32, 32.61, 27.92, 25.33, 25.01, 22.19, 9.08; ESI MS: m/z 527.3 (M+H)+; HR ESI MS for C32H39N4O3 required: 527.3022, found: 527.3021; Anal. (C32H38N4O3·1.0HCl·1.0CF3COOH·0.6H2O) C, H, N.
(3S,6S,11S)-6-((S)-2-Amino-butyrylamino)-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid ((S)-naphthalen-1-yl-phenyl-methyl)-amide (19)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1HNMR (300 MHz, D2O) δ 7.58 (m, 1H), 7.20-6.50 (12H), 4.56 (m, 1H), 4.33 (m, 1H), 3.87 (m, 1H), 3.74 (m, 1H), 1.82-0.90 (m, 12H), 0.80 (t, J = 7.4 Hz, 3H); 13CNMR (75 MHz, D2O) δ 172.22, 172.16, 169.64, 141.38, 141.19, 139.58, 137.49, 134.18, 131.30, 129.21, 128.69, 128.18, 127.00, 125.97, 125.34, 124.57, 123.90, 62.01, 60.87, 54.62, 54.18, 51.13, 36.32, 33.58, 32.51, 27.74, 25.37, 25.04, 22.30, 9.10; ESI MS: m/z 527.3 (M+H)+; HR ESI MS for C32H39N4O3 required: 527.3022, found: 527.3013; Anal. (C32H38N4O3·1.0HCl·1.0CF3COOH·0.8H2O) C, H, N.
(3S,6S,11S)-6-((S)-2-Methylamino-propionylamino)-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (20)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1H NMR (300 M Hz, D2O) δ 7.30-7.18 (m, 10H), 5.95 (s, 1H), 4.74 (m, 1H), 4.34 (m, 1H), 4.24 (m, 1H), 3.83 (m, 1H), 2.57 (s, 3H), 2.21-1.50 (m, 11H), 1.40 (d, J = 7.0 Hz, 3H), 1.38 (m, 1H); 13C NMR (75 MHz, 173.77, 172.62, 169.95, 141.56, 141.28, 129.54, 129.44, 128.40, D2O) δ 128.35, 128.01, 127.82, 62.50, 61.40, 58.16, 57.49, 51.42, 36.16, 33.30, 32.64, 31.61, 28.07, 25.36, 22.14, 15.95; Anal. (C28H36N4O3·1.0HCl·1.0CF3COOH) C, H, N.
(3S,6S,11S)-6-((S)-2-Methylamino-butyrylamino)-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (21)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The putridity was checked by analytical HPLC to be over 98%. 1H NMR (300 M Hz, D2O) δ 7.32-7.19 (m, 10H), 5.95 (s, 1H), 4.75 (m, 1H), 4.36 (m, 1H), 4.25 (m, 1H), 3.71 (m, 1H), 2.56 (s, 3H), 2.18-1.40 (m, 14H), 0.87 (t, J = 7.6 Hz, 3H); 13C NMR (75 MHz, D2O) δ 173.76, 172.39, 168.73, 141.60, 141.27, 129.54, 129.44, 128.37, 128.05, 127.76, 62.96, 62.41, 61.44, 58.21, 51.51, 36.18, 33.40, 32.66, 32.04, 28.10, 25.38, 24.08, 22.11, 8.81; ESI MS: m/z 491.3 (M+H)+; HR ESI MS for C29H39N4O3 required: 491.3022, found: 491.3026; Anal. (C29H38N4O3·1.0HCl·1.0CF3COOH) C, H, N.
(3S,6S,11S)-6-((S)-(2-pyrrolidin-1-yl-butyrylamino))-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (22)
To a solution of 17 (salt with HCl, 260 mg, 0.5 mmol) in 10 mL of methanol was added 1,4-dibromobutane (430 mg, 2 mmol) and N,N-diisopropylethylamine (0.5 mL). The solution was refluxed overnight and then condensed. The residue was dissolved in 5 mL of methanol. To this solution was added 0.5 mL of TFA. After condensation, the residue was purified by C18 reverse phase semipreparative HPLC to give 22 (215 mg, 67%). The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 40 min. The purity was checked by analytical HPLC to be over 98%.1H NMR (300 M Hz, D2O) δ 7.30-7.19 (m, 10H), 5.95 (s, 1H), 4.77 (m, 1H), 4.36 (m, 1H), 4.25 (m, 1H), 3.68 (m, 1H), 3.50-2.70 (brs, 4H), 2.23-1.39 (m, 18H), 0.85 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, D2O) δ 173.78, 172.23, 168.82, 141.60, 141.26, 129.54, 129.45, 128.38, 128.06, 127.74, 69.12, 62.40, 61.45, 58.22, 51.52, 36.17, 33.45, 32.66, 28.12, 26.40, 25.38, 23.64, 23.23, 22.10, 8.98; ESI MS: m/z 531.3 (M+H)+; HR ESI MS for C32H43N4O3 required: 531.3335, found: 531.3334; Anal. (C32H42N4O3·1.0HCl·1.3H2O) C, H, N.
(3S,6S,11S)-6-((S)-2-Morpholin-4-yl-butyrylamino))-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (23)
To a solution of 17 (salt with HCl, 256 mg, 0.5 mmol) in 10 mL of methanol was added 2-bromoethyl ether (580 mg, 2.5 mmol) and N,N-diisopropylethylamine (0.5 mL). The solution was refluxed overnight and then condensed. The residue was dissolved in 5 mL of methanol. To this solution was added 0.5 mL of TFA. After condensation, the residue was purified by C18 reverse phase semipreparative HPLC to give 23 (178 mg, 54%). The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 40 min. 1H NMR (300 M Hz, D2O) δ 7.30-7.19 (m, 10H), 5.95 (s, 1H), 4.79 (m, 1H), 4.37 (m, 1H), 4.25 (m, 1H), 3.86 (brs, 4H), 3.67 (m, 1H), 3.31-3.20 (m, 4H), 2.18-1.39 (m, 14H), 0.85 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, D2O) δ 173.77, 172.16, 167.95, 141.61, 141.26, 129.54, 129.45, 128.38, 128.06, 127.74, 70.21, 64.28, 62.40, 61.45, 58.22, 51.58, 51.02, 36.17, 33.44, 32.66, 28.12, 25.37, 22.09, 21.22, 9.14; ESI MS: m/z 547.3 (M+H)+; HR ESI MS for C32H43N4O4 required: 547.3284, found: 547.3298; Anal. (C32H42N4O4·1.0HCl·2.5H2O) C, H, N.
(3S,6S,11S)-6-((S)-2-Hydroxy-propionylamino))-5-oxo-decahydro-pyrrolo[1,2-α]azocine-3-carboxylic acid benzhydryl-amide (24)
To a solution of 28 (320 mg, 1 mmol) in 10 mL of CH2Cl2 was added aminodiphenylmethane (220 mg, 1.2 mmol), EDC (230 mg, 1.2 mmol), HOBt (160 mg, 1.2 mmol) and N,N-diisopropylethylamine (1.2 mL) at 0 °C. The mixture was stirred at room temperature overnight. After condensation, the residue was purified by chromatography to give an amide. To a solution of this amide in 5 mL of methanol was added 2 mL of HCl solution (4 N in 1,4-dioxane). The solution was stirred at room temperature overnight and then condensed to give an ammonium salt. To a mixture of this salt in 10 mL of CH2Cl2 was added L-lactic acid (110 mg, 1.2 mmol), EDC (232 mg, 1.2 mmol), HOBt (164 mg, 1.2 mmol) and N,N-diisopropylethylamine (1.2 mL) at 0 °C. The mixture was stirred at room temperature overnight and then condensed. The residue was purified by chromatography to give compound 24 (328 mg, 71% over three steps). 1H NMR (300 M Hz, CDCl3, TMS) δ 7.78 (brd, J = 8.4 Hz, 1H), 7.37-7.20 (m, 11H), 6.22 (brd, J = 8.5 Hz, 1H), 4.88 (m, 1H), 4.64 (m, 1H), 4.23 (m, 2H), 3.80 (brs, 1H), 2.50 (m, 1H), 2.12 (m, 1H), 2.08-1.75 (m, 4H), 1.74-1.39 (m, 5H), 1.41 (d, J = 7.2 Hz, 3H), 1.27 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 174.17, 172.08, 169.77, 141.63, 141.47, 128.60, 128.52, 127.52, 127.36, 127.22, 68.27, 60.32, 59.48, 57.10, 49.55, 36.59, 35.36, 32.16, 25.03, 24.70, 22.94, 20.88; ESI MS: m/z 464.3 (M+H)+; HR ESI MS for C27H34N3O4 required: 464.2549, found: 464.2544; Anal. (C27H33N3O4·1.6H2O) C, H, N.
(3S,6S,11S)-6-((S)-2-Methylamino-propionylamino)-5-oxo-decahydro-pyrrolo[1,2-a]azocine-3-carboxylic acid ((R)-(1,2,3,4-tetrahydro-naphthalen-1-yl)-amide (25)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1HNMR (300 MHz, D2O) δ 7.22-7.04 (m, 4H), 4.87 (m, 1H), 4.70 (m, 1H), 4.32-4.16 (m, 2H), 3.90-3.78 (m, 1H), 2.68-2.60 (m, 2H), 2.56 (s, 3H), 2.16-1.42 (m, 16), 1.40 (d, J = 7.2 Hz, 3H); 13CNMR (75 MHz, D2O) δ 173.62, 172.70, 169.86, 138.61, 136.40, 129.76, 128.93, 128.06, 126.85, 62.76, 61.44, 57.49, 51.48, 48.62, 36.26, 33.30, 32.65, 31.61, 30.08, 29.19, 28.18, 25.44, 22.18, 20.47, 15.94; ESI MS: m/z 441.3 (M+H)+; HR ESI MS for C25H37N4O3 required: 441.2866, found: 441.2862; Anal. (C25H36N4O3·1.0HCl·1.0CF3COOH) C, H, N.
(3S,6S,11S)-6-((S)-2-Methylamino-propionylamino)-5-oxo-decahydro-pyrrolo[1,2-a]azocine-3-carboxylic acid ((S)-(1,2,3,4-tetrahydro-naphthalen-1-yl)-amide (26)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1HNMR (300 MHz, D2O) δ 7.20-7.02 (m, 4H), 4.88 (m, 1H), 4.22 (m, 1H), 4.16 (m, 1H), 3.83 (m, 1H), 2.69-2.60 (m, 2H), 2.57 (s, 3H), 2.20-1.52 (m, 15 H), 1.44 (m, 1H), 1.40 (d, J = 7.2 Hz, 3H); 13CNMR (75 MHz, D2O) δ 173.65, 172.61, 169.88, 138.67, 136.46, 129.89, 128.74, 128.13, 126.84, 63.01, 61.39, 57.48, 51.45, 48.44, 36.30, 33.30, 32.71, 31.64, 30.16, 29.10, 28.15, 25.43, 22.21, 20.22, 15.97; ESI MS: m/z 442.3 (M+H)+; HR ESI MS for C25H37N4O3 required: 441.2866, found: 441.2864; Anal. (C25H36N4O3·1.0HCl·1.0CF3COOH) C, H, N.
(3S,6S,11S)-6-((S)-2-Methylamino-propionylamino)-5-oxo-decahydro-pyrrolo[1,2-a]azocine-3-carboxylic acid ((R)-indan-1-ylamide)-amide (27)
The gradient ran from 70% of solvent A and 30% of solvent B to 50% of solvent A and 50% of solvent B in 30 min. The purity was checked by analytical HPLC to be over 98%. 1HNMR (300 MHz, D2O) δ 7.23-7.06 (m, 4H), 5.19 (t, J = 7.8 Hz, 1H), 4.75 (m, 1H), 4.32-4.18 (m, 2H), 3.83 (m, 1H), 2.98-2.69 (m, 2H), 2.56 (s, 3H), 2.36 (m, 1H), 2.22-1.50 (m, 12 H), 1.43 (m, 1H), 1.40 (d, J = 7.0 Hz, 3H); 13CNMR (75 MHz, D2O) δ 174.18, 172.66, 169.87, 144.29, 143.14, 128.66, 127.35, 125.45, 124.66, 62.78, 61.45, 57.48, 55.33, 51.48, 36.25, 33.39, 33.30, 32.65, 31.60, 30.27, 28.20, 25.44, 22.17, 15.94; ESI MS: m/z 427.3 (M+H)+; HR ESI MS for C24H35N4O3 required: 427.2709, found: 427.2707; Anal. (C24H34N4O3·1.0HCl·1.0CF3COOH) C, H, N.
II. Molecular modeling
The crystal structure of XIAP BIR3 in a complex with the Smac protein15 (PDB reference:1G73) was used to predict the binding models of XIAP BIR3 bound to designed compounds. We used the Amber program suite40 (version 7) to perform all molecular dynamics (MD) simulations. A recent version of the Amber force field (ff96)41 was used for the natural amino acids in the complex and the TIP3P model42 was used for water molecules. There is one Zn2+ ion covalently bound to C300, C303, H320, and C327 in the XIAP BIR3 domain. This Zn2+ ion, while important for structural integrity, has no direct interaction with the ligands. We used parameters developed by Ryde43 for the Zn2+ ion and its coordination with the neighboring four residues to model this chelating structure in our simulation. All the MD simulations were carried out at NTP. The SHAKE algorithm44 was used to fix the bonds involving hydrogen. The PME method45 was used to account for long range electrostatic interactions and the non-bonded cutoff distance was set at 10 Å. The time step was 2 fs, and neighboring pairs list was updated after every 20 steps. For the refinement of the structure between compound 20 and XIAP BIR3, the protocol is as follows: A 500-step minimization of the solvated system was performed followed by 6 ps of MD simulation to gradually heat the system from 0K to 298K. The system was then equilibrated by another 34 ps simulation at 298K. Finally, the 1 ns production simulation was run and the snapshots of conformations (typically 2000), evenly spaced in time, were collected for structural analysis.
All the initial binding poses of designed ligands with XIAP BIR3 were predicted using the GOLD program46 (version 2.2). The center of the binding site for XIAP BIR3 was set at Thr308 and the radius of the binding site was defined as 13 Å, large enough to cover the binding pocket. For each genetic algorithm run, a maximum number of 200,000 operations were performed on a population of 5 islands of 100 individuals. Operator weights for crossover, mutation and migration were set to 95, 95 and 10 respectively. The docking simulations were terminated after 20 runs for each ligand. GoldScore implemented in Gold 2.2 was used as the fitness function to evaluate the docked conformations. The 20 conformations ranked highest by each fitness function were saved for analysis of the predicted docking modes. For the docking poses reported, these were the highest ranked conformations from the docking simulations.
III. Protein expression and purification, binding assays to XIAP BIR3 and cell-free functional assays for the activity of caspase-9
Protein expression and purification
An N-terminal 6xHis-tagged BIR3 domain from human XIAP expressed in abundance of 50 mg of protein per liter of culture in Escherichia coli and was readily purified from soluble extracts. The sequence encoding the BIR3 domain (residues 241 – 356) of the human XIAP gene was cloned into the pET28a expression vector. High protein expression levels were achieved by growing transformed BL21(DE3) cells at 37 °C in 2xYT media containing 30 μg/ml kanamycin to an optical density (600 nm) of 0.6 and inducing with 0.4 mM isopropyl-D-thiolgalactoside for 4 hours at 25 °C. Cells pelleted at 5000 g were frozen at − 80 °C and resuspended in lysis buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 50 mM Zn acetate, 3 mM β-mercaptoethanol and protease inhibitors. The cells were subjected to sonication and the cellular debris pelleted by centrifugation at 30,000 g for 20 min. The soluble fraction was applied to Ni-NTA agarose (Qiagen) pre-equilibrated with wash buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 50 mM Zn acetate and 25 mM imidazole. The BIR3 domain protein was then eluted from the column using wash buffer containing 250 mM imidazole. To reduce the number of protein aggregates, a final concentration of 1 mM DTT was added to the elute before applying it to a Superdex-75 column (Pharmacia) equilibrated with 20 mM Tris pH 7.5, 200 mM NaCl, 50 mM Zn acetate and 1 mM DTT. The resulting protein was judged to be greater than 98% pure by the presence of a single band on an SDS-PAGE gel.
Fluorescence-polarization-based binding for XIAP BIR3 protein
A sensitive in vitro binding assay using the fluorescence polarization (FP)-based method32 was used to determine the binding affinity of Smac mimetics to XIAP BIR3 protein. In this assay, 5-carboxyfluorescein was coupled to the lysine side chain of a mutated Smac peptide with the sequence (AbuRPFK-Fam). This fluorescently tagged peptide, named SM5F, was used as the fluorescent tracer in the FP-based binding assay of different compounds to the XIAP BIR3 protein (residues 241–356). The Kd value of SM5F peptide to XIAP BIR3 protein was determined to be 17.9 nM.32
Dose-dependent binding experiments were carried out with serial dilutions of the tested compounds. An aliquot of the samples and preincubated XIAP BIR3 protein (0.030 μM) and SM5F peptide (5 nM) in the assay buffer (100 mM potassium phosphate, pH 7.5; 100 μg/ml bovine gamma globulin; 0.02% sodium azide, purchased from Invitrogen Life Technology), were added to Dynex 96-well, black, round-bottom plates (Fisher Scientific). For each assay, the controls included XIAP BIR3 protein and SM5F (equivalent to 0% inhibition), and SM5F only (equivalent to 100% inhibition). The polarization values were measured after 3 hours of incubation using an ULTRA READER (Tecan U.S. Inc., Research Triangle Park, NC). IC50 values, the inhibitor concentration at which 50% of the bound tracer is displaced, were determined from a plot using nonlinear least-squares analysis. Curve fitting was performed using GRAPHPAD PRISM software (GraphPad Software, Inc., San Diego, CA).
Cell-free caspase-9 functional assay
MDA-MB-231 cell lysates were prepared by solubilizing cells in ice cold buffer (pH 7.5), which contained 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM DTT, 0.2% CHAPS and 50 mM HEPES, and cocktail protease inhibitors and incubated on ice for 10 minutes. Cytochrome c and dATP were added into the cell lysates, and incubated at 30 °C in a water bath for 60 minutes to activate caspase-9. To inhibit the activity of caspase-9, recombinant human XIAP BIR3 protein was added and was found to dose-dependently inhibit the activity of caspase-9. To determine caspase-9 activity, 25 μM of caspase-9 substrate (Z-LEHD-AFC) purchased from BioVision was added. Fluorescence detection of substrate cleavage by caspase-9 was carried out with an ULTRA READER using an excitation wavelength of 400 nm and an emission wavelength of 505 nm and the reaction was monitored for 60–120 minutes. To determine the antagonism of a Smac mimetic, different concentrations of a tested Smac mimetic (from 100 μM to 1 nM) were added into the reaction mixture together with XIAP BIR3 protein.
IV. Assays for cell growth, apoptosis and cell viability
Cell lines
MDA-M-231 and SK-OV-3 cancer cell lines were purchased from the American Type Culture Collection (Manassas, Virginia, USA), maintained in RPMI 1640 (Invitrogen), supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin, at 37°C in 5% CO2. Both cell lines were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin in the presence of puromycin from EMD Biosciences (2 μg/ml).
Cell growth assay
The effect of Smac mimetics on cell growth was evaluated by a WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt] assay (Dojindo Molecular Technologies, Inc). Cells (3000–4000 cells in each well) were cultured in 96-well tissue culture plates in medium (200 μl) containing various concentrations of Smac mimetics for the indicated time. At the end of incubation, WST-8 dye (20 μl) was added to each well and incubated for an additional 1–3 h, then the absorbance was measured in a microplate reader (Molecular Devices) at 450 nm. Cell growth inhibition was evaluated as the ratio of the absorbance of the sample to that of the control.
Apoptosis assay
The apoptosis assay was performed with an annexin-V/propidium iodide (PI) apoptosis detection kit (Roche) according to the manufacturer’s instructions. Briefly, cells were harvested, washed with ice-cold PBS and then stained with annexin-V-FITC and PI for 15 minutes at room temperature in the dark. Stained cells were analyzed in a FACS calibur flow cytometer. Annexin-V (−)/PI (−) cells were classified as live cells; Annexin-V (+)/PI (−) cells were classified as early apoptotic cells; Annexin-V (+)/PI (+) cells were classified as late apoptotic cells; Annexin V (−) and P.I. (+) cells were classified as dead cells.
Cell viability assay
Cell viability was determined using a trypan blue exclusion assay. Approximately 10,000 MDA-MB-231 cells were seeded in 24-well plates and incubated at 37 °C in an atmosphere of 95% air and 5% CO2 for 24 hours before drug treatments. Cells were treated with different concentrations of compounds 20 and 25 for 48 hours. To examine the role of caspase-3/-7 in cell death induction by Smac mimetics, a cell-permeable caspase-3/-7 inhibitor, Z-DEVD-FMK (Calbiochem, San Diego, CA) was employed and cells were treated with a combination of Z-DEVD-FMK with compound 20 or 25 concurrently. To determine cell viability, 1:1 dilution of the suspension of 0.4% Trypan blue reagent (Invitrogen Corporation) was used. Blue cells were counted as non-viable cells. At least 100 cells were counted in the control well and three independent countings were performed.
IV. Crystallographic studies
Crystallization of XIAP BIR3:compound 21 complex
Bipyramidal crystals of the BIR3 domain of human XIAP (residues 241 – 356) in complex with compound 21 were grown at 20 °C from hanging drop vapor diffusion experiments by mixing 2 ml of protein (10 mg/ml pre-incubated with 10 mM compound 21) in 20 mM Tris pH 7.5, 200 mM NaCl, 50mM Zn acetate and 1 mM DTT with 2 ml of precipitant (20 % polyethylene glycol 8000, 0.2 M MgCl2 and 0.1 M Tris-HCl pH 8.5). Crystals were cryoprotected with paratone-N and data collected at a wavelength of 1.0 Å at the Advanced Photon Source on the COMCAT 32-ID beam line equipped with a MAR165 CCD and processed with DENZO/SCALEPACK.35 The complex crystallized in space group P6522 with a solvent content of 66%36 resulting in one molecule per asymmetric unit. The data are 99.3% complete to 2.8 Å resolution with a Rsym of 7.2%.
Structure Determination and Refinement
The BIR3:compound 21 complex was solved to 2.8 Å resolution by molecular replacement methods in CNS37 using the crystal structure of XIAP BIR3 (residues 256 – 346) with another Smac mimetic solved in our laboratory as the search model. Difference fourier maps (Fo – Fc) calculated after CNS rigid-body refinement of the BIR3 domain against BIR3:compound 21 diffraction data showed the presence of compound 21 and six additional residues at the N-terminus (250 - 255). Multiple rounds of CNS refinement and model building using O38 resulted in a BIR3:compound 21 structure refined to 2.8 Å resolution (Table I). Simulated annealing omit maps calculated in CNS were used to check for the correct placement of atoms in the structure. The final structure was analyzed in PROCHECK.39 All residues are in the allowed regions of the Ramanchandran plot. The N-terminal residues (237 – 249) and the C-terminal residues (346 – 358) were disordered in the structure.
Supplementary Material
Elemental analysis data for compounds 16–27. This material is available free of charge via the Internet at http://pubs.acs.org.
Table 2.
Data collection, phasing and refinement statistics of the crystal structure of XIAP BIR3:compound 21.
| Data Collection Statistics | |
| Data Set | XIAP BIR3:21 |
| Space Group | P6522 |
| Unit Cell | a = b = 115.76 Å; c = 61.79 Å |
| Wavelength | 1.0000 Å |
| Resolution (Å)1 | 2.8 (2.9 -2.8) |
| Rsym (%)2 | 7.2 (30.8) |
| <I/σI>3 | 20 (3) |
| Completeness (%)4 | 99.3 (100) |
| Redundancy | > 9 |
| Refinement Statistics | |
| Resolution (Å) | 2.8 |
| R-Factor (%)10 | 22.8 (37.5) |
| Rfree (%)11 | 26.9 (46.3) |
| Protein atoms12 | 784 |
| Water Molecules | 38 |
| Unique Reflections | 6181 |
| R.m.s.d.13 | |
| Bonds | 0.007 |
| Angles | 1.203 |
Statistics for highest resolution bin of reflections in parentheses.
Rsym = ΣhΣj l Ihj-<Ih> l/ΣhΣjIhj, where Ihj is the intensity of observation j of reflection h and <Ih> is the mean intensity for multiply recorded reflections.
Intensity signal-to-noise ratio.
Completeness of the unique diffraction data.
Resolution cut-off used during heavy-atom refinement and phase calculations.
Riso =Σ l lFphI − IFpI I/Σ IFpI, where Fp and Fph are the native and heavy-atom structure factor amplitudes, respectively.
Rcullis =Σh/I IFph − FpI − FhI/ΣhIFph − FpI for centric data, where Fp and Fph are the native and heavy-atom structure for factor amplitudes, respectively. The MLPHARE program from CCP4 slightly modifies this equation for acentric and anomalous data.
Phasing power for isomorphic derivatives (PPiso) = <IFhI>/<E>, where Fh is the heavy-atom structure factor amplitude and E is the residual lack of closure error. Phasing power for anomalous-scattering derivatives (PPano) = <2IFH”I>/<E>, where FH” is the calculated anomalous-scattering structure factor.
Number of heavy-atom sites per asymmetric unit.
R-factor = Σh I IFoI − IFcI I/ΣhIFoI, where Fo and Fc are the observed and calculated structure factor amplitudes for reflection h.
Rfree is calculated against a 10% random sampling of the reflections that were removed before structure refinement.
Total number of protein atoms refined in the asymmetric unit.
Root mean square deviation of bond lengths and bond angles.
Acknowledgments
The financial support from the Breast Cancer Research Foundation, the National Cancer Institute, NIH (R01CA109025), the Prostate Cancer Foundation, the Department of Defense Prostate Cancer Program (W81XWH-04-1-0213) and Ascenta Therapeutics are greatly appreciated. We thank Dr. G.W.A. Milne and Ms. Karen Kreutzer for critical reading of the manuscript.
Abbreviations
- IAP
inhibitor of apoptosis protein
- XIAP
X-linked IAP
- Smac
second mitochondria-derived activator of caspases
- DIABLO
direct IAP binding protein with low pI
- BIR
baculoviral IAP Repeats (BIR) domain
- BIR2
the second BIR domain
- BIR3
the third BIR domain
- FP
fluorescence polarization
Footnotes
Coordinates of compound 21 in complex with XIAP BIR3 and structural factors have been deposited in the RCSB protein Data Bank (access code 2JK7).
References
- 1.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407:810–816. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 4.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1:111–121. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 5.Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev. 1999;1:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 6.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 7.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 8.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17:3247–3259. doi: 10.1038/sj.onc.1202569. [DOI] [PubMed] [Google Scholar]
- 9.Holcik M, Gibson H, Korneluk RG. XIAP: Apoptotic brake and promising therapeutic target. Apoptosis. 2001;6:253–261. doi: 10.1023/a:1011379307472. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 11.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, Andreeff M. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–1803. [PubMed] [Google Scholar]
- 12.Fulda S. Inhibitor of apoptosis proteins as targets for anticancer therapy. Expert Rev Anticancer Ther. 2007;7:1255–64. doi: 10.1586/14737140.7.9.1255. [DOI] [PubMed] [Google Scholar]
- 13.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 14.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 15.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z, Sun C, Olejniczak ET, Meadows R, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 18.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11:519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Rich RL, Myszka DG, Wu H. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. J Biol Chem. 2003;278:49517–22. doi: 10.1074/jbc.M310061200. [DOI] [PubMed] [Google Scholar]
- 20.Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nature Med. 2002;8:808–815. doi: 10.1038/nm735. [DOI] [PubMed] [Google Scholar]
- 21.Arnt CR, Chiorean MV, Heldebrant MP, Gores GJ, Kaufmann SH. Synthetic Smac/DIABLO Peptides Enhance the Effects of Chemotherapeutic Agents by Binding XIAP and cIAP1 in Situ. J Biol Chem. 2002;277:44236–44243. doi: 10.1074/jbc.M207578200. [DOI] [PubMed] [Google Scholar]
- 22.Yang L, Mashima T, Sato S, Mochizuki M, Sakamoto H, Yamori T, Oh-Hara T, Tsuruo T. Predominant suppression of apoptosome by inhibitor of apoptosis protein in non-small cell lung cancer H460 cells: therapeutic effect of a novel polyarginine-conjugated Smac peptide. Cancer Res. 2003;63:831–837. [PubMed] [Google Scholar]
- 23.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Liu M, Tomita Y, Pan H, Yoshioka Y, Krajewski K, Roller PP, Wang S. Structure-Based Design of Potent, Conformationally Constrained Smac Mimetics. J Am Chem Soc. 2004;126:16686–16687. doi: 10.1021/ja047438+. [DOI] [PubMed] [Google Scholar]
- 24.Sun H, Nikolovska-Coleska Z, Yang CY, Xu L, Tomita Y, Krajewski K, Roller PP, Wang S. Structure-based design, synthesis, and evaluation of conformationally constrained mimetics of the second mitochondria-derived activator of caspase that target the X-linked inhibitor of apoptosis protein/caspase-9 interaction site. J Med Chem. 2004;47:4147–50. doi: 10.1021/jm0499108. [DOI] [PubMed] [Google Scholar]
- 25.Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H, Fesik SW. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem. 2004;47:4417–26. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 26.Sun H, Nikolovska-Coleska Z, Lu J, Qiu S, Yang CY, Gao W, Meagher J, Stuckey J, Wang S. Design, Synthesis, and Evaluation of a Potent, Cell-Permeable, Conformationally Constrained Second Mitochondria Derived Activator of Caspase (Smac) Mimetic. J Med Chem. 2006;49:7916–7920. doi: 10.1021/jm061108d. [DOI] [PubMed] [Google Scholar]
- 27.Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO, Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother WJ, Deshayes K. Design, Synthesis, and Biological Activity of a Potent Smac Mimetic That Sensitizes Cancer Cells to Apoptosis by Antagonizing IAPs. ACS Chem Biol. 2006;1:525–533. doi: 10.1021/cb600276q. [DOI] [PubMed] [Google Scholar]
- 28.Chauhan D, Neri P, Velankar M, Podar K, Hideshima T, Fulciniti M, Tassone P, Raje N, Mitsiades C, Mitsiades N, Richardson P, Zawel L, Tran M, Munshi N, Anderson KC. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM) Blood. 2007;109:1220–7. doi: 10.1182/blood-2006-04-015149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A Small Molecule Smac Mimic Potentiates TRAIL- and TNF-Mediated Cell Death. Science. 2004;305:1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 30.Sun H, Nikolovska-Coleska Z, Lu J, Meagher JL, Yang CY, Qiu S, Tomita Y, Ueda Y, Jiang S, Krajewski K, Roller PP, Stuckey JA, Wang S. Design, Synthesis, and Characterization of a Potent, Nonpeptide, Cell-Permeable, Bivalent Smac Mimetic That Concurrently Targets Both the BIR2 and BIR3 Domains in XIAP. J Am Chem Soc. 2007;129:15279–15294. doi: 10.1021/ja074725f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duggan HME, Hitchcock PB, Young DW. Synthesis of 5/7-, 5/8- and 5/9-bicyclic lactam templates as constraints for external beta-turns. Org Biomol Chem. 2005;3:2287–2295. doi: 10.1039/b503014e. [DOI] [PubMed] [Google Scholar]
- 32.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–73. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 33.Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Struct Biol. 2000;7:602–608. doi: 10.1038/76838. [DOI] [PubMed] [Google Scholar]
- 34.Franklin MC, Kadkhodayan S, Ackerly H, Alexandru D, Distefano MD, Elliostt LO, Flygare JA, Mausisa G, Okawa DC, Ong D, Vucic D, Deshayes K, Fairbrother WJ. Structure and Function Analysis of Peptide Antagonists of Melanoma Inhibitor of Apoptosis (ML-IAP) Biochemistry. 2003;42:8223–8231. doi: 10.1021/bi034227t. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology, Volume 276: Macromolecular Crystallography, part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 36.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 37.Brunger AT, Admas PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewsji J, Nigles N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 40.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER7. University of California; San Francisco: 2002. [Google Scholar]
- 41.Kollman PA, Dixon R, Cornell W, Fox T, Chipot C, Pohorille A. The development/application of a ‘minimalist’ organic/viochemical molecular mechanics force field using a combination of ab initio calculations and experimental data. In: Wilkinson A, Weiner P, van Gunsteren WF, editors. Computer Simulation of Biomolecular Systems. Vol. 3. Elsevier; 1997. pp. 83–96. [Google Scholar]
- 42.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 43.Ryde U. Molecular Dynamics Simulations of Alcohol Dehydrogenase with a four- or five-coordinate catalytic Zinc ion. Proteins. 1995;21:40–56. doi: 10.1002/prot.340210106. [DOI] [PubMed] [Google Scholar]
- 44.Rychaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 45.Darden TA, York DM, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 46.Jones G, Willett P, Glen RC, Leach AR, Talylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis data for compounds 16–27. This material is available free of charge via the Internet at http://pubs.acs.org.