

Abstract
 A five step-synthesis of fused bis-tetrahydrofurans and attached bis-tetrahydrofurans from butadiene diepoxide is described. Two sequential Pd-catalyzed carboetherification reactions between protected 1,2-diols and aryl/alkenyl bromides, each of which form both a C–O bond and a C–C bond, are used to generate the heterocyclic rings with >20:1 dr. Installation of different R1 and R2 groups is achieved in a straightforward fashion through use of different aryl or alkenyl bromide coupling partners.
A five step-synthesis of fused bis-tetrahydrofurans and attached bis-tetrahydrofurans from butadiene diepoxide is described. Two sequential Pd-catalyzed carboetherification reactions between protected 1,2-diols and aryl/alkenyl bromides, each of which form both a C–O bond and a C–C bond, are used to generate the heterocyclic rings with >20:1 dr. Installation of different R1 and R2 groups is achieved in a straightforward fashion through use of different aryl or alkenyl bromide coupling partners.
A large number of interesting compounds contain attached-ring or fused-ring tetrahydrofuran scaffolds (Figure 1). The 2,6-dioxabicyclo[3.3.0]octane framework (1) is found in both naturally occurring1 and synthetic molecules that are relevant to human health (e.g., 3; antitumor activity)2 and agriculture (e.g., 4; herbicide).3 Attached-ring tetrahydrofurans (2) are displayed in a vast number of natural products, including the annonaceous acetogenins,4 of which asimicin (5) is a member.
Figure 1.

Examples of Biologically Active bis-Tetrahydrofurans
A variety of different approaches have been developed for the construction of these useful compounds.4,5 Many of these strategies involve generation of the bis-tetrahydrofuran framework via sequential (or tandem) ring-closing reactions of 1,2-diols bearing pendant functional groups such as alkenes, epoxides, alcohols, or allylic acetates/halides.6 Although these methods effectively form the heterocyclic ring, they do not allow the simultaneous construction of a C–C bond. Thus, substituents attached to the tetrahydrofuran C2-position, such as the side chains present in 3–5, must be installed in separate steps either prior to or following ring-closure.
We felt that an alternative approach to the construction of substituted fused-ring tetrahydrofurans with the general structure 8 could be developed using sequential Pdcatalyzed carboetherification reactions7,8 of unsaturated 1,2-diols such as 6. As shown in Scheme 1, treatment of 6 with an aryl or alkenyl halide in the presence of NaOtBu and a palladium catalyst should provide 7, which could be converted to 8 in a second catalytic transformation. This strategy could also be applied to the synthesis of attached-ring tetrahydrofurans (e.g., 9 to 10) by simply extending the tether between the alcohols and the alkenes by one methylene unit. Importantly, each carboetherification reaction would generate both a C–O bond (to form the heterocylic ring) and a C–C bond, thus providing a more concise approach to substituted bis-tetrahydrofurans compared to currently available methods.
Scheme 1.

Synthetic Strategy
In order to examine the feasibility of the strategy outlined above, we elected to examine the selective monocyclization of known diols 66i and 9,9 which can be generated by Cu-catalyzed addition of vinylmagnesium bromide or allylmagnesium bromide to commercially available butadiene diepoxide. We also prepared mono-TBS-protected10 derivatives 11 and 12 (Figure 2), as our prior studies indicated that carboetherification reactions of mono-protected 1,2-diols are often more efficient than transformations of the corresponding unprotected diols.8
Figure 2.

Protection of 1,2-Diols
Preliminary attempts to effect selective monocyclization of unprotected diols 6 and 9 provided unsatisfactory results (Figure 3). Treatment of 6 with one equivalent of bromobenzene under our standard carboetherification conditions (NaOtBu, cat. Pd2 (dba)3/Dpe-Phos)11 afforded mixtures of bis-cyclized product 13 and unreacted starting material. Treatment of 6 with four equivalents of bromobenzene led to complete consumption of starting material and the formation of 13 with >20:1 dr, albeit in only 30% yield. Efforts to achieve monocyclization of 9 did lead to the formation of desired tetrahydrofuran 14, but yields were low and isomerization of the second alkene was problematic. Use of excess aryl halide in this reaction failed to generate significant amounts of the bis-tetrahydrofuran target, and instead provided an 81% combined yield of 14 and inseparable alkene isomers.
Figure 3.

Attempted Monocyclization of 1,2-Diols
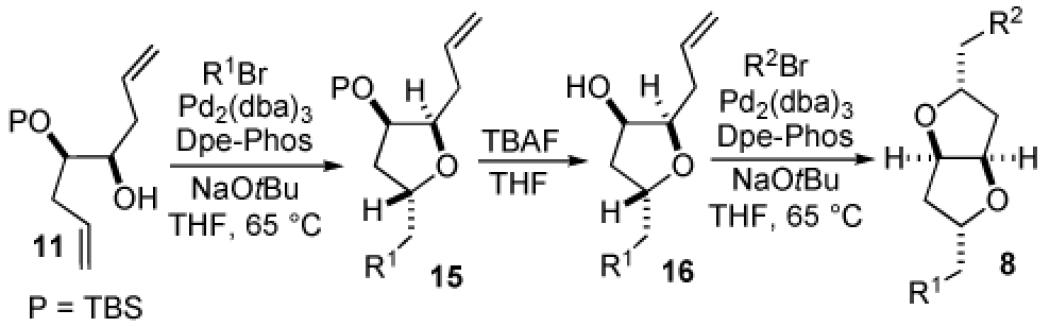
Although carboetherification reactions of 6 and 9 were generally ineffective, transformations of TBS-protected substrates 11 and 12 proceeded smoothly. As shown in Table 1, treatment of 11 with an aryl bromide in the presence of NaOtBu and a catalyst composed of Pd2(dba)3 and Dpe-Phos provided tetrahydrofurans 15 in good yields with excellent diastereoselectivities. Cleavage of the silyl ether protecting group was achieved under standard conditions, and carboetherification of the resulting alcohols 16 provided fused tetrahydrofurans (8) as single stereoisomers (>20:1 dr). Both cyclizations led to products that are trans−2,5-disubstituted around the tetrahydrofuran ring(s), which is consistent with our previously reported observations and stereochemical models for the conversion of γ-hydroxy alkenes to tetrahydrofurans.8
Table 1.
Stepwise Synthesis of Fused Tetrahydrofuransa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | yield 15 (%)b |
yield 16 (%)b |
R2 | yield 8 (%)b |
overall yield (%)c |
| 1 | Ph | 86 | 92 | p-F3CPh | 86 | 68 |
| 2 | p-PhC(O)Ph | 94 | 74 | |||
| 3 | p-Tol | 91 | 91 | 3-pyridyl | 91 | 75 |
| 4 | m-MeOPh | 81 | 67 | |||
| 5 | p-tBuPh | 71 | 90 | p-PhPh | 87 | 43 |
| 6 | 2-naphthyl | 85 | 54 | |||
| 7 | p-PhC(O)Ph | 44 | 92 | (E)-β-styryl | 67 | 27 |
| 8 | 6-MeO-2-naphthyl | 89 | 36 | |||
Conditions: Steps 1 and 3: 1.0 equiv 11 or 16, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 2 mol % Pd2(dba)3, 4 mol % Dpe-Phos, THF, 65 °C. Step 2: 1.0 equiv 15, 10 equiv TBAF, THF, rt.
Isolated yields (average of two or more experiments). All products were obtained with >20:1 dr.
Yield obtained over the three step sequence from 11 to 8.
The first carboetherification reaction in this sequence (11 to 15) was sensitive to the electronic properties of the aryl bromide, and the best yields were obtained with electron-neutral substrates (entries 1–6). However, the scope of the second carboetherification reaction (16 to 8) was much broader, and a number of different aryl bromides were effectively coupled. In addition, use of β-bromostyrene in the second transformation was also successful (entry 7). Diastereoselectivities were uniformly high in all of the carboetherification reactions (>20:1 dr), and in many cases the overall yield of 8 exceeded 50% over the three-step sequence.
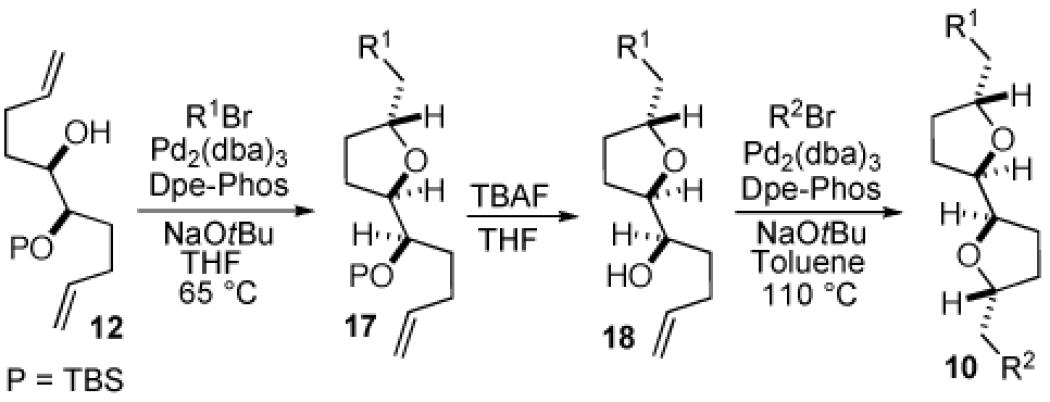
The synthesis of attached-ring bis-tetrahydrofurans was achieved by subjecting protected diol 12 to an analogous sequence of carboetherification (12 to 17), deprotection (17 to 18), and carboetherification (18 to 10). As observed in the transformations of 11, the scope of the second carboetherification step is considerably broader than the first (with respect to the aryl bromide component). Yields of attached-ring tetrahydrofurans were slightly lower than the corresponding fused-ring products described above. However, diastereoselectivities were excellent, and all products were obtained with >20:1 dr favoring 2,5-trans-stereochemistry around both tetrahydrofuran rings.
To further probe the synthetic utility of these transformations, we sought to determine if nonracemic starting materials could be converted to bis-tetrahydrofuran products without loss of enantiomeric purity. To this end, (–)−12 was prepared in 96% ee via asymmetric dihydroxylation of commercially available trans−1,5,9-decatriene12 followed by mono-TBS-protection of the resulting diol. This substrate was converted to (–)−10h using a sequence of reactions identical to that shown in Table 2, entry 8, and the product was obtained with >20:1 dr and 95% ee (Figure 4).
Table 2.
Stepwise Synthesis of Attached Tetrahydrofuransa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | yield 17 (%)b |
yield 18 (%)b |
R2 | yield 10 (%)b |
overall yield (%)c |
| 1 | p-tBuPh | 73 | 89 | p-Tol | 64 | 42 |
| 2 | p-PhPh | 63 | 41 | |||
| 3 | Ph | 72 | 91 | 3-pyridyl | 54 | 35 |
| 4 | 2-naphthyl | 61 | 40 | |||
| 5 | m-MeOPh | 64 | 91 | p-F3CPh | 75 | 44 |
| 6 | p-PhC(O)Ph | 72 | 42 | |||
| 7 | p-MeOPh | 55 | 96 | 3,5-Cl2Ph | 71 | 37 |
| 8 | 6-MeO-2-naphthyl | 53 | 28 | |||
Conditions: Steps 1 and 3: 1.0 equiv 12 or 18, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 2 mol % Pd2(dba)3, 4 mol % Dpe-Phos, Toluene or THF, 65 °C or 110 °C. Step 2: 1.0 equiv 17, 10 equiv TBAF, THF, rt.
Isolated yields (average of two or more experiments). All products were obtained with >20:1 dr.
Yield obtained over the three step sequence from 12 to 10.
Figure 4.

Synthesis of an Enantioenriched bis-Tetrahydrofuran
The synthesis of more elaborate tetrahydrofuran products is also feasible using this method. For example, protected tetraol derivative 19 was generated from Dmannitol using standard transformations.13 This substrate was converted to bis-tetrahydrofuran 22 with >20:1 dr using the same reaction sequence described above (Scheme 2).
Scheme 2.

Synthesis of a Highly Substituted bis-Tetrahydrofuran
In conclusion, we have developed a concise approach to the construction of both attached-ring and fused-ring bis-tetrahydrofurans using sequential Pd-catalyzed carboetherification reactions. This strategy allows for preparation of derivatives bearing different substituents at the 2-position of each tetrahydrofuran ring, and provides access to derivatives that could not be easily generated with existing methods. Further studies on the application of these reactions to the synthesis of natural products and molecules of medicinal interest are currently underway.
Supplementary Material
Acknowledgment
The authors thank the NIH-NIGMS (GM071650) for financial support of this work. Additional support was provided by the Camille and Henry Dreyfus Foundation (Camille Dreyfus Teacher Scholar Award), GlaxoSmithKline, Eli Lilly, Amgen, and 3M. The authors also thank Ms. Jody Canapp for performing preliminary experiments in this area.
Footnotes
Supporting Information Available. Experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra for all new compounds reported in the text (128 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Suzuki T, Koizumi K, Suzuki M, Kurosawa E. Chem. Lett. 1983:1639–1642. [Google Scholar]; (b) Suzuki M, Kurosawa E. Phytochemistry. 1985;24:1999–2002. [Google Scholar]
- 2.Pinacho Cisostomo FR, Padron JM, Martin T, Villar J, Martin VS. Eur. J. Org. Chem. 2006:1910–1916. [Google Scholar]
- 3.Kakimoto T, Koizumi F, Hirase K, Arai K. J. Pestic. Sci. 2006;31:380–389. [Google Scholar]
- 4.For recent reviews, see: McLaughin JL. J. Nat. Prod. 2008;71:1311–1321. doi: 10.1021/np800191t. Bermejo A, Figadere B, Zafra-Polo M-C, Barrachina I, Estornell E, Cortes D. Nat. Prod. Rep. 2005;22:269–303. doi: 10.1039/b500186m. Alali FQ, Liu X-X, McLaughlin JL. J. Nat. Prod. 1999;62:504–540. doi: 10.1021/np980406d.
- 5.For general reviews on the synthesis of tetrahydrofurans, see: Wolfe JP, Hay MB. Tetrahedron. 2007;63:261–290. doi: 10.1016/j.tet.2006.08.105. Harmange J-C, Figadere B. Tetrahedron: Asymmetry. 1993;4:1711–1754.
- 6.For representative examples of attached-ring tetrahydrofuran synthesis, see: Li P, Wang T, Emge T, Zhao K. J. Am. Chem. Soc. 1998;120:7391–7392. Hoye TR, Ye Z. J. Am. Chem. Soc. 1996;118:1801–1802. Wysocki LM, Dodge MW, Voight EA, Burke SD. Org. Lett. 2006;8:5637–5640. doi: 10.1021/ol062390l. Marshall JA, Sabatini JJ. Org. Lett. 2006;8:3557–3560. doi: 10.1021/ol061352z. Carlisle J, Fox DJ, Warren S. Chem. Commun. 2003:2696–2697. doi: 10.1039/b308609g. Sinha A, Sinha SC, Keinan E. J. Org. Chem. 1999;64:2381–2386. Beauchamp TJ, Powers JP, Rychnovsky SD. J. Am. Chem. Soc. 1995;117:12873–12874. For representative examples of fused-ring tetrahydrofuran synthesis, see: Reference 2. Wang J, Pagenkopf BL. Org. Lett. 2007;9:3703–3706. doi: 10.1021/ol701797e. Duclos A, Fayet C, Gelas J. Synthesis. 1994:1087–1090.
- 7.For reviews see: Wolfe JP. Eur. J. Org. Chem. 2007:571–582. Wolfe JP. Synlett. 2008:2913–2937.
- 8.(a) Wolfe JP, Rossi MA. J. Am. Chem. Soc. 2004;126:1620–1621. doi: 10.1021/ja0394838. [DOI] [PubMed] [Google Scholar]; (b) Hay MB, Hardin AR, Wolfe JP. J. Org. Chem. 2005;70:3099–3107. doi: 10.1021/jo050022+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hay MB, Wolfe JP. J. Am. Chem. Soc. 2005;127:16468–16476. doi: 10.1021/ja054754v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hay MB, Wolfe JP. Tetrahedron Lett. 2006;47:2793–2796. doi: 10.1016/j.tetlet.2006.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baylon C, Heck M-P, Mioskowski C. J. Org. Chem. 1999;64:3354–3360. doi: 10.1021/jo982098u. [DOI] [PubMed] [Google Scholar]
- 10.Essenfeld AP, Gillis HR, Roush WR. J. Org. Chem. 1984;49:4674–4682. [Google Scholar]
- 11.Dpe-Phos = bis(2-diphenylphosphinophenyl)ether.
- 12.Braddock DC, Cansell G, Hermitage SA, White AJP. Tetrahedron: Asymmetry. 2004;15:3123–3129. [Google Scholar]
- 13.(a) Kajiwara M, Miyazawa M, Nakazawa M, Shimayama T, Takatori K, Takahashi T, Yamada H. Tetrahedron Lett. 1992;33:5973–5976. [Google Scholar]; (b) Depezay JC, Duréault A, LeMerrer Y, Greck C, Micas-Languin D, Gravier C. Heterocycles. 1987;25:541–548. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.