

Abstract
Genome-wide association studies are revolutionizing the search for the genes underlying human complex diseases. The main decisions to be made at the design stage of these studies are the choice of the commercial genotyping chip to be used and the numbers of case and control samples to be genotyped. The most common method of comparing different chips is using a measure of coverage, but this fails to properly account for the effects of sample size, the genetic model of the disease, and linkage disequilibrium between SNPs. In this paper, we argue that the statistical power to detect a causative variant should be the major criterion in study design. Because of the complicated pattern of linkage disequilibrium (LD) in the human genome, power cannot be calculated analytically and must instead be assessed by simulation. We describe in detail a method of simulating case-control samples at a set of linked SNPs that replicates the patterns of LD in human populations, and we used it to assess power for a comprehensive set of available genotyping chips. Our results allow us to compare the performance of the chips to detect variants with different effect sizes and allele frequencies, look at how power changes with sample size in different populations or when using multi-marker tags and genotype imputation approaches, and how performance compares to a hypothetical chip that contains every SNP in HapMap. A main conclusion of this study is that marked differences in genome coverage may not translate into appreciable differences in power and that, when taking budgetary considerations into account, the most powerful design may not always correspond to the chip with the highest coverage. We also show that genotype imputation can be used to boost the power of many chips up to the level obtained from a hypothetical “complete” chip containing all the SNPs in HapMap. Our results have been encapsulated into an R software package that allows users to design future association studies and our methods provide a framework with which new chip sets can be evaluated.
Author Summary
Genome-wide association studies are a powerful and now widely-used method for finding genetic variants that increase the risk of developing particular diseases. These studies are complex and must be planned carefully in order to maximize the probability of finding novel associations. The main design choices to be made relate to sample sizes and choice of commercially available genotyping chip and are often constrained by cost, which can currently be as much as several million dollars. No comprehensive comparisons of chips based on their power for different sample sizes or for fixed study cost are currently available. We describe in detail a method for simulating large genome-wide association samples that accounts for the complex correlations between SNPs due to LD, and we used this method to assess the power of current genotyping chips. Our results highlight the differences between the chips under a range of plausible scenarios, and we demonstrate how our results can be used to design a study with a budget constraint. We also show how genotype imputation can be used to boost the power of each chip and that this method decreases the differences between the chips. Our simulation method and software for comparing power are being made available so that future association studies can be designed in a principled fashion.
Introduction
The International HapMap project [1],[2] documented the strong correlations between alleles at polymorphic loci in close physical proximity along human chromosomes. As a consequence it is necessary to genotype only a subset of loci to capture much of the common variation in the genome. Combined with recent technological innovations this observation has made the concept of genome-wide association (GWA) studies a reality [3],[4]. Over the few last years these studies have been very successful in uncovering new disease genes for many different complex diseases [5]. Well over 300 such loci have already been published and many more studies are currently being planned.
In the design of such studies two fundamental decisions have to be made: which loci to genotype, and in how many individuals. Both decisions have practical constraints. For example it is currently not possible to assay all known variation in the human genome at a reasonable cost and choices must be made between a set of commercially available genotyping chips. Similarly, sample sizes are often limited by the number of well characterized clinical samples. Therefore, ultimately, the researcher and funding bodies must ask how to use the financial and practical resources available in order to best further the understanding of the genetics of the disease or trait of interest. A primary consideration should be the power of the study: the probability of detecting a variant assumed to be causal.
In comparing chips for GWA studies it has been common to ask what proportion of SNPs not directly genotyped are “captured” or “tagged” by the chip, i.e. are well predicted, via LD, by a SNP, or combination of SNPs, on the chip. To do so it is necessary to define the level of prediction required, or equivalently to set a threshold for the required level of correlation. Although arbitrary, this has often been set at 0.8 [6],[7],[8]. The resulting proportion of SNPs captured at this level is often referred to as the coverage of the chip. Having specified the threshold it is possible to estimate the coverage of a particular chip from HapMap data, although we note that some care is required to account for SNPs not in HapMap [8].
Here we focus instead on the power of particular chips to detect causal variants of different effect sizes, and the way in which this varies with study size and/or study cost and when using genotype imputation methods. Although coverage is straightforward to estimate, power is a complicated function of the set of SNPs on the chip, effect size, and sample size, and can only be assessed by simulation.
It turns out that differences in coverage between chips are often not reflected in substantial differences in power and that the use of genotype imputation further reduces these differences. Study power is routinely used throughout science in experimental design and we argue that it should be the primary consideration in designing GWAs. This approach was used in settling several design questions in the Wellcome Trust Case Control Consortium [5]. Our results have been encapsulated in a user-friendly R package that allows the power of different chip and sample size combinations to be assessed given a total budget for the study.
Knowledge of study power is also invaluable when analysing data from a study. Assessment of whether positive results at a particular significance level are “real” or due to chance requires knowledge of power [5], and the practical decision of how far down the list of potential associations one should go in replication studies should be informed by power considerations.
Other comparisons of chips have been carried out but have either focussed exclusively on estimating coverage [8], have been limited in scope of which chips have been evaluated [9] or have used analytical calculations that do not properly take into account the complex LD structure of the human genome [10],[11] or failed to assess the impact of imputation correctly [11]. A recent paper [12] has used chip data to assess the performance of the chips but the small sample size (N = 359) means that these results cannot be used to assess power of new study designs of more realistic sizes. In addition, the simulations of quantitative phenotypes used the Signal to Noise Ratio (SNR) to measure effect size of the causal SNP which is non-standard and difficult to interpret. For binary traits, simulations assumed a disease prevalence of 25%, a relative risk of 3 and a sample size of only 75 cases and 75 controls. These parameter settings are not realistic for genome-wide association studies or useful when designing new studies.
Results
Theoretical Results
Study power depends on assumptions about the underlying disease model, in addition to effect sizes and sample sizes. When the true causative SNP is not on the genotyping chip there will typically be several SNPs on the chip which are correlated with it. One or more of these could give a signal of significant association and hence allow detection of the locus. The LD structure of the human genome is sufficiently complicated that this effect cannot be captured analytically. It must be assessed via simulation studies.
Nonetheless, there is one very simple situation for which analytical calculation is possible and helpful: that of the simplest disease model in which only a single SNP, correlated with the causal variant, is genotyped. For a design with the same number of cases and controls, under the disease model in which disease risk changes multiplicatively with the number of copies of the risk allele carried by an individual (this model is often referred to as the additive model because risk increases additively on the log scale), there is a known analytical relationship [13]:
| (1) |
where χ
2 is the chi-squared test statistic,  the number of cases and controls, γ the effect size, p the allele frequency of the risk variant and γ
2 is the correlation between the marker and causal SNP.
the number of cases and controls, γ the effect size, p the allele frequency of the risk variant and γ
2 is the correlation between the marker and causal SNP.
Although the real problem is much more complicated than this setting, Equation 1 does provide some useful intuition. Firstly, when the relative effect size is large ( ) the correlation between the marker and causal SNP may only need to be weak (r
2≪0.8) for the association to be detected (the expected test statistic is big). Equally, if the relative effect size is small (
) the correlation between the marker and causal SNP may only need to be weak (r
2≪0.8) for the association to be detected (the expected test statistic is big). Equally, if the relative effect size is small ( ) then even strong or complete association (0.8<r
2≤1) may not generate sufficient power to reject the null hypothesis of no association.
) then even strong or complete association (0.8<r
2≤1) may not generate sufficient power to reject the null hypothesis of no association.
Simulating Case-Control Samples – HAPGEN
Assessment by simulation of the power of a particular chip requires simulation of large sets of case and control samples which mimic the LD patterns in human populations (see Figure S1 for an example). The approach we use, implemented in a software package called HAPGEN is conceptually simple and is illustrated in Figure 1. We have previously used this approach to compare different analysis methods and has been briefly describe before [14]. In this paper we provide full details of the approach and these are given in the Methods section.
Figure 1. Schematic of how power is estimated.

At the top of the figure is the recombination map and haplotypes estimated from the HapMap project [1]. Using this population genetic information we simulate a case-control sample (grey lines) where the red dots indicate the disease locus and blue dots indicate linked genetic variation. By performing a test of association at each SNP on the genotyping chip we can estimate power by counting the number of simulation for which a test statistic exceed a significance threshold (dotted line). We compare genotyping chips by changing the set of SNP at which we carry out a test. See Methods.
Informally, the required samples are built up from the known haplotypes in HapMap. Consider first the simulation of control samples in a region of the genome. A particular control individual is simulated by separately simulating its two haplotypes in the region. Each of these haplotypes is made up as mosaics of the known haplotypes in HapMap, with the mechanism for constructing these mosaic haplotypes based on population genetics theory. Fine-scale estimates of recombination rates are used to calculate the probability of breaks in the mosaic pattern as one moves along the region.
For a given SNP assumed to be causal under a particular disease model and effect sizes, it is straightforward to calculate the genotype frequencies in cases at that SNP. Case samples are simulated separately by first simulating the genotype of the case at the causative SNP and then working outwards in each direction to construct the haplotypes carrying the alleles simulated at the causative SNP. Loosely, this process will result in oversampling of HapMap chromosomes which carry the risk allele, with the effect dropping off as one moves away (in genetic distance) from the causative locus (see Figures S2, S3, and S4 for examples).
We apply the method here to an assessment of the power of different chips, but we note that there are many other settings which require simulations of large case-control samples. These include comparisons of analysis methods [14] and tagging approaches [15], assessments of parameter estimates, and design questions for follow-on studies such as resequencing and fine mapping of associated regions.
The Power of GWAS Using Commercially Available Chips
We assessed the power of commercially available chips via simulation. Each simulation assumed a particular SNP in HapMap was causative, with a given effect size and used the HAPGEN package to simulate case and control samples of different sizes. In the simulated data we then restrict attention to the genotypes at only the SNPs on the chip in question and ask whether analysis of these would yield a significant result for any of the SNPs on the chip. An estimate of power is obtained by repeating the simulation over a large number of putative disease SNPs across the genome and using the proportion of simulations in which we find a significant test statistic.
For definiteness, in the results presented below we simulate data under the additive disease model, and in analysis of the data consider each SNP separately and apply the so-called trend, or Cochran-Armitage test [16], a chi-squared test with one degree of freedom. We fix a significance level of 5×10−7, and vary the number of cases and controls in the simulated study. There are various other versions of these assumptions which could be made. We explicitly look at one set of multi-marker tests below and also carry-out a limited set of simulations to assess the impact of genotype imputation.
There are two somewhat different perspectives that could be adopted regarding genome-wide association studies. One is to regard the GWAS as a self-contained experiment in its own right with the statistical inference being a formal hypothesis test of the null hypothesis of no association. From this perspective, the goal at the conclusion of the GWAS is to decide whether particular SNPs are, or are not, associated with the phenotype of interest.
But this is not what happens in practice. There is a strong consensus in the field that the results of association studies should not be relied upon without additional (statistically significant) evidence from analyses in independent replication samples [17] , and many major journals have policies which preclude publication of GWAS studies by themselves, without such replication evidence. Common practice is thus to regard the GWAS as an experiment to highlight SNPs of interest, and then to take as many as possible of the interesting SNPs into replication studies.
We adopt the second perspective throughout this paper, and our power calculations are for the probability that for each of the genotyping chips considered, there will be SNPs reaching a prespecified, low, p-value, under specific assumptions about the underlying genetic effects. Given current practice, we believe the right quantity to calculate would be the probability, for the respective chips, effect sizes, and sample sizes, that the experiment would give rise to SNPs showing enough signal to be taken forward for replication. This is (inevitably) ill-posed, so we focus instead on a surrogate for it, namely the probability that at least one SNP will have a p-value below a very stringent threshold. In this context there is nothing special about the choice of p-value threshold, and it is now well understood, for example from meta-analyses, that SNPs well down any ranked list of hits from the GWAS associations can still be genuine associations. For definiteness, we focus throughout on the threshold of p<5×10−7). This is deliberately set so that false positive rates will be low – for example, most SNPs with trend test p-values passing this threshold in GWAS studies, including all of those in the WTCCC experiment, have had associations confirmed in replication studies (see [18] and the NHGRI Catalog of Published Genome-Wide Association Studies at http://www.genome.gov/GWAStudies/). Choice of a different p-value threshold changes the numerical value of the power we calculate, but does not affect the relative performance of the chips, or the relative effect of sample size (data not shown).
If one were to adopt the first of the two perspectives on a GWAS study, namely that it is a formal statistical hypothesis test in its own right, then power comparisons become more complicated, at least under a frequentist statistical perspective: for a given nominal per-SNP significance level, the overall GWAS experiment will have somewhat different false positive rates for the different commercial chips, because they have different SNP sets, or when some SNP genotypes are imputed, depending on the number of imputed SNPs, for the same reason. Actually, even for a fixed chip, overall false positive rates will differ depending on the population in which the GWAS is conducted, because of differing patterns of LD between the SNPs on the chip (and hence different effective numbers of independent tests).
We do not pursue this approach here, principally because it does not reflect the way GWAS experiments are typically used in practice: regardless of the genotyping chip used, whether or not genotype imputation is employed, and the population studied, researchers tend to focus on the most significant SNPs after the GWAS and try to confirm that they are real in replication studies. In addition, as noted above, overall GWAS false positive rates are low, for any of the commercial chips, at the very low per-SNP significance level we consider. Nonetheless, in what follows, readers should be aware that we are comparing power, defined here as the probability that at least one SNP reaches a fixed p-value threshold under specific assumptions about design and effect sizes, across settings in which these very low false positive rates will differ between chips (and across populations).
In calculating power, as thus defined, we simulate data under the assumption that a particular allele is causal and then look to see whether any SNPs on the respective genotyping chip, within a large region around the causal SNP attain the specified significance level. In ignoring the SNPs on the chip elsewhere in the genome, this approximation will underestimate the probability of there being a SNP meeting the significance threshold, but at the very low threshold, the probability of there being a SNP elsewhere in the genome meeting the threshold is extremely small, so that effect of this approximation will be minimal and our power calculations based on only on SNPs within the 1Mb region containing the causal SNP will be very close to the true values.
We simulated putative disease loci at SNPs in phase II of the HapMap within twenty-two one megabase regions on each of the autosomes, a total of nearly 50,000 SNPs, which together are typical for the genome in terms of SNP coverage and recombination rates (see Figure S5 and Text S1 for details).
We investigated the power afforded by seven different genotyping chips: the 100 k, 500 k and 6.0 chips from Affymetrix (www.affymetrix.com) and the 300 k, 610 k, 650 k and 1 M chips from Illumina (www.illumina.com). These chips sets differ in the way in which the SNPs are chosen and the total number of SNPs assayed.
As technology develops and genotyping chips become denser it is a natural question to ask how much power would be gained by genotyping additional SNPs or by using genotyping imputation methods [14]. To facilitate such comparisons we evaluated the performance of a hypothetical chip that contains all the SNPs in HapMap to act as a point of reference in our results. The performance of this ‘complete’ chip is shown as a solid black line in all of the figures showing power. Since the simulations we carry out only use HapMap SNPs as causal SNPs this analysis approximates the scenario in which we have a chip which types all possible SNP variation.
We return below to consideration of results for studies in the Yoruban population. Focussing now on the power curves in the top row of Figure 2 several features are evident. The first is the profound effect of sample size. Effect sizes of 1.5 or smaller might be typical of what would now be expected for most variants affecting susceptibility to common human diseases [5]. For effect sizes at the top of this range (1.3–1.5) very large studies (say 2,000–3,000 cases and the same number of controls) are needed to have reasonable power, while for smaller effect sizes even studies of 5000 cases and 5000 controls have very little power. This ties in with growing empirical evidence. For example, for Crohn's disease, the WTCCC study, of 2000 cases and 3000 controls found 9 loci with p<5×10−7, whereas several smaller studies published around the same time each found only one or two of the loci, with little overlap across these smaller studies, consistent with each having modest power for the larger set of loci. Further, recent meta-analyses of 4,539 cases for type 2 diabetes and 3,230 for Crohn's disease have been needed to discover further loci with estimated effect sizes in the range 1.1–1.2. Even for a disease not previously studied by GWA, studies with fewer than 2000 cases and 2000 controls will have low power, except in special circumstances, for example if there are loci with larger effect sizes than has been typical across many other diseases.
Figure 2. Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of cases and controls (x-axis).

From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. Results are for single-marker test of association and for simulations where the risk allele frequency of the causal allele is >0.05. The top row shows power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. The bottom row relates to case-control studies simulated from the YRI HapMap panel.
A second general feature of the power curves for Caucasian studies in Figure 2 is that aside from the Affymetrix 100 K chip (which is no longer available), there are not major differences in power across the other seven chips. For Caucasian samples the chips are typically ordered (with decreasing power): Illumina 1M, Illumina 650 k, Illumina 610 k, Affymetrix 6.0, Illumina 300 k, Affymetrix 500 k, but the absolute difference in power between the best and worst of these chips is often no more than around 10%. Put another way, for effect sizes in the range 1.3–1.5, a study with the Affymetrix 500 K chip would have the same power as one with the Illumina 1 M chip if its sample size were larger by 10–20%, with smaller increases in sample sizes giving studies with other chips the same power. Further, in Caucasian studies, power for all chips other than the Affymetrix 100 K chip is quite close to the best which could be obtained, namely by directly genotyping the causative SNP.
Rare Alleles and Small Effect Sizes
Equation 1 makes clear the dependence of power on the frequency of the risk allele. The results in Figure 2 are averaged over putative causative SNPs with a risk allele frequency (RAF) in the range 5–95%. Figure 3 shows that this hides quite different behaviour depending on whether the putative disease SNP is rare or common, and that the conclusions in the preceding subsection apply principally for common causative SNPs. The Figure shows a substantial difference in power for common and rare alleles with the same effect size and that power is minimal for the rare alleles when the effect size is small. These results refer to single-SNP analyses. While there are definitely more powerful analysis methods for rare alleles [14], this is not a major factor in the loss of power, and neither is the incomplete coverage of the SNPs on the commercially available chips: even using a sample size of 3000 cases and controls and genotyping the causal locus directly (black line) is unlikely to lead to a test statistic which will reach the small levels of significance thought appropriate for GWAS.
Figure 3. Power for Common versus Rare alleles.

Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of cases and controls (x-axis). From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. Results are for single-marker test of association. The top two rows show the power for rare risk alleles (RAF<0.1) and the bottom two rows show the power for common risk alleles (RAF>0.1). Rows 1 and 3 show power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. Rows 2 and 4 relate to case-control studies simulated from the YRI HapMap panel.
There is an open question as to whether rarer causal alleles might have larger effect sizes than common causal alleles. If this were though plausible, then in assessing power overall for a particular chip, one could focus in Figure 3 on particular ranges of effect sizes for common causative alleles and a different range of effect sizes for rarer causative alleles.
It is becoming clear that many loci harbouring common alleles affecting common diseases will have effect sizes in the range 1.1–1.2, and our simulations demonstrate that there is almost no power to detect these in studies of the size currently underway. As has already been shown empirically [19],[20] these loci can be found by meta-analyses and follow up in larger samples of GWA findings. Slightly larger relative risks do become detectable in large samples. For example the power to detect an effect of size 1.3 jumps from almost zero with 1000 cases and 1000 controls to over 50% in a study three times the size.
Figure 3 also demonstrates that chip sets differ in the power they offer to detect associations at different frequencies. Most noticeably, when averaged over common alleles the Illumina 300 k chip set offers more power than Affymetrix 500 k. For rare alleles, the opposite is true with the Affymetrix 500 k chip having more power than the Illumina 610 k chip. This is most likely due to the way in which the Illumina SNP sets have been designed to specifically tag the common variation present in the HapMap panels.
Power of Chips Compared to a ‘Complete Chip’
Immediately apparent is how close, for studies in Caucasian populations, the genotyping chips track the power afforded by the ideal “Complete chip” in a given study design and disease model. Figure 3 illustrates that the potential benefits of increasing SNP density on the chips or from using imputation [14] are greatest for low frequency SNPs. When focusing on common alleles, the potential benefits are greatest for the Affymetrix 100 k and 500 k chips and the Illumina 300 k chip and we show this when specifically consider imputation below (see Table 1). However, a clear consequence of these results is that for any of the chips in current use, increasing sample size is likely to have a bigger effect on power than increasing SNP density.
Table 1. The table shows the power for each chip with a sample size of 2000 cases and 2000 controls and a relative risk at the causal SNP of 1.3 using a p-value threshold of 5×10−7.
| Chip | Chip SNP Tests | MultiMarker Tests | IMPUTE |
| Affy100 k | 0.178 | 0.212 | 0.242 |
| Affy500 k | 0.363 | 0.378 | 0.450 |
| Illu300 k | 0.392 | 0.424 | 0.467 |
| Illu610 k | 0.439 | 0.455 | 0.488 |
| Illu650 k | 0.443 | 0.458 | 0.492 |
| Affy6.0 | 0.420 | 0.433 | 0.478 |
| Illu1M | 0.457 | 0.461 | 0.493 |
| Complete | 0.499 | 0.499 | 0.499 |
Three different methods of analyzing the genotype data from each chip are shown: (a) testing just the SNPs on each chip, (b) using MultiMarker Tests in addition to the tests at each chip SNP, and (c) carrying out imputation using IMPUTE and testing all imputed SNPs in addition to those on each chip. The last line of the table shows the power that woud be obtained using the ‘Complete’ chip.
Power versus Coverage
A striking feature of Figures 2 and 3 is that substantial differences in coverage between different chips do not translate into big differences in power. Put another way, coverage is often a poor surrogate for power. As an example, the coverage in the CEU HapMap population (r 2≥0.8) provided by the Affymetrix 500 k and Illumina 610 k chips are 65% and 87% respectively, a difference of 22%. On the other hand, the difference in power e.g. for relative risk 1.5 and 1500 cases and controls, is only 7% (66% and 73% respectively).
In one sense this shouldn't be surprising. Coverage is measured to a hard threshold: so if SNP has r 2 of 0.85 to its best proxy on one chip and 0.75 to its best proxy on another chip, it will be counted as “covered” by one chip but not by the other, whereas the difference in power is small. Coverage statistics also do not depend on study size or disease model.
Figure 4 illustrates the differences in correlation structure for two chips. For each HapMap SNP we found it's best “tag” (the SNP on the chip with which it has the highest r 2) and generated a histogram of these maximized r 2 values. To recover coverage we simply count the proportion of SNPs for which the best tag r 2 is ≥0.8, coloured red in the bottom row of figure 4. In this sense, informally, it is useful to think of coverage as assuming that there is power one for every “tagged” SNP and no power for every other SNP. This is of course false, in ways which help to explain why coverage differences do not translate into power differences. When a SNP is common and the effect size is moderate or large, there will still be good power to detect it even if the best SNP on the chip only has r 2 = 0.5 or less. At the other extreme, for rare SNPs, unless the effect size is very large, power would be low even if the SNP had a perfect proxy on the chip. Thus even if these SNPs were well covered by one chip and completely missed by another they would not contribute to a difference in power between the chips because both chips would have power close to zero for them. The top row of Figure 4 shows the average power for SNPs in each LD bin. For the Affymetrix 500 K chip, there is a greater contribution to power from the sets of SNPs which are not well “covered”, than for the Illumina chip, and hance a smaller difference in power than in coverage.
Figure 4. Histograms of the proportion of SNPs in the 22 1Mb regions (see Methods) in HapMap Phase II for which the maximum r2 with a SNP on the genotyping chip in in one of eleven bins (increasing in correlation (LD) from left to right).

The same histograms are coloured in two ways. The top row shows in red the percentage of the SNPs in each bin detected (See Methods and text) when selected to be the causal SNP in our simulations (the proportion of the total volume of the bars coloured red is therefore an estimate of power). In the bottom row all r2 bins above 0.8 are coloured red (the proportion of the total volume of all the bars is therefore an estimate of coverage). Note that the use of HapMap data in choosing SNPs for the Illumina chip leads to a higher proportion of SNPs in high r2 bins.
Case-Control Population
For several reasons it is of interest to study the power of commercially available chips in different populations. Firstly the Illumina 100 k, 300 k and 610 k chips are aimed at capturing variation in the CEU population, whereas the Affymetrix 500 k chip is not designed with a specific population in mind. Furthermore the Illimina 650 k chip has a subset of SNPs targeted at capturing variation in the HapMap YRI (Yoruba, Africa) population. LD will not extend as far in the YRI collection [1] as in the CEU, reducing the coverage of a given set of SNPs.
Figures 2 and 3 show the results of power calculation using the distribution of diversity in both the HapMap CEU and YRI populations. The results show that the increased ancestral recombination leads to a loss of power and coverage across all chips for a range of study designs. The difference between the power available from commercial genotyping chips and that achievable by exhaustively assaying all SNPs shows that increasing marker density may yield a better return than a similar approach in non-African populations. The Illumina 650 k chip, with the YRI fill-in illustrates these potential benefits, showing a marked increase in power over the 610 k. However the performance of the Illumina 300 k chip, designed using the CEU HapMap, falls below the Affymetrix 500 k when genetic diversity is modelled on the YRI HapMap panel.
It is not yet clear how closely patterns of diversity and LD in other African populations mimic those in the Yoruba, and hence to what extent the power results will translate to studies in other populations. One general point is that the Illumina 650 k chip was designed specifically to capture common Yoruban variation, so one might expect power for this chip to decrease in other African populations, for which it is not specifically designed. On the other hand, the Affymetrix 500 k chip was not designed using this data, so there would be not a systematic effect changing power estimates for other African populations. As a consequence, differences in power between the Illumina 650 k chip and Affymetrix chips may well be smaller in other African populations.
The Gain from Using Multi Marker Methods and Genotype Imputation
Multi-marker methods, which use combinations of SNPs, have been suggested as an efficient way to increase both coverage and power [15]. Figures S6 and S7 show the results of simulations that implement the multi-marker tests. In these figures the dotted lines, which represent coverage, are higher for all chips in comparison to single marker approaches (Figures 2 and 3) consistent with previous observations. We find that multi-marker approaches also increase statistical power to detect disease loci, but that the increase is modest relative to coverage, and the broad conclusions above are not much affected. Interestingly, when comparing across genotyping platforms, we find for example that the Affymetrix 500 k chip gains more by combining SNPs than the Illumina 300 k chip.
Genotype imputation methods [21],[14] are now being widely used in the analysis of genome-wide association studies [5] and meta-analysis of such studies [22],[20]. These methods can be thought of as a more sophisticated version of Multi Marker tests but are relatively much more computationally demanding. We carried out an evaluation of the boost in power that can be gained by imputation using the program IMPUTE [14]. For our simulations with a sample size of 2000 cases and 2000 controls and a relative of the causal SNP or 1.3 we ran IMPUTE on the genotype data from each of the chips under study using the CEU HapMap as the basis for imputation. We then carried out a test of association at all the imputed SNPs in addition to the SNPs on each chip. We used our program SNPTEST to carry out tests of association at imputed SNPs to properly account for the uncertainty that can occur at such SNPs[14]. The results of the simulations are shown in Table 1 and shows that the use of IMPUTE provides a noticeable boost in power over testing just the SNPs on each chip or using Multi Marker tests (as defined in [15]). This agrees with our previous results [14]. It is also very noticeable that imputation reduces the differences in power between the chips and that the use of imputation produces a level of power that is almost as high as our hypothetical ‘complete’ chip.
We also note that the boost in power is more substantial than that estimated in another recent study [11]. A close look at the details of this other study shows that the only imputed SNPs used were those (a) which had real genotype data from one of the other chips, and (b) the imputed and real data at the SNP agreed with an r 2>0.8. So for example, for the Affy 500 k chip only genotypes at 427,838 imputed SNPs were used, rather than all those available from HapMap (approximately 2.5 milion SNPs), as normal practice when carrying out imputation. Using such a filter clearly creates a bias towards imputed SNPs that are almost perfect tags for SNPs on the chip so it is not surprising that this study shows such small increases in power when using imputation.
Unequal Case Control Sample Sizes
One option open to researchers who would like to increase power in the context of limited case series is just to increase the control collection. This strategy might include using cases for one disease as extra controls for another (assuming suitably different disease aetiologies and similar population history). We investigated the utility of such an approach by performing simulations with 1000 cases and an increasing number of control (Figure S8). Although the gains are not as strong as increasing both the case and control sample sizes (Figure 2), the ability to reject the null hypothesis of no association increase considerably with the size of the control panel. For example, adding an extra 2000 controls to a case-control study with sample size 1000–1000 increases power to detect an effect of 1.5 typically by 20%. Subject to care in their use, the growing availability of genotyped sets of controls promises to make this a possibility worth investigating for many studies.
Designing a New Study
The results of our simulations can be used to assess the power of a range of possible designs for a given budget and have been encapsulated in a user friendly R package for this purpose (see Software section). Table 2 shows the study size and power that can be achieved on a budget of $2,000,000 for each of the chips assuming the disease causing allele of has a relative risk of 1.5, a risk allele frequency of at least 0.05 and that a p-value threshold of 5×10−7 is used to define power. Since the different chips vary in their prices and their per sample processing costs we obtained quotes from service providers for the various chips and averaged them (see Text S1). The prices were based on quotes for 4000 chips and quotes were converted to US dollars using current exchange rates where necessary. We obtained 5 different quotes for the Affymetrix chips and 6 different quotes for the Illumina chips.
Table 2. The table shows the power that can be achieved by each chip with a total budget of $2,000,000.
| Chip | Average Price ($) | Number of cases/controls | Power |
| Affy500 k | 420 | 2381 | 0.767 |
| Illu300 k | 377 | 2653 | 0.821 |
| Illu610 k | 452 | 2212 | 0.818 |
| Affy6.0 | 505 | 1980 | 0.772 |
| Illu1M | 750 | 1257 | 0.635 |
| Complete | - | 2653 | 0.881 |
These results were calculated assuming a disease causing allele with a relative risk of 1.5, a minor allele frequency of at least 0.05, that a p-value threshold of 5×10−7 is used to define power and that the study should consist of an equal number of cases and controls. The second column shows the prices that we were able to obtain for these products at the time of submission. The last line of the table shows the power that woud be obtained using the ‘Complete’ chip using the sample size equal to that of the most powerful design.
The results show that in this scenario the Illumina 300 k chip produces the most powerful design (82.1%) primarily due to its relatively cheap price compared to the other chips. Using the same sample size (2653 cases and controls) the ‘Complete’ chip has a power of 88.1%. It is also notable that the power of thie Illumina 300 k chip is nearly 17% greater than the power that can be achieved by the Illumina1 M chip (63.5%) which has approximately 3 times the SNP density. These result further illustrate the deficiencies in using coverage as a measure of chip performance as sample size is not factored into the calculation. Although these results are interesting we advise against using them directly in the design of a new study. There were noticeable variations in the quotes we obtained from the service providers and prices are likely to change through time. We encourage new studies to re-calculate power of various designs based on a set of up to date and competitive prices and to take into account the general effect that genotype imputation can have on these power estimates.
Discussion
Because of the complexity of human LD patterns, many questions of interest cannot be addressed analytically. We have described in detail our simulation method, HAPGEN, for generating large samples of case and control data at every HapMap SNP, which mimic the patterns of diversity and LD present in the HapMap data. The software can simulate case data under a single causal disease SNP model for specified genotypic relative risks. We have used the method here to assess the power of various commercially available genotyping chips for case-control genome-wide association studies, but note that it could be utilised to assess other design questions, in the evaluation of analytical methods, and in considering follow-on studies such as resequencing and fine-mapping.
In Caucasian populations the differences in power afforded by current-generation genotyping chips are not large, and the power of these chips is close to that of an optimal chip which always directly genotyped the causal SNP. Listed in order of decreasing power for the CEU population, averaged over all potential disease SNPs with RAF ≥5%, the chips we considered were: Illumina 1M, Illumina 650 k, Illumina 610 k, Affymetrix 6.0, Illumina 300 k, Affymetrix 500 k and Affymetrix 100 k. In line with our previous work we have shown that imputation can boost the power of each chip substantially and that the resulting power will approach that which could be obtained by a hypothetical ‘complete’ chip that types all the SNPs in HapMap.
One limitation of the approach we (and others [9],[10],[12],[11]) have used is that the causal SNP is assumed to be one of those SNPs in the HapMap panel and this will not always be true. Other studies [1] have shown that the majority of SNPs not in HapMap will be highly correlated with the SNPs that are in HapMap and this is especially true for the more common SNPs. This means there is a slight bias in our power results for each chip and for the use of imputation but we do not expect it to be large. A consequence of this point is that the power we estimate for the ‘complete’ chip approximates the power we might obtain if we had a chip which typed all the SNPs that exist in the human genome.
A main conclusion from our analysis is that study size is a crucial determinant of the power to detect a causal variant. Increasing study size typically has a larger effect on power than increasing the number or coverage of SNPs on the chip, at least amongst chips currently available. Even for effect sizes at the larger end of those estimated to date for common human diseases (RRs of 1.3–1.5) quite large sample sizes, at least 2000 cases and 2000 controls and ideally more, are needed to give good power to detect the causal variant. When case numbers are limited, there are still non-trivial gains in power available from increasing just the number of controls. Care is needed in assessing the appropriateness of a set of controls, but as larger sets of control genotypes are made publicly available this strategy has considerable appeal, whatever the number of available cases. SNPs with smaller effect sizes are unlikely to be detected even in studies of the sizes currently undertaken, but as has been shown empirically for several diseases, these can be found by meta-analyses which combine different GWAs, or by follow-up in large samples of SNPs which look promising in the original GWA but fail to meet the low levels of significance thought appropriate for GWAS.
When the causal SNP is rare (MAF<10%), all chips have low power unless its effect is large and sample sizes are large. This conclusion would hold even if the chip directly genotyped the causal SNP. The relative ordering of different chips, on the basis of power, also changes in this context.
As would be expected, power is also lower for all chips for samples which match the patterns of LD seen in the Yoruba HapMap sample, and again the relative ordering of chips changes in this setting. It is not yet clear how well the results for the Yoruba would extend to other African populations.
An often-quoted metric in assessing chips is the coverage of each chip: an estimate of the proportion of SNPs which have r 2>0.8 with at least one SNP on the chip. Although relatively simple to calculate (and even simpler to miscalculate), not least because it does not depend on study size, our results show that coverage can be a poor surrogate for power, and that relatively large differences between chips in coverage do not translate to large differences in power.
The sets of SNPs on Illumina chips are chosen in part to maximize particular criteria, such as coverage, for certain populations, typically those in HapMap. One difficulty of analyses such as those in this paper is that these resources are also the natural ones with which to assess properties of the chips. Thus when Illumina chips “tuned” to one population (say the 610 K chip for CEU) are used in other populations, power might be systematically lower than the levels assessed here. In contrast, SNP sets of Affymetrix chips are chosen largely in a non-population specific way. While power is likely to vary in populations other than those we have considered here, there is not the same systematic effect which would lead to a decrease in power. A quantitative assessment of this phenomena will be possible when dense genotype data is available for other populations, such HapMap Phase 3.
We have assumed here that accurate genotypes are available for all SNPs on each chip. In practice some SNPs on each chip will fail QC tests and not be available for analyses. As a consequence, our study will overestimate power, though this effect is unlikely to be large. We are only able to use SNPs in HapMap as potential disease SNPs. These may not be systematically representative of all potential disease SNPs. HapMap SNPs have systematically higher MAFs than do arbitrary SNPs [2], but for SNPs within a particular range of MAF, it seems unlikely that their LD properties will differ systematically, so, for example, we would expect our results for common SNPs to extend beyond those in HapMap.
We have focussed on the most common GWA design, namely of a single-stage study, and the simplest disease model. The flexibility of the simulation approach allows many other practical aspects of study design to be incorporated into power calculations. These include more complex disease models, two-stage strategies (the starting point for our work was a comparison of power for one- and two-stage designs in the context of the WTCCC study [5]), genotyping errors, QC filters, misidentification of cases as controls and simple types of population structure. The HAPGEN software also provides a useful tool for the development and comparison of more sophisticated multi-marker approaches to detecting disease association (e.g. imputation [14]). We therefore believe that simulations are an essential tool in the design of association studies by allowing a focus on study power and an assessment of the affect on power of following a given study design. We hope that this method will continue to find use and can be extended to new catalogs of genetic variation such as the 1000 Genomes Project http://www.1000genomes.org/.
As in other areas of science, power seems a central consideration in study design and choice of genotyping chip. But other issues may also play a role. These include coverage of particular genes, or genomic regions of interest; the utility of GWA data for directing downstream studies such as resequencing and fine mapping; data quality for particular chips; and the extent to which a chip reliably assays other forms of genetic variation such as copy number polymorphisms. Adding data to existing studies is straightforward if the same chip is used, but the success of imputation methods, in particular in meta-analyses [19],[20] means that this is not essential.
In general, Affymetrix chips have more redundancy than do Illumina chips, in the sense of containing sets of SNPs which are correlated with each other. The immediate consequence of this is lower coverage and lower power for the same number of SNPs, but there can be advantages to this redundancy: loss of a particular SNP to QC filters may not be as costly; and signals of association are likely to include more SNPs, thus making them easier to distinguish from genotyping artefacts.
Ultimately power can only be calculated under an alternative model. Thus on a practical level the optimal choice of assays and sample sizes will actually depend on the researcher's belief regarding the unknown distribution of effect sizes and models relating genotype and phenotype. In particular we show that one might adopt different strategies depending on the expected frequency of disease causing variant, the effect size and even the population from which cases and controls are sampled (Figure 3).
In the continuing search to better understand the genetic basis of common human diseases, numerous study designs can be adopted which may involve combining data sets, imputing missing SNPs [14], distilling signals of association over multiple experimental stages, and so on. In this complex setting study power will remain a central criterion in study design, and the kinds of approaches developed here will continue to allow informed decision making by experimenters.
Methods
HAPGEN
We adopt the model introduced by [23] (denoted LS from now on), who described a new model for linkage disequilibrium, which enjoys many of the advantages of coalescent-based methods (e.g. it directly relates LD patterns to the underlying recombination rate) while remaining computationally tractable for huge genomic regions, up to entire chromosomes. Their model relates the distribution of sampled haplotypes to the underlying recombination rate, by exploiting the identity
| (2) |
where h
1
,…,hn denote the n sampled haplotypes, and ρ denotes the recombination parameter (which may be a vector of parameters if the recombination rate is allowed to vary along the region). This identity expresses the unknown probability distribution on the left as a product of conditional distributions on the right. LS substitute an approximation for these conditional distributions  into the right hand side of (3), to obtain an approximation to the distribution of the haplotypes h given ρ
into the right hand side of (3), to obtain an approximation to the distribution of the haplotypes h given ρ
| (3) |
If h
1
,…,hn are n sampled haplotypes typed at S bi-allelic loci (SNPs) LS modelled the distribution of the first haplotype as independent of ρ, i.e. all 2S possible haplotypes are equally likely, so  . For the conditional distribution of h
k+1 given h
1
,…,hk, LS modelled h
k+1 as an imperfect mosaic of h
1
,…,hk through the use of a Hidden Markov Model (HMM). That is, at each SNP, h
k+1 is a (possibly imperfect) copy of one of h
1
,…,hk at that position where where the transition rates between the hidden copying states are parameterized in terms of the underlying recombination rate. The transition rates are different for each of the conditional distributions in such a way so as to mimic the property that as we condition on an increasingly larger number of haplotypes we expect to see fewer novel recombinant haplotypes. A parameterisation for the mutation rate (or emission probabilities of the HMM) is used that has similar properties (see [23] for more details).
. For the conditional distribution of h
k+1 given h
1
,…,hk, LS modelled h
k+1 as an imperfect mosaic of h
1
,…,hk through the use of a Hidden Markov Model (HMM). That is, at each SNP, h
k+1 is a (possibly imperfect) copy of one of h
1
,…,hk at that position where where the transition rates between the hidden copying states are parameterized in terms of the underlying recombination rate. The transition rates are different for each of the conditional distributions in such a way so as to mimic the property that as we condition on an increasingly larger number of haplotypes we expect to see fewer novel recombinant haplotypes. A parameterisation for the mutation rate (or emission probabilities of the HMM) is used that has similar properties (see [23] for more details).
The simulation of a new set of haplotypes for 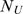 control and
control and 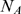 case individuals is proceeds using the following algorithm.
case individuals is proceeds using the following algorithm.
1. Pick a locus from the set of markers in the real dataset as the disease locus. The disease locus is chosen at random from all those loci with a minor allele frequency (MAF) within some specified range [l,u]. We use  to denote the disease locus, a and A to denote the major and minor alleles at the disease locus and use p denote the sample minor allele frequency at this locus.
to denote the disease locus, a and A to denote the major and minor alleles at the disease locus and use p denote the sample minor allele frequency at this locus.
2. For a given disease model simulate the alleles at the disease locus of the new individual conditional upon case-control status. At the disease locus we use a general genotype model in which the frequencies of the genotypes aa, Aa and AA in control individuals are given by (1−p)2, 2p(1−p) and p2 respectively. This assumes that the control individuals are so-called population controls (as used by the WTCCC study [5]) rather than individuals who have been selected to specifically not have the disease. For case individuals the genotype frequencies are determined by specification of the two relative risks
| (4) |
where  denotes the probability that an individual is a case conditional upon having genotype g. Under this model
denotes the probability that an individual is a case conditional upon having genotype g. Under this model
| (5) |
where γ = (1−p)2+2αp(1−p)+βp2. As an example, if p = 0.1, α = 2 and β = 4 the control and case genotype frequencies are (0.81, 0.18, 0.01) and (0.67, 0.30, 0.03) respectively.
Assuming we have a set of k known haplotypes, the generation of a case (control) starts by simulating a genotype g using the case (control) genotype frequencies. This simulated genotype specifies the alleles on the the two haplotypes of the new individual at the disease locus. For example, if g = Aa then h k+1,d = 1 and h k+2,d = 0.
3. This step involves the simulation of two new haplotypes for the individual conditional upon the alleles simulated at the disease locus in Step 2 and conditional upon the fine-scale recombination map across the region. This involves simulating the rest of hk+1 and hk+2. We only describe the generation of sites right flanking of the disease locus as the generation of the left flanking markers is virtually identical. Also the simulation of h k+2 follows directly from our description of how the rest of h k+1 is simulated.
Let Xj be the hidden state of the HMM that denotes which haplotype h
k+1 copies at site j (so that  ). This state variable is initialized at the disease locus as follows
). This state variable is initialized at the disease locus as follows
 |
(6) |
The value of  , as with LS, is Watterson's point estimate (Watterson, 1975)
, as with LS, is Watterson's point estimate (Watterson, 1975)
| (7) |
Simulation of the hidden state of the HMM then proceeds using the following transition rule
 |
(8) |
where zj is the physical distance between markers j and j+1 (assumed known); and  , where
, where  is the effective (diploid) population size, and cj is the average rate of crossover per unit physical distance, per meiosis, between sites j and j+1 (so that cjzj is the genetic distance between sites j and j+1). This transition matrix captures the idea that, if sites j and j+1 are a small genetic distance apart (i.e. cjzj is small) then they are highly likely to copy the same chromosome (i.e. Xj
+1 = Xj).
is the effective (diploid) population size, and cj is the average rate of crossover per unit physical distance, per meiosis, between sites j and j+1 (so that cjzj is the genetic distance between sites j and j+1). This transition matrix captures the idea that, if sites j and j+1 are a small genetic distance apart (i.e. cjzj is small) then they are highly likely to copy the same chromosome (i.e. Xj
+1 = Xj).
To mimic the effects of mutation the copying process may be imperfect: with probability k/(k+θ) the copy is exact, while with probability θ/(k+θ) a mutation will be applied to the copied haplotype. Specifically,
 |
4. Return to step 2 to generate another individual or terminate.
Illustrations of the HAPGEN method in practice and details of the testing the method against coalecent simulations are given in Text S1.
Details of SNP Sets Used in the Study
We used release 21 of the HapMap data for which phased haplotypes are available in NCBI b35 coordinates. The SNPs that occur on each genotyping chip were obtained from the websites of Affymetrix and Illumina respectively. Some of the SNPs in these sets do not occur in the HapMap phased haplotype data due to QC measures applied to the raw genotype data. For the Affymetrix 6.0 and Illumina 1 M chips 90.8% and 88.1% of the SNPs on these chips respectively are in this release in HapMap. This will have the effect of making our estimates of power slight underestimates of the true power.
We simulated data for twenty-two one megabase regions chosen at random, one from each autosome. To ensure that the regions used to approximate genome-wide power were representative of the genome at large we their SNP density. Figure S5 plots the distribution of inter-SNP distances within the 22 analysis regions and across the whole genome for three of the genotyping chips analyzed. The close match between the distribution, both on the physical scale and in terms of genetic distance suggests that our results are insensitive to the regions we chose to simulate, and can be used to make comparisons of genotyping chips genome-wide.
Calculating Coverage and Testing for Association
We used data from the HapMap project Phase II to estimate coverage. Single marker coverage was defined to be the proportion of all variation (with minor allele frequency greater than 5%) in r 2 with a SNP on the genotyping chip above 0.8. Using this definition we achieved very similar estimates to previous studies which used the whole genome (we use twenty two representative megabases). Multi-marker coverage was calculated by an aggressive search of all 2-SNP and 3 SNP haplotypes within 250kb of the SNP being tagged [6]. The SNP was tagged if any of these multi-marker tags had r2 above 0.8, the rule defining the haplotype was also stored and added to the list of multi-marker tests.
Single marker tests (Cochran-Armitage test) were performed at each SNP on the genotyping chip where information were simulated from the relevant HapMap panel. Multi-marker tests of association were performed in an identical fashion with the marker being formed by the multi-marker haplotypes known to tag HapMap variation. To avoid over estimation of power, multi-marker tags chosen to tag the current putative disease SNP in the simulations were excluded from the test set. Tests at imputed SNPs took account of the uncertainty in genotypes through a missing data likelihood as described in [14].
Software
The HAPGEN software is freely available for academic use from the website http://www.stats.ox.ac.uk/˜marchini/software/gwas/gwas.html.
In addition, the results of the power calculations for the 7 commercially available genotyping chips have been included in an R package called GWASpower available from http://www.stats.ox.ac.uk/˜marchini/#software.
This package allows the user to determine the most powerful study design for a given budget. As new commercial genotyping chips become available we will update the package to include results of new chips. The package works by fitting a Generalised Linear Model to the results of the simulation study and using the model fit to predict the power for a given number of cases and controls.
Supporting Information
Top plot : Linkage disequilibrium plots across the region : D′ (top left), r2 (bottom right). Bottom plot : Fine scale recombination map ρ across the region.
(0.21 MB TIF)
For simulated dataset A (α = 1.3, β = 1.69) the top plot shows D′ and r2 LD measures. The bottom plot shows the χ2 statistic for association across the region. The vertical blue line shows the location of the disease locus.
(0.28 MB TIF)
For simulated dataset B (α = 1.5, β = 2.25) the top plot shows D′ and r2 LD measures. The bottom plot shows the χ2 statistic for association across the region. The vertical blue line shows the location of the disease locus.
(0.29 MB TIF)
For simulated dataset (α = 1.7, β = 2.89) the top plot shows D′ and r2 LD measures. The bottom plot shows the χ2 statistic for association across the region. The vertical blue line shows the location of the disease locus.
(0.30 MB TIF)
Representativeness of the 22 1Mb regions used in the simulation study. Bar plots are shown of the proportion of SNPs which fall into increasing inter-SNP distances for three of the genotyping chips used in this study. These distribution are measured on the physical scale (left column) and genetic map (right column).
(1.37 MB EPS)
Power of Multi-marker tests. Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of cases and controls (x-axis). From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. The results are based on simulations where the risk allele frequency of the causal allele is >0.05. The top row shows power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. The bottom row relates to case-control studies simulated from the YRI HapMap panel.
(1.71 MB EPS)
Power of Multi-marker tests for common versus rare alleles. Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of cases and controls (x-axis). From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. Results are for single-marker test of association. The top two rows show the power for rare risk alleles (RAF<0.1) and the bottom two rows show the power for common risk alleles (RAF>0.1). Rows 1 and 3 show power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. Rows 2 and 4 relate to case-control studies simulated from the YRI HapMap panel.
(2.90 MB EPS)
Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of controls (x-axis). The number of case individuals is fixed at 1000. From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. Results are for single-marker test of association and for simulations where the minor allele frequency of the causal allele is >0.05. The top row shows power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. The bottom row relates to case-control studies simulated from the YRI HapMap panel.
(1.73 MB EPS)
Supplementary text associated with the main article.
(0.06 MB PDF)
Acknowledgments
We would like to thank Dr Rory Bowden for his help and advice in comparing the different genotyping chips. This research was supported by funding and grants from the Wellcome Trust (PD and CS), a Wolfson-Royal Society Merit Award (PD) and the EPSRC (ZS).
Footnotes
The authors have declared that no competing interests exist.
This research was supported by funding and grants from the Wellcome Trust (PD and CCAS), The Wolfson Foundation (PD), a Wolfson-Royal Society Merit Award (PD) and the EPSRC (ZS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.The International HapMap Consortium. A Haplotype Map of the Human Genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 4.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 5.The Wellcome Trust Case-Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pe'er I, de Bakker PI, Maller J, Yelensky R, Altshuler D, et al. Evaluating and improving power in whole-genome association studies using fixed marker sets. Nat Genet. 2006;38:663–7. doi: 10.1038/ng1816. [DOI] [PubMed] [Google Scholar]
- 7.de Bakker PI, Burtt NP, Graham RR, Guiducci C, Yelensky R, et al. Transferability of tag snps in genetic association studies in multiple populations. Nat Genet. 2006;38:1298–303. doi: 10.1038/ng1899. [DOI] [PubMed] [Google Scholar]
- 8.Barrett JC, Cardon LR. Evaluating coverage of genome-wide association studies. Nat Genet. 2006;38:659–62. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- 9.Eberle M, Ng P, Kuhn K, Zhou L, Peiffer D, et al. Power to detect risk alleles using genome-wide tag SNP panels. PLoS Genet. 2007;3:1827–1837. doi: 10.1371/journal.pgen.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Li M, Long J, Cai Q, Zheng W. Evaluating cost efficiency of SNP chips in genome-wide association studies. Genet Epidemiol. 2008;32:387–395. doi: 10.1002/gepi.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson C, Pettersson F, Barrett J, Zhuang J, Ragoussis J, et al. Evaluating the effects of imputation on the power, coverage, and cost efficiency of genome-wide SNP platforms. Am J Hum Genet. 2008;83:112–119. doi: 10.1016/j.ajhg.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao K, Schadt E, Storey J. Calibrating the performance of SNP arrays for whole-genome association studies. PLoS Genet. 2008;4:e1000109. doi: 10.1371/journal.pgen.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman JM, Cooper JD, Todd JA, Clayton DG. Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Hum Hered. 2003;56:18–31. doi: 10.1159/000073729. [DOI] [PubMed] [Google Scholar]
- 14.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 15.de Bakker P, Yelensky R, Pe'er I, Gabriel S, Daly M, et al. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 16.Clayton D. Population Association. In: D Balding MB, Cannings C, editors. Handbook of Statistical Genetics. Wiley: Chichester; 2001. chapter 19. [Google Scholar]
- 17.Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 18.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeggini E, Scott L, Saxena R, Voight B, Marchini J, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett J, Hansoul S, Nicolae D, Cho J, Duerr R, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeggini E, Scott L, Saxena R, Voight B, Marchini J, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–33. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Top plot : Linkage disequilibrium plots across the region : D′ (top left), r2 (bottom right). Bottom plot : Fine scale recombination map ρ across the region.
(0.21 MB TIF)
For simulated dataset A (α = 1.3, β = 1.69) the top plot shows D′ and r2 LD measures. The bottom plot shows the χ2 statistic for association across the region. The vertical blue line shows the location of the disease locus.
(0.28 MB TIF)
For simulated dataset B (α = 1.5, β = 2.25) the top plot shows D′ and r2 LD measures. The bottom plot shows the χ2 statistic for association across the region. The vertical blue line shows the location of the disease locus.
(0.29 MB TIF)
For simulated dataset (α = 1.7, β = 2.89) the top plot shows D′ and r2 LD measures. The bottom plot shows the χ2 statistic for association across the region. The vertical blue line shows the location of the disease locus.
(0.30 MB TIF)
Representativeness of the 22 1Mb regions used in the simulation study. Bar plots are shown of the proportion of SNPs which fall into increasing inter-SNP distances for three of the genotyping chips used in this study. These distribution are measured on the physical scale (left column) and genetic map (right column).
(1.37 MB EPS)
Power of Multi-marker tests. Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of cases and controls (x-axis). From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. The results are based on simulations where the risk allele frequency of the causal allele is >0.05. The top row shows power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. The bottom row relates to case-control studies simulated from the YRI HapMap panel.
(1.71 MB EPS)
Power of Multi-marker tests for common versus rare alleles. Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of cases and controls (x-axis). From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. Results are for single-marker test of association. The top two rows show the power for rare risk alleles (RAF<0.1) and the bottom two rows show the power for common risk alleles (RAF>0.1). Rows 1 and 3 show power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. Rows 2 and 4 relate to case-control studies simulated from the YRI HapMap panel.
(2.90 MB EPS)
Plots of power (solid lines) and coverage (dotted line) for increasing sample sizes of controls (x-axis). The number of case individuals is fixed at 1000. From left to right plots are given for increasing effect sizes (relative risk per allele). Both power and coverage range from 0 to 1 and are given on the y-axis. Results are for single-marker test of association and for simulations where the minor allele frequency of the causal allele is >0.05. The top row shows power for case-control studies simulated in a Caucasian population based on the CEU HapMap panel. The bottom row relates to case-control studies simulated from the YRI HapMap panel.
(1.73 MB EPS)
Supplementary text associated with the main article.
(0.06 MB PDF)