

Abstract
TorsinA (TorA) is an AAA+ ATPase in the endoplasmic reticulum (ER) lumen that is mutated in early onset DYT1 dystonia. TorA is an essential protein in mice and is thought to function in the nuclear envelope (NE) despite localizing throughout the ER. Here, we report that transient interaction of TorA with the ER membrane protein LULL1 targets TorA to the NE. FRAP and Blue Native PAGE indicate that TorA is a stable, slowly diffusing oligomer in either the absence or presence of LULL1. Increasing LULL1 expression redistributes both wild-type and disease-mutant TorA to the NE, while decreasing LULL1 with shRNAs eliminates intrinsic enrichment of disease-mutant TorA in the NE. When concentrated in the NE, TorA displaces the nuclear membrane proteins Sun2, nesprin-2G, and nesprin-3 while leaving nuclear pores and Sun1 unchanged. Wild-type TorA also induces changes in NE membrane structure. Because SUN proteins interact with nesprins to connect nucleus and cytoskeleton, these effects suggest a new role for TorA in modulating complexes that traverse the NE. Importantly, once concentrated in the NE, disease-mutant TorA displaces Sun2 with reduced efficiency and does not change NE membrane structure. Together, our data suggest that LULL1 regulates the distribution and activity of TorA within the ER and NE lumen and reveal functional defects in the mutant protein responsible for DYT1 dystonia.
INTRODUCTION
Early-onset (DYT1) torsion dystonia is a neurological movement disorder characterized by twisting movements of the limbs and an absence of neuropathology or neurodegeneration (Fahn, 1988; Breakefield et al., 2008). The disease is caused by autosomal dominant inheritance of a glutamic acid deletion in the protein TorsinA (TorA), frequently referred to as the ΔGAG mutation because of the deleted codon (Ozelius et al., 1997). Although TorA is expressed ubiquitously (Ozelius et al., 1998), the only abnormalities described so far in animals lacking this essential protein are in neurons (Goodchild et al., 2005). The failure of ΔGAG-mutant TorA to rescue TorA knockout animals from perinatal lethality suggests that the ΔGAG mutant lacks whatever essential activity TorA normally provides (Dang et al., 2005; Goodchild et al., 2005).
The specific cellular functions ascribed to TorA vary widely despite the fact that it has been a decade since the protein was first described and linked to dystonia (Breakefield et al., 2008). TorA resides in the lumen of the endoplasmic reticulum (ER; Hewett et al., 2000, 2003; Kustedjo et al., 2000). Based on its membership in the AAA+ (ATPases associated with a variety of cellular activities) family of ATPases (Ozelius et al., 1997; Hanson and Whiteheart, 2005), it is likely that TorA disassembles or changes the conformation of a protein or protein complex within the ER or nuclear envelope (NE) lumen. Although typically found throughout the ER, several things point to a function for TorA in the NE. Reducing TorA activity by gene knockout in mice (Goodchild et al., 2005) or expressing a dominant negative form of the enzyme in cultured cells (Naismith et al., 2004) selectively perturbs NE structure. The NE is the favored binding site for hydrolysis deficient “substrate trap” TorA mutants (Goodchild and Dauer, 2004; Naismith et al., 2004; Kock et al., 2006). Finally, the outer nuclear membrane protein nesprin-3 (Wilhelmsen et al., 2005) is abnormally distributed in fibroblasts from TorA knockout mice, and these cells move slower than controls in a polarized cell migration assay (Nery et al., 2008). Because nesprin-3 participates in linker of the nucleoskeleton and cytoskeleton (LINC) complexes (Wilhelmsen et al., 2005; Ketema et al., 2007; Crisp and Burke, 2008; Stewart-Hutchinson et al., 2008; Starr, 2009), these data suggest that TorA activity may help regulate NE structure and connections between nucleus and cytoskeleton.
Previously identified binding partners for TorA include the transmembrane proteins LAP1 (also known as TOR1AIP1) in the NE and LULL1 (also known as TOR1AIP2 or NET9) in the ER, which control the distribution of hydrolysis deficient “substrate trap” TorA between NE and ER in direct proportion to their relative abundance (Goodchild and Dauer, 2005). Here, we describe the surprising finding that increasing expression of LULL1 induces TorA to redistribute from throughout the ER into the NE, whereas decreasing it reverses concentration there. Once in the NE, TorA displaces a subset of LINC complex components and gradually promotes structural changes in the NE. The disease-associated ΔGAG mutant is less efficient at enacting these changes. We conclude that LULL1 dynamically tunes the distribution of TorA between the ER and NE and is thereby likely to regulate its function.
MATERIALS AND METHODS
Plasmids
Green fluorescent protein (GFP) in previously described TorA-GFP plasmids (Naismith et al., 2004) was changed to monomeric GFP (mGFP) by site-directed mutagenesis of L221K in GFP (Snapp et al., 2003b). TorA-TagRFP was made by transferring TorA mutants excised with XhoI and EcoRI from EGFP-N1 into a RFP-N1 vector containing TagRFP (Merzlyak et al., 2007). The TorA Δ26-43 deletion was created by QuikChange mutagenesis (Stratagene, La Jolla, CA). LULL1-myc was made by amplifying LULL1 (NM_145034, NP_659471) from human cDNA with primers containing HindIII and EcoRI restriction sites and ligating it into pcDNA4/TO/MycHis B (Invitrogen, Carlsbad, CA). LULL1-mCherry was made by excising LULL1 from EGFP-N1 using HindIII and EcoRI restriction sites and ligating into pmCherry-N1 (Clontech, Mountain View, CA). ER-RFP was from (Snapp et al., 2006). Sun2-GFP was from Hodzic et al. (2004). Nesprin-3α-GFP was described in Ketema et al. (2007) and was obtained from Arnoud Sonnenberg (the Netherlands Cancer Institute). The sequences of all constructs were verified by nucleotide sequencing.
Cell Culture and Cell Line Generation
U2OS cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and l-glutamine. Transient transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Clonal LULL1-myc tetracycline-inducible U2OS cell lines were made as described previously (Dalal et al., 2004), with selection in hygromycin (50 μg/ml) and zeocin (100 μg/ml). LULL1-myc/Sun2-GFP dual-expressing cells were made by transfecting Sun2-GFP into LULL1-myc cells and selecting with 400 μg/ml G418. LULL1 was depleted by lentiviral transduction of short hairpin RNAs (shRNAs) directed against human LULL1 (Sigma Mission shRNA, RefSeq NM_145034; St. Louis, MO). Lentiviruses were produced by cotransfecting hairpins in pLKO with the pCMV 8.2 ΔR packaging plasmid into 293T cells. U2OS cells were then transduced with viral particles according to manufacturer's instructions. Cells depleted of LULL1 were enriched for by selecting transduced cells with puromycin (10 μg/ml).
Immunofluorescence
Cells were fixed with 3% paraformaldehyde in PBS for 10 min, followed by permeabilization in 0.2% Triton X-100 for 10 min. Coverslips were blocked in 2% goat serum in PBS for 1 h before incubation with primary and Alexa Fluor–conjugated secondary antibodies. For epifluorescence imaging, coverslips were mounted in Mowiol (Calbiochem, San Diego, CA) and imaged with a Diaplan microscope (Leica Microsystems, Bannockburn, IL) using a 63× 1.4 NA objective and a Zeiss AxioCam MRm camera (Thornwood, NY). Brightness and contrast were adjusted in Adobe Photoshop (Adobe Systems, San Jose, CA). For confocal imaging, coverslips were mounted in VectaShield (Vector Laboratories, Burlingame, CA), sealed with nail polish, and imaged with a Bio-Rad Radiance 2000 microscope (Hercules, CA) with a 63× 1.4 NA oil objective, using the 488- and 543-nm laser lines. Confocal z-series were acquired with a pinhole of 1.7 at 0.3-μm spacing. All confocal images were processed using the smooth function in Image J (v1.4, NIH; http://rsb.info.nih.gov/ij/), which replaces each pixel with the average in its 3 × 3 neighborhood. Maximum intensity projections of confocal z-series were made in ImageJ. Composite figures were prepared using Photoshop and Illustrator software (Adobe).
NE–ER Ratio Analysis
U2OS or LULL1 knockdown cells were transiently transfected with TorA-mGFP and ER-RFP as indicated. Confocal images were acquired as above. Fluorescence intensity was quantified using Metamorph 6.0 software (Molecular Devices, Downingtown, PA). For each image, average intensities in the GFP and red fluorescent protein (RFP) channels were quantified in four regions each of the ER (∼400 square pixels each) and NE (∼200 square pixels each). Regions of interest were delineated as boxes for ER and as hand-drawn outlines for NE and any overlapping perinuclear ER. These four values were averaged for each cell's ER and NE and then used to compute the NE–ER ratio for each channel. The data shown represent the average of >20 cells per condition.
Live Imaging
For experiments involving coexpression of LULL1 and TorA, LULL1-myc expression was induced with tetracycline (1 μg/ml) for 6 h before adding Lipofectamine 2000/TorA-mGFP mixtures to initiate transfection. One to 2 h later, coverslips were transferred to Bioptechs Delta T imaging dishes (Bioptechs, Butler, PA) and overlaid with phenol-red–free media containing 1% FBS. The dishes were placed on an enclosed stage (custom made by A. Czirok, Department of Biological Physics, Eotvos University, Budapest, Hungary) attached to a CO2 pump to maintain 5% CO2 and to a temperature control device set to 37°C. Images were obtained with a 20× 0.4 NA objective on an inverted epifluorescence/DIC microscope (DMI6000B; Leica Microsystems). Images (696 × 520 pixels, 12-bit intensity resolution) were recorded with a Retiga Exi camera (QImaging, Surrey, BC, Canada) using 2 × 2 binning and predetermined 1–2-s exposures. Image acquisition and microscope settings were controlled by software based on Czirok et al. (2002). Briefly, up to 16 preselected fields were visited in each scanning cycle, and epifluorescence images were taken in one or two channels. For each field and channel, five images were acquired in z positions centered around the chosen focal plane. Cells were imaged every 15 min for the duration of the experiment. Images were processed using software based on Czirok et al. (2002). For each time point, the software chooses the most in-focus image of the “z-stack” for each 64 × 64-pixel block of the image. These best-focused pixel blocks are then collapsed into a single image. The software also corrects for small shifts in x position. The resulting images were further processed using “Subtract Background” and “Enhance Contrast” tools in ImageJ. Time-lapse imaging (TiLa) code was created by the Computational Imaging Group at the University of Kansas Medical Center, under the direction of Drs. C. Little, B. Rongish, and A. Czirok. Dr. Czirok devised the original code for image acquisition and processing, which has been further developed and modified by Alan Petersen, Michael Filla, and Dr. Evan Zamir. A current version of this open source code is available from the Computational Imaging Group upon request (e-mail: clittle@kumc.edu).
Fluorescence Recovery after Photobleaching Analyses
Cells were grown in eight-well Labtek chambers (Thermo Fisher Scientific, Rochester, NY) and imaged in phenol red–free RPMI supplemented with 10 mM HEPES and 10% FBS. Live cells were imaged on a 37°C environmentally controlled chamber of a confocal microscope system (Duoscan; Carl Zeiss MicroImaging) with a 63× 1.4 NA oil objective and a 489-nm 100 mW diode laser with a 500–550 nm bandpass filter for GFP. Fluorescence recovery after photobleaching (FRAP) experiments were performed by photobleaching a region of interest at full laser power of the 489-nm line and monitoring fluorescence recovery by scanning the whole cell at low laser power. No photobleaching of the cell or adjacent cells during fluorescence recovery was observed. Diffusion (D) measurements were calculated as described previously (Siggia et al., 2000; Snapp et al., 2003a). Composite figures were prepared using Photoshop CS2 and Illustrator CS software (Adobe). Plotting of diffusion coefficients was performed with Prism 4.0c (GraphPad Software, La Jolla, CA).
Immunoblotting
Western blots were developed as described (Dalal et al., 2004). For quantitative analysis using the Odyssey system (Li-Cor Biosciences, Lincoln, NE), samples were separated, blocked, and blotted according to manufacturer's instructions. Secondary antibodies conjugated to IRDye-680 or -800 were used (Li-Cor Biosciences).
Antibodies
The following antibodies were used: mouse anti-myc (9E10, Developmental Studies Hybridoma Bank, University of Iowa), rabbit anti-Sun1 (Sigma), rabbit anti-Sun2 (Hodzic et al., 2004), chicken anti-nesprin2-Giant (Brian Burke, University of Florida, Gainesville), mouse anti-NPCs (mAb414, Covance, Princeton, NJ), rabbit anti-calreticulin (Assay Designs, Ann Arbor, MI), mouse anti-torsinA DM2A8 (Hewett et al., 2003), mouse anti-α-tubulin (Sigma), and rabbit anti-Sec61β (Joe Bass, Northwestern University). We generated an affinity-purified rabbit antibody against residues 1-217 of human LULL1 fused to glutathione S-transferase (GST). Protein purified from Escherichia coli was sent to Sigma Genosys for injection into rabbits. The resulting sera were depleted of GST-reactive antibodies by incubation with GST protein and subsequently affinity-purified on immobilized antigen. Secondary goat anti-mouse, goat anti-rabbit, and goat anti-chicken antibodies conjugated to Alexa 488, 555, or 595 were purchased from Invitrogen. Goat anti-mouse and goat anti-rabbit antibodies conjugated to HRP were purchased from Bio-Rad.
Triton X-114 Phase Separation
Cells from a 6-cm plate of U2OS cells transfected with the indicated construct were collected and resuspended in 250 μl buffer containing 2% Triton X-114 (Pierce Biotechnologies, Rockford, IL) and 200 mM NaCl. Samples were incubated at 4°C for 30 min and then centrifuged for 10 min at 10,000 × g to remove insoluble material. The soluble extract was incubated at 37°C for 10 min followed by centrifugation at 1000 × g for 10 min at room temperature to separate phases. The top “aqueous” phase was transferred to a new tube, and the bottom “detergent” phase brought to the same volume as the aqueous phase. Equal volumes were boiled and analyzed by SDS-PAGE and immunoblotting.
Blue Native PAGE
Ten-centimeter plates of U2OS cells or U2OS cells with tetracycline-inducible LULL1-myc were induced with tetracycline and/or transfected with constructs as indicated. Cells were resuspended in sample buffer (50 mM imidazole, 50 mM NaCl, 2 mM EDTA, 2 mM ATP, 2 mM aminocaproic acid, 4 mM MgCl2, PMSF, protease inhibitors, and dodecylmaltoside or digitonin detergent at indicated concentrations). The samples were incubated at 4°C for 15 min with agitation and then centrifuged for 30 min at 60,000 rpm at 4°C, followed by 15 min at 60,000 rpm at 4°C in fresh tubes. The protein concentration of the supernatant was determined by Bradford assay, and equivalent amounts of protein (∼20 μg per sample) were supplemented with Coomassie G-250 at 0.125% wt/vol final concentration and glycerol to 5% final concentration. Samples were then loaded onto 7.5% Blue Native (BN)-PAGE gels, run, and transferred to PVDF for Western blot detection. Samples were run alongside a native PAGE molecular weight marker (High Molecular Weight Calibration Kit for native electrophoresis, GE Healthcare, Piscataway, NJ). BN-PAGE gels, buffer, and PAGE protocols were as previously described (Wittig et al., 2006).
FACS Isolation of a LULL1, TorA-mGFP–positive Cell Population
Fifteen-centimeter plates of U2OS cells with tetracycline-inducible LULL1-myc were induced with tetracycline 6 h before transfection of TorA-mGFP. Either 12 or 22 h later cells were trypsinized, washed in PBS, and resuspended in phenol red–free DMEM plus 1% FBS and 0.1 mM EDTA at 4°C. Cells were diluted to a concentration of 5–10 × 106 cells/ml for sorting. Cells were collected by GFP fluorescence to obtain cells exclusively expressing LULL1-myc- and TorA-mGFP. FACS was performed at a Washington University School of Medicine core facility on a FACS Vantage Sorter (Becton Dickinson, Franklin Lakes, NJ).
Immunoprecipitation
U2OS-LULL1 myc cells in 6-cm plates were induced to express LULL1-His6myc by adding 1 mg/ml tetracycline 6 h before adding Lipofectamine 2000 and the indicated torA-mGFP plasmid. On the following day, cells were collected and solubilized in 20 mM HEPES, pH 7.2, 25 mM NaCl, 2 mM Mg2+ATP, 0.5% CHAPS, and complete protease inhibitor cocktail. LULL1-His6myc was then immunoprecipitated with 9E10 anti-myc mAb and protein G Sepharose (GE Healthcare). Samples were resolved by SDS-PAGE and the distribution of mGFP-tagged torsinA proteins monitored by immunoblotting with a GFP antibody.
RESULTS
Dynamic Regulation of TorA Distribution by LULL1
In an effort to understand how and when TorA functions within the NE, we studied the subcellular localization of TorA with mGFP fused to its C-terminus in transfected U2OS cells. As expected, TorA-mGFP is distributed evenly throughout the ER (Figure 1A, left). The dystonia-causative mutant, ΔGAG-TorA, also localizes to the ER but is enriched in the contiguous NE as expected based on earlier studies (Figure 1A, middle; Goodchild and Dauer, 2004; Naismith et al., 2004; Giles et al., 2008). TorA's binding partner LULL1-myc is also found throughout the ER (Figure 1A, right).
Figure 1.

LULL1 promotes relocalization of TorA to the NE. (A) Distribution of TorA-mGFP, ΔGAG-TorA-mGFP, and LULL1-myc expressed individually in U2OS cells. (B) Distribution of coexpressed TorA-mGFP and LULL1-myc, shown in maximum intensity projections (B and B″) and a confocal slice (B′). (C) Distribution of coexpressed ΔGAG-TorA-mGFP and LULL1-myc. (D) Distribution of coexpressed Sec61γ-mGFP and LULL1-myc. (E) Distribution of coexpressed Sec61γ-mGFP and TorA-myc. (A–C and E) are all maximum intensity projections of confocal z-series. (D) An epifluorescence image. Scale bars, 10 μm.
To our surprise, however, expressing TorA-mGFP together with LULL1-myc shifted TorA-mGFP almost entirely into the NE, where it displayed patterns ranging from concentration in a portion of the NE (Figure 1B, left-hand cell) to coverage of the entire nucleus (Figure 1B, right-hand cell). In the latter cells, the TorA-mGFP containing NE was often distorted (Figure 1B″). A confocal slice from the z-stack used to generate the projected image in Figure 1B confirms that TorA-mGFP concentrates at the nuclear periphery and that the LULL1-myc containing ER appears generally normal (Figure 1B′). Overall, 65% of cells (n > 150) expressing both proteins for 18 h had TorA-mGFP concentrated in the NE. A smaller, more variable proportion of LULL1-myc also relocalized to the NE (cf. Figure 1, A with B), leading us to hypothesize that LULL1 changes the subcellular targeting of TorA rather than vice versa. ΔGAG-TorA-mGFP similarly responded to coexpressed LULL1-myc by further concentrating in the NE (Figure 1C).
To assess the specificity of this phenomenon, we asked whether overexpressing LULL1 would cause another ER protein to redistribute, and conversely, whether introducing another ER protein would change the localization of TorA. For this analysis, we used Sec61γ tagged with GFP, which, similarly to LULL1, is a mobile single-pass transmembrane protein of the ER (Snapp et al., 2003b). We found that expressing LULL1-myc did not change the distribution of mGFP-Sec61γ between bulk ER and NE (Figure 1D) and that introducing mGFP-Sec61γ did not cause TorA to redistribute to the NE (Figure 1E). To ensure that the effect of LULL1 on TorA is not tag-dependent, we examined different forms of the proteins and found that untagged TorA and TorA-TAG-RFP responded to LULL1-myc and that LULL1-mGFP caused TorA-myc to redistribute (Supplemental Figure S1).
The variable distribution of wild-type TorA in fixed LULL1- and TorA-expressing cells suggested a dynamic process that we further explored in living cells. Shortly after transfecting TorA-mGFP into LULL1-expressing cells, we transferred coverslips to an imaging chamber and collected pictures every 15 min for up to 24 h. A typical field of cells is shown 2 h after transfection of TorA-mGFP and again 5 h later (Figure 2A). These images demonstrate that as TorA-mGFP expression begins, it is predominantly localized to the ER, recognizable at low magnification as a crescent of fluorescence surrounding a comparatively dark nucleus (Figure 2A, top panel). Over time, TorA-mGFP shifts to the NE in many of the expressing cells (Figure 2A, bottom panel). A pair of cells viewed at higher magnification in Figure 2B show the typical manner in which TorA redistributes. TorA starts to concentrate in the NE at one or a few points and proceeds to surround the whole nucleus in an average of 45 min (Figure 2B). This confirms that partially covered nuclei in fixed cells represent intermediates in the redistribution of TorA into the entire NE. In most cells, TorA remained concentrated in the NE for the duration of the experiment (Figure 2B, right-hand cell). Rarely, it later returned to the ER, demonstrating that the redistribution is reversible and implying that NE-localized TorA is still responsive to changes in its environment (Figure 2B, left-hand cell). Mitosis was occasionally observed, demonstrating that NE breakdown and reformation remain possible in the presence of NE-localized TorA (data not shown). Initiation of TorA's redistribution to the NE occurred randomly with respect to time of observation and position in the viewing field, suggesting that the redistribution is a cell-autonomous process initiated by factor(s) that could include a threshold amount of LULL1, TorA, or something else. Imaging of ΔGAG-TorA-mGFP in LULL1-myc–expressing cells showed that it too enriched in the NE with time, as expected based on the immunofluorescence of fixed cells (Supplemental Figure S2A). However, the mutant enzyme never progressed with polarity into the NE (and we never saw partially covered nuclei in fixed samples). To be sure that LULL1 was responsible for the enhanced enrichment of ΔGAG-TorA-mGFP in the NE, we also imaged it in U2OS cells with only endogenous levels of LULL1. In this case, its partitioning between NE and ER did not change over time (Supplemental Figure S2B).
Figure 2.

Live imaging and FRAP analysis of LULL1-directed TorA relocalization to the NE. (A) Selected images from time-lapse observation of TorA-mGFP in cells expressing unlabeled LULL1-myc. (B) Selected images from zoomed-in portion of a field similar to A. TorA-mGFP fills the NE over an average time of 45 min (±26 min, n = 52 cells). (C) Time series showing FRAP of LULL1-mGFP in U2OS cells. (D) Comparable time series showing FRAP of TorA-mGFP in U2OS cells. (E) Time series showing FRAP of TorA-mGFP in a cell coexpressing LULL1-mCherry. (E′) Corresponding time series showing FRAP of LULL1-mCherry in a cell expressing TorA-mGFP. (F) Diffusion coefficient values for LULL1 and TorA–mGFP, determined as described in Siggia et al. (2000) and Snapp et al. (2003a) from recovery curves such as those shown in Supplemental Figure S3. LULL1's diffusion coefficient averages ∼0.4 μm2/s in U2OS cells and in cells coexpressing TorA-mGFP, whereas TorA's diffusion coefficient ranges between 0.09 and 0.13 μm2/s in both U2OS and LULL1-mCherry–expressing cells. For comparison, luminal ER-GFP has a Deff of ∼10 μm2/s (Snapp et al., 2003a), an ER-localized transmembrane protein has a Deff of ∼0.4 μm2/s (Snapp et al., 2003a), and a polysome-associated translocon has a Deff of ∼0.05 μm2/s (Nikonov et al., 2002). Scale bars, 10 μm.
We next asked whether the LULL1-dependent shift of TorA into the NE could be explained by selective immobilization there, analogous to the behavior of NE-resident proteins such as the lamin B receptor (Ellenberg et al., 1997). To address this, we carried out quantitative FRAP experiments (Snapp et al., 2003a). We found that expressed individually, TorA-mGFP recovered slowly from photobleaching (Figure 2, D and F), whereas LULL1-mGFP recovered an order of magnitude more quickly (Figure 2, C and F). Coexpressing TorA-mGFP with LULL1-mCherry did not change the slow mobility of TorA in either the NE or ER (Figure 2, E and F). Conversely, coexpression with TorA-mGFP did not detectably change LULL1-mCherry diffusion (Figure 2, E′ and F). Notably, TorA-mGFP is readily solubilized by mild detergents (data not shown, but see Figure 3 below), indicating that its slow diffusion and localization to the NE are not consequences of aggregation.
Figure 3.

BN-PAGE separation of TorA oligomers. (A) BN-PAGE separation of untagged TorA expressed in U2OS cells. TorA is detectable as a species of approximately hexameric size in 0.25% wt/vol DDM; this species is decreased in abundance in the presence of higher concentrations of DDM. (B) 2D PAGE of untagged TorA expressed in U2OS cells. TorA was solubilized in 0.5% wt/vol digitonin, run on first dimension BN-PAGE and then separated in a second dimension by SDS-PAGE. (C) BN-PAGE separation of untagged TorA expressed alone or with LULL1-myc, solubilized in 1% wt/vol digitonin.
The diffusive behavior of TorA suggested that it participates in a large protein complex in the ER and NE. Furthermore, AAA+ proteins typically function as higher order oligomers, typically either hexamers or dodecamers (Hanson and Whiteheart, 2005). However, previous analyses of TorA's oligomeric state had not identified any such large assemblies (Kustedjo et al., 2003), although TorA can self-associate as judged by coimmunoprecipitation (Torres et al., 2004; Giles et al., 2008). To reconcile these two lines of data, we hypothesized that TorA oligomers might be disrupted by the detergents used to prepare previous samples for analysis. We turned to BN-PAGE, which separates native membrane proteins by size and shape in the presence of mild detergent and the protein binding dye Coomassie Brilliant Blue (Wittig et al., 2006). BN-PAGE of dodecylmaltoside (DDM)-solubilized TorA reveals the presence of an oligomeric species of approximately hexameric size in samples containing 0.25% wt/vol DDM (Figure 3A, left lane). This oligomer is destabilized by higher concentrations of DDM (Figure 3A), indicating that it represents a complex of folded proteins rather than an aggregate. A comparable oligomer is detected in samples solubilized with 0.5% wt/vol digitonin (Figure 3B, top panel). Further, the oligomeric species is completely dispersed on a second-dimension denaturing SDS-PAGE gel (Figure 3B, bottom panel), where all immunoreactivity corresponds to the size of monomeric TorA (∼38 kDa). This suggests that TorA forms a hexameric unit similar to other AAA+ ATPases (Hanson and Whiteheart, 2005), and together with earlier suggestions that TorA is peripherally associated with the ER lumenal membrane (Liu et al., 2003; Callan et al., 2007) provides a likely explanation for its slow mobility in the FRAP experiments described above. Slow diffusion may explain why it takes an average of 45 min (Figure 2B) for TorA-mGFP to relocalize from the ER to the NE. Finally, TorA is a stable oligomeric species of approximately hexameric size regardless of whether or not LULL1 is coexpressed (Figure 3C). The fact that LULL1 changes neither the oligomeric state nor the apparent mobility of TorA suggests that LULL1 changes the enzyme's targeting but not its fundamental organization.
Features of TorA Involved in Its Redistribution to the NE
LULL1-promoted redistribution of TorA to the NE was unexpected based on the earlier finding that hydrolysis deficient “substrate trap” mutant TorA (containing a Walker B motif E171Q mutation) accumulates together with LULL1 in the peripheral ER and efficiently coimmunoprecipitates with it (Goodchild and Dauer, 2005). Because wild-type TorA only inefficiently coimmunoprecipitates with LULL1 (Goodchild and Dauer, 2005), we hypothesized that a transient, ATP-dependent interaction between active enzyme and LULL1 changes something, most likely in TorA, that in turn targets it to the NE. To learn more about how TorA responds to LULL1, we explored the effects of mutations in defined motifs within TorA. Under conditions in which wild-type enzyme efficiently relocalizes to the NE, we found that, as expected (Goodchild and Dauer, 2005), TorA(E171Q) was distributed throughout the ER (Figure 4B). Interestingly, TorA with a mutation in its Walker A ATP-binding motif (K108A) also did not redistribute into the NE (Figure 4B), establishing that TorA needs to be able to engage ATP to move in response to LULL1.
Figure 4.

Structural requirements for LULL1-directed TorA redistribution. (A) Schematic of TorA structure. (B) Representative epifluorescence images of TorA-mGFP containing indicated mutation in LULL1-myc (not shown) expressing U2OS cells. Scale bars, 10 μm. (C) Quantitation of redistribution. n > 150 cells for each mutant. (D) Immunoblots of equal fractions of aqueous and hydrophobic phases from a Triton X-114 phase-partitioning assay show that wild-type TorA partitions with the hydrophobic phase, but shifts to the aqueous phase after deleting the protein's N-terminal hydrophobic domain (amino acids 26–43). (E) E171Q-TorA-mGFP immunoprecipitates efficiently with LULL1-myc, and deletion of the N-terminal hydrophobic domain (amino acids 26–43) does not abolish binding.
We next wondered whether peripheral association of TorA with the lumenal membrane is important for its targeting to the NE. We deleted the hydrophobic sequence (Δ26-43) implicated in membrane association (Liu et al., 2003; Callan et al., 2007) and confirmed that TorA no longer behaved as a hydrophobic protein in Triton X-114 phase partitioning experiments (Figure 4D). Interestingly, this hydrophilic TorA did not move into the NE in cells expressing LULL1 (Figure 4B). Because deleting the hydrophobic sequence does not affect binding of “substrate trap” E171Q mutant TorA to LULL1 (Figure 4E), this mutant's failure to redistribute suggests that the N-terminus of TorA plays an essential role in LULL1-dependent NE targeting.
Removing LULL1 Reduces Enrichment of ΔGAG-TorA in the Nuclear Envelope
The above experiments indicate that overexpressing LULL1 shifts a large proportion of TorA into the NE. This, together with the fact that ΔGAG-TorA (and in some cell types, also wild-type TorA) is known to be intrinsically enriched in the NE (Figure 1A; Gonzalez-Alegre and Paulson, 2004; Goodchild and Dauer, 2004; Naismith et al., 2004), led us to hypothesize that distribution of TorA between ER and NE may normally be controlled by interaction with endogenous LULL1. To explore this possibility, we used RNA interference (RNAi) to deplete LULL1 (Figure 5A) and compared the localization of ΔGAG-TorA-mGFP to that of a cotransfected ER lumenal marker consisting of a prolactin signal sequence and a KDEL ER-retrieval sequence fused to mRFP (ER-RFP; Snapp et al., 2006). Representative confocal sections show that ΔGAG-TorA-mGFP is more concentrated around the nucleus than is ER-RFP in U2OS cells (Figure 5B), but not in LULL1 knockdown cells (Figure 5C). Wild-type TorA, meanwhile, is distributed similarly to ER-RFP in both cell types (images not shown, Figure 5D). Quantitative analysis (Figure 5D; see Materials and Methods) confirms that wild-type TorA has no preference for the NE and/or perinuclear ER in U2OS cells. ΔGAG-TorA-mGFP, in contrast, is enriched in the perinuclear region in U2OS cells but not in cells depleted of LULL1 (p = 0.003). The distribution of ΔGAG-TorA-mGFP in the absence of LULL1 was indistinguishable from that of ER-RFP (p > 0.05). Analysis of ΔGAG-TorA-mGFP in the presence of another RNA hairpin directed against LULL1 showed the same effect (data not shown). This demonstrates that enrichment of ΔGAG-TorA-mGFP in the NE requires LULL1. Together with the finding that high levels of LULL1 shift TorA into the NE, these experiments suggest that LULL1 has a general role in regulating localization of both wild-type and disease-mutant TorA.
Figure 5.

Removing LULL1 by RNAi reverses enrichment of ΔGAG-TorA in NE. (A) Immunoblot of lysates from cells transduced with the indicated shRNA probed for LULL1. (B and C) Representative confocal images of ΔGAG-TorA-mGFP and ER-RFP coexpressed in control U2OS cells (B) versus in U2OS cells transduced with shRNA no. 2088 (C). (D) Relative fluorescence intensity of ER-RFP, wt-TorA-mGFP, and ΔGAG-TorA-mGFP in NE versus ER. ΔGAG-TorA-mGFP has a significantly higher NE–ER ratio in U2OS cells than in LULL1 knockdown cells (p = 0.003). The NE–ER ratio of ΔGAG-TorA-mGFP is not significantly different from that of ER-RFP in LULL1 knockdown cells (p = 0.19). n >21 cells for each condition. Scale bars, 10 μm.
Redistributed TorA Displaces a Subset of LINC Complex Components from the NE
To explore TorA's effects on the NE, we examined specific NE components by immunostaining. In cells containing NE-localized TorA, the distribution and staining of nuclear pores did not change indicating that the overall integrity of the NE is maintained in these cells (Figure 6A). We next examined TorA's effects on the inner nuclear membrane proteins Sun1 and Sun2. Although Sun1 was not affected by redistributed TorA (Figure 6B), Sun2 immunostaining was notably diminished in regions of the NE where TorA was concentrated, suggesting that TorA might displace Sun2 as it moves into the NE (Figure 6C). Orthogonal views of confocal z-series confirmed that Sun1 remains at the nuclear periphery in cells with TorA in the NE (Figure 6B′), whereas Sun2 is decreased (Figure 6C′). The normal distribution of nuclear pores, known to colocalize with Sun1 but not Sun2 (Liu et al., 2007), could explain a selective effect of TorA on Sun2. A survey of Sun2 immunoreactivity in cells containing LULL1-redistributed TorA-mGFP indicated that changes in Sun2 were widespread; 60% of these cells lacked a clearly delineated nuclear rim of Sun2 compared with only 4% of untransfected cells (n > 110 for each). To rule out the alternative possibility that redistributed TorA masked the lumenal Sun2 antibody epitope (Hodzic et al., 2004), we made a tetracycline-inducible LULL1-myc cell line that also constitutively expressed Sun2-GFP and assessed the effect of introducing TorA fused to TagRFP (Merzlyak et al., 2007). Direct examination of protein fluorescence showed that Sun2-GFP was displaced by TorA-TagRFP (Figure 6D), paralleling the effect of TorA on endogenous Sun2. Importantly, despite being somewhat concentrated in the NE, E171Q-TorA and ΔGAG-TorA expressed individually in U2OS cells had little or no effect on Sun2 (Supplemental Figure S4). Further, expression of wild-type TorA on its own had no detectable effect. Sun2 in the NE thus appears to decrease specifically in response to LULL1-redistributed TorA. The overall decrease in Sun2 intensity in cells with redistributed TorA in the NE suggests that displaced Sun2 may be unstable and possibly degraded. Western blot analysis of Sun2 in such cells confirms a decrease in the overall level of Sun2 but not nucleoporins or a general ER marker (Figure 7). Increased degradation of INM proteins has been reported when lamin anchors are absent (Muchir et al., 2006); it is possible that TorA could similarly destabilize Sun2 as a consequence of altering its association with the nuclear lamina.
Figure 6.

Redistributed TorA displaces LINC complex components from the NE. (A) Nuclear pore components stained with mAb414 in cells expressing LULL1-myc (not shown) and TorA-mGFP, epifluorescence image. (B) Sun1 in cells expressing LULL1-myc (not shown) and TorA-mGFP. Shown are maximum intensity projections of a confocal z-series. (B′) XZ orthogonal view: top, TorA-mGFP; bottom, Sun1. Approximate position of xz slice is marked by asterisk in B. (C) Sun2 immunostaining in the same cells. Sixty percent of cells containing TorA-mGFP in the NE lack a nuclear rim of Sun2, compared with 4% of untransfected cells (n >110 for each). (C′) XZ orthogonal view: top, TorA-mGFP; bottom, Sun2. Approximate position of xz slice is marked by asterisk in C. (D) Sun2-GFP and TorA-TagRFP in cells also expressing LULL1-myc, projected z-series. (E) Nesprin2-Giant and TorA-mGFP in cells also expressing LULL1-myc (not shown), imaged by epifluorescence microscopy. Fifty-six percent of cells containing TorA-mGFP in the NE lack NE-localized nesprin2-Giant, compared with 1% of untransfected cells (n >35 for each). (F) Transfected nesprin3α-GFP in cells also expressing TorA-TagRFP and LULL1-myc (not shown), projected z-series. Scale bars, 10 μm.
Figure 7.
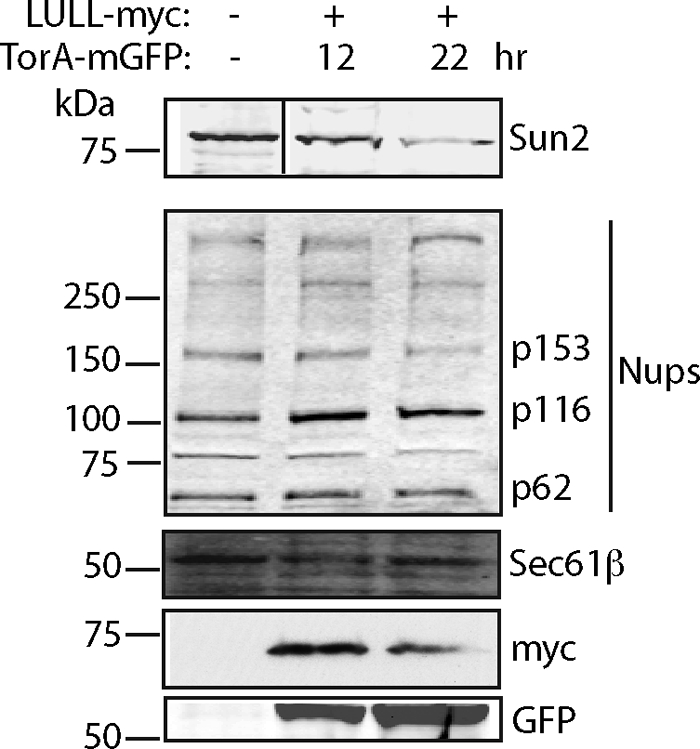
Sun2 protein levels are decreased as a consequence of LULL1-directed NE localization of TorA. Western blot analysis of cells coexpressing LULL1-myc and TorA-mGFP for 12 or 22 h and sorted by GFP fluorescence indicates that Sun2 protein levels decrease, whereas NPCs (mAb 414) and a representative ER protein (Sec61β) are not affected.
SUN proteins participate in NE-spanning LINC complexes (Crisp et al., 2006), which consist of SUN proteins in the inner nuclear membrane (INM) and nesprins in the outer nuclear membrane (ONM; Starr, 2009). Conserved domains of these proteins interact in the perinuclear space to physically connect the nucleus to the cytoskeleton (Stewart-Hutchinson et al., 2008; Starr, 2009). We therefore next asked whether redistributed TorA affects the ONM-localized nesprins. Cells expressing LULL1-myc and TorA-mGFP were stained with an antibody for nesprin2-Giant, revealing that it too was missing from the NE when TorA was concentrated there (Figure 6E). This displacement occurred in 56% of cells expressing LULL1-myc and TorA-mGFP compared with only 1% of untransfected cells (n > 35 for each). Similarly, transiently transfected nesprin3a-GFP was absent from regions of the NE containing TorA-TagRFP (Figure 6F). These findings demonstrate that TorA changes the behavior of a subset of LINC complex components, including Sun2 in the INM and nesprin2-Giant and nesprin-3 in the ONM. Indeed, some of the changes in NE membranes caused by redistributed wild-type TorA (Figure 1B″; see also Figure 8A) are reminiscent of changes induced by manipulating components of LINC complexes (Crisp et al., 2006). A recent report of interaction between TorA and nesprin-3 suggests that these effects could be direct (Nery et al., 2008), but further studies are needed to understand the molecular changes involved. Because live imaging analysis (Figure 2B) indicated that TorA moves across the NE within an hour, the coincident patterns of Sun2 and nesprin-3 imply that their displacement is temporally related to the arrival of TorA in the NE and that they could be its direct targets.
Figure 8.

Comparison of wild-type and ΔGAG-TorA-mGFP effects on the NE. (A) Comparison of NE in cells expressing LULL1-myc (not shown) with redistributed wild-type TorA-mGFP (left) and ΔGAG-TorA-mGFP (right). NE membrane distortions occur in 68% of cells with NE-localized wild-type TorA (n = 136) and in 7% of cells with NE-localized ΔGAG-TorA (n = 74). Shown are maximum intensity projections of confocal z-series, with orthogonal xz views in A′. Approximate position of xz orthogonal slice marked by asterisk in A. (B) Effect of LULL1-redistributed ΔGAG-TorA-mGFP on Sun2. Projected z-series, with orthogonal views of ΔGAG-TorA (top) and Sun2 (bottom) in B′. Approximate position of xz slice is marked by asterisk in B. (C) Time-lapse imaging of Sun2-GFP and TorA-TagRFP in LULL1-myc–expressing cells. (D) Time-lapse imaging of Sun2-GFP and ΔGAG-TorA-TagRFP in LULL1-myc–expressing cells. The median delay between the onset of TorA-TagRFP redistribution and initiation of Sun2-GFP loss was <15 min (between consecutive frames) for wild type (±20 min, n = 32 cells) and 420 min for ΔGAG-TorA (±378 min, n = 33 cells). Scale bars, 10 μm.
The DYT1 ΔGAG Mutation Impairs the Effects of TorA on the NE
ΔGAG-TorA has consistently been found to be more enriched in the NE than is wild-type TorA (Gonzalez-Alegre and Paulson, 2004; Goodchild and Dauer, 2004; Naismith et al., 2004; Giles et al., 2008), leading to the hypothesis that mislocalization to the NE might contribute to the development of dystonia (Goodchild and Dauer, 2004). At the same time, ΔGAG-TorA is thought to be less functional and less stable than wild-type TorA such that loss of normal enzyme function could underlie the disease (Goodchild et al., 2005; Giles et al., 2008; Gordon and Gonzalez-Alegre, 2008). As shown in Figure 1 and Supplemental Figure S2, coexpressing LULL1 with ΔGAG-TorA-mGFP caused it to enrich further in the NE, albeit without the distinctive polarity that characterized the shift of wild-type protein. We therefore compared the behavior of ΔGAG- and wild-type TorA in more detail. Maximum intensity projections of nuclei in cells in which these proteins were concentrated for up to 18 h revealed that wild-type TorA distorted NE membranes into both the nucleus and the cytoplasm (Figure 8, Aand A′, left) in 68% of cells with NE-localized TorA (n = 136). In contrast, ΔGAG-TorA-mGFP left the NE largely unchanged (Figure 8, A and A′, right), causing NE distortions in only 7% of cells containing NE-localized protein (n = 74). These differences suggest that ΔGAG-TorA interacts differently than wild-type enzyme with components of the NE.
To determine how redistributed ΔGAG-TorA affects specific NE components, we looked for changes in Sun2. In cells expressing LULL1-myc and ΔGAG-TorA-mGFP Sun2 was diminished (Figure 8, B and B′). However, a difference between mutant and wild-type enzyme became apparent when we used time-lapse imaging to compare the effects of wild-type or ΔGAG TorA-TagRFP on Sun2-GFP. Representative sequential images show that transiently transfected wild-type TorA-TagRFP displaces Sun2-GFP as it appears in the NE (Figure 8C). In the cells shown, Sun2-GFP decreases at one end of the nucleus, whereas TorA-TagRFP is too dim to see, but as TorA-TagRFP becomes visible it is apparent that the boundary between increasing TorA-TagRFP and decreasing Sun2-GFP remains closely apposed (see especially 90- and 120-min frames of Figure 8C). In contrast, time-lapse imaging of ΔGAG-TorA-TagRFP revealed a much slower effect of ΔGAG-TorA on Sun2-GFP (Figure 8D). Although Sun2-GFP started to decrease within 15 min of when wild-type TorA began to concentrate in the NE, it took ∼7 h for this to happen in cells expressing ΔGAG-TorA. Together with the lack of structural abnormalities in the NE of cells expressing ΔGAG-TorA, this suggests that ΔGAG-TorA is less efficient than wild-type TorA at changing NE components.
DISCUSSION
Controlling the localization of enzymes within the cell is an important general mechanism for regulating their activity. Posttranslational modifications and conformational changes are known to retarget many cytoplasmic enzymes from one place to another, but dynamic changes in the distribution of enzymes within the ER lumen are less well characterized. We find that the TorA-binding partner LULL1—an ER transmembrane protein—drives redistribution of this lumenal AAA+ ATPase from throughout the ER network specifically into the NE (Figure 9), where previous in vitro and in vivo studies have suggested a function (Goodchild and Dauer, 2004; Naismith et al., 2004; Goodchild et al., 2005). The idea that regulated distribution between the ER and NE might control TorA's activity toward spatially restricted substrate(s) arose initially from studies showing that so-called “substrate trapped” TorA mutants accumulated in the NE (Gerace, 2004; Goodchild and Dauer, 2004; Naismith et al., 2004). The wild-type enzyme, in contrast, is diffusely distributed throughout the ER except in a few cell types (Giles et al., 2008) and after certain pharmacological manipulations (Hewett et al., 2003, 2006; Nery et al., 2008). We hypothesized that additional factor(s), which perhaps are more abundant in cell types where TorA exhibits NE preference (Giles et al., 2008), were therefore likely to control the targeting and activity of wild-type TorA. Extrapolating between the extremes of LULL1 overexpression (Figures 1, 2, and 6) and LULL1 silencing (Figure 5) leads us to propose that transient interaction with LULL1 positively regulates the targeting and activity of TorA in the NE. The abundance of LULL1 thus emerges as a critical regulator of TorA activity. Future studies will address whether variations in endogenous LULL1 levels explain previously described cell type–specific differences in TorA distribution (Giles et al., 2008) and whether there are peaks in the expression or stabilization of LULL1 that correlate with important events in neuronal development or plasticity.
Figure 9.

Model of LULL1-dependent TorA function at the NE. LULL1 interacts with the TorA hexamer in the peripheral ER, which promotes an activating change in TorA that requires catalytic residues and association with the membrane. TorA then moves into the NE, where it displaces components of LINC complexes, including Sun2, nesprin2-Giant, and nesprin3. This gives TorA the ability to alter contacts between the nuclear lamina (or nuclear contents), the nuclear envelope membranes, and the cytoskeleton.
LULL1 is a ∼70-kDa single-pass transmembrane protein with no defined functional motifs and no known binding partners other than TorA (Goodchild and Dauer, 2005). Several features suggest that LULL1 may be regulated at both the transcriptional and posttranslational levels, making it an attractive potential regulatory protein. There are significant variations in the level of LULL1 message in different cells and tissues (Goodchild and Dauer, 2005; Chen et al., 2006) and over the course of in vitro muscle cell differentiation (Chen et al., 2006). Further, the extralumenal domain of LULL1 has a preponderance of charged residues and a dearth of hydrophobic residues, both of which are likely to predispose it to rapid and potentially regulated turnover (Fink, 2005). Consistently, secondary structure prediction algorithms indicate a lack of stably folded structures in this region, and the PESTfind algorithm (http://www.at.embnet.org/toolbox/pestfind) finds two high-scoring sequences that may correlate with rapid protein turnover (Rechsteiner and Rogers, 1996). Finally, as a transmembrane protein, LULL1 provides a way to directly or indirectly transmit signals across the ER membrane to TorA.
Using LULL1 overexpression to concentrate TorA in the NE, we were able to uncover molecular and structural changes that are likely to represent TorA's normal activity, including alterations in a subset of NE proteins that participate in NE-bridging LINC complexes (Figures 6 and 7). LINC complexes are formed when INM-localized Sun proteins and ONM-localized nesprin proteins interact within the NE lumen and have recently attracted attention as important connectors between the cytoskeleton, NE membranes, and elements within the nucleus including the lamina and telomeres (Starr, 2009). Although much remains to be learned about the cell biology of LINC complexes, it is clear that they have important roles in such diverse processes as nuclear anchorage, cell migration, and regulating gene expression (Worman and Gundersen, 2006; Crisp and Burke, 2008). Our data showing that some LINC complex proteins—Sun2, nesprin-2G, and nesprin-3—are destabilized by TorA as it accumulates in the NE suggest possible roles for TorA in these same processes. In support of this, a recent study showed that fibroblasts from TorA knockout mice migrate more slowly than controls in a polarized cell migration assay (Nery et al., 2008).
Although LINC complex proteins may help recruit TorA to the NE, the subset that TorA displaces (i.e., at least Sun2, nesprin-2G, and nesprin-3) seem unlikely to be directly responsible for retaining it there because TorA remains in the NE even after these proteins are gone. Future work will need to explore possible roles for other NE proteins in this process, perhaps especially remaining LINC complex proteins such as Sun1 and the known TorA-binding partner LAP1.
The mechanism by which LULL1 induces TorA retargeting is an area of ongoing interest. Here, we present several observations about TorA that constrain the possible explanations. Using BN-PAGE (Figure 3) and FRAP (Figure 2), we established that TorA assembles into a large, membrane-associated oligomer—probably a hexamer—within the ER lumen. The oligomeric state of TorA did not change after interaction with LULL1, and LULL1 remained highly mobile whether or not it was transiently engaging and affecting TorA. These results are the first demonstration that TorA indeed assembles into the kind of oligomer expected of an AAA+ protein and establish that LULL1 changes TorA targeting without affecting its fundamental structure. Our mutagenesis experiments indicate that an N-terminal hydrophobic sequence distinct from TorA's core AAA+ domain (Kock et al., 2006; Zhu et al., 2008) is required for this retargeting (Figure 4). It is therefore attractive to hypothesize that interaction with LULL1 causes a conformational change involving this N-terminal domain, enhancing TorA's affinity for something within the NE. Future work will focus on defining these states and further delineating the mechanism(s) responsible for controlling the transition between the ER-distributed and NE-enriched forms of TorA.
Importantly, we found that DYT1-associated ΔGAG-TorA is also redirected to the NE by LULL1 (Figures 1 and 8), but once there it is less effective at enacting changes in NE structure and protein composition (Figure 8). These results suggest a molecular loss-of-function that may correlate with the previously described inability of ΔGAG-TorA to rescue the lethality of TorA knockout in the mouse (Dang et al., 2005; Goodchild et al., 2005) and could ultimately contribute to the development of DYT1 dystonia. At a structural level, comparison of TorA's AAA+ domain to that of ClpB or ClpA suggests that the ΔGAG deletion falls in a position that could perturb a helix preceding an ATP-interacting loop known as the sensor-2 motif thereby leading to a loss-of-function (Kock et al., 2006; Zhu et al., 2008). The fact that TorA is now established to be an oligomeric enzyme (Figure 3) supports the possibility that mixed oligomers containing wild-type and mutant subunits could turn a loss-of-function mutation into a dominantly inherited trait. Separately, data from other groups have shown that overexpressing ΔGAG-TorA can have toxic effects on the function of the secretory pathway (Hewett et al., 2007), raising the possibility that a combination of the loss-of-function shown here and gain-of-function shown elsewhere might explain the dominant inheritance of DYT1 dystonia. The discovery that LULL1 regulates the distribution and activity of TorA within the ER and the NE paves the way for future exploration of how changes in its activity correlate with the development of disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Seema Dalal for early work cloning LULL1 and creating stable cell lines, Soomin Shim for assistance with LULL1 shRNA experiments, P. J. Stewart-Hutchison for technical assistance and helpful discussions, and members of the Hanson lab for helpful discussions. Robert Mecham and members of his laboratory at Washington University, specifically Jessica Wagenseil and Thomas Broekelmann, are gratefully acknowledged for assistance with the time-lapse imaging. This work was supported by grants from the National Institute of Neurological Disorders and Stroke Grant R01 NS050717 (P.I.H.), Ellison Medical Foundation (E.L.S.), the Muscular Dystrophy Association (D.H.), and the Bachman Strauss Parkinson and Dystonia Foundation (P.I.H.).
Abbreviations used:
- AAA+
ATPases associated with a variety of cellular activities
- BN-PAGE
Blue Native-PAGE
- DDM
dodecylmaltoside
- ER
endoplasmic reticulum
- FRAP
fluorescence recovery after photobleaching
- INM
inner nuclear membrane
- LINC
linker of the nucleoskeleton and cytoskeleton
- mGFP
monomeric green fluorescent protein
- NE
nuclear envelope
- ONM
outer nuclear membrane
- RFP
red fluorescent protein
- TorA
TorsinA.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-01-0094) on April 1, 2009.
REFERENCES
- Breakefield X. O., Blood A. J., Li Y., Hallett M., Hanson P. I., Standaert D. G. The pathophysiological basis of dystonias. Nat. Rev. Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Callan A. C., Bunning S., Jones O. T., High S., Swanton E. Biosynthesis of the dystonia-associated AAA+ ATPase torsinA at the endoplasmic reticulum. Biochem. J. 2007;401:607–612. doi: 10.1042/BJ20061313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. H., Huber M., Guan T., Bubeck A., Gerace L. Nuclear envelope transmembrane proteins (NETs) that are up-regulated during myogenesis. BMC Cell Biol. 2006;7:38. doi: 10.1186/1471-2121-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M., Burke B. The nuclear envelope as an integrator of nuclear and cytoplasmic architecture. FEBS Lett. 2008;582:2023–2032. doi: 10.1016/j.febslet.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Crisp M., Liu Q., Roux K., Rattner J. B., Shanahan C., Burke B., Stahl P. D., Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 2006;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirok A., Rupp P. A., Rongish B. J., Little C. D. Multi-field 3D scanning light microscopy of early embryogenesis. J. Microsc. 2002;206:209–217. doi: 10.1046/j.1365-2818.2002.01032.x. [DOI] [PubMed] [Google Scholar]
- Dalal S., Rosser M. F., Cyr D. M., Hanson P. I. Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell. 2004;15:637–648. doi: 10.1091/mbc.E03-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang M. T., Yokoi F., McNaught K. S., Jengelley T. A., Jackson T., Li J., Li Y. Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Ellenberg J., Siggia E. D., Moreira J. E., Smith C. L., Presley J. F., Worman H. J., Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. Concept and classification of dystonia. Adv. Neurol. 1988;50:1–8. [PubMed] [Google Scholar]
- Fink A. L. Natively unfolded proteins. Current Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Gerace L. TorsinA and torsion dystonia: Unraveling the architecture of the nuclear envelope. Proc. Natl. Acad. Sci. USA. 2004;101:8839–8840. doi: 10.1073/pnas.0402441101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles L. M., Chen J., Li L., Chin L. S. Dystonia-associated mutations cause premature degradation of torsinA protein and cell type-specific mislocalization to the nuclear envelope. Hum. Mol. Genet. 2008;17:2712–2722. doi: 10.1093/hmg/ddn173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alegre P., Paulson H. L. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J. Neurosci. 2004;24:2593–2601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild R. E., Dauer W. T. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. USA. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild R. E., Dauer W. T. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 2005;168:855–862. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild R. E., Kim C. E., Dauer W. T. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Gordon K. L., Gonzalez-Alegre P. Consequences of the DYT1 mutation on torsinA oligomerization and degradation. Neuroscience. 2008;157:588–595. doi: 10.1016/j.neuroscience.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson P. I., Whiteheart S. W. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- Hewett J., Gonzalez-Agosti C., Slater D., Ziefer P., Li S., Bergeron D., Jacoby D. J., Ozelius L. J., Ramesh V., Breakefield X. O. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum. Mol. Genet. 2000;9:1403–1413. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- Hewett J., et al. TorsinA in PC12 cells: localization in the endoplasmic reticulum and response to stress. J. Neurosci Res. 2003;72:158–168. doi: 10.1002/jnr.10567. [DOI] [PubMed] [Google Scholar]
- Hewett J. W., Tannous B., Niland B. P., Nery F. C., Zeng J., Li Y., Breakefield X. O. Mutant torsinA interferes with protein processing through the secretory pathway in DYT1 dystonia cells. Proc. Natl. Acad. Sci. USA. 2007;104:7271–7276. doi: 10.1073/pnas.0701185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewett J. W., Zeng J., Niland B. P., Bragg D. C., Breakefield X. O. Dystonia-causing mutant torsinA inhibits cell adhesion and neurite extension through interference with cytoskeletal dynamics. Neurobiol. Dis. 2006;22:98–111. doi: 10.1016/j.nbd.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Hodzic D. M., Yeater D. B., Bengtsson L., Otto H., Stahl P. D. Sun2 is a novel mammalian inner nuclear membrane protein. J. Biol. Chem. 2004;279:25805–25812. doi: 10.1074/jbc.M313157200. [DOI] [PubMed] [Google Scholar]
- Ketema M., Wilhelmsen K., Kuikman I., Janssen H., Hodzic D., Sonnenberg A. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J. Cell Sci. 2007;120:3384–3394. doi: 10.1242/jcs.014191. [DOI] [PubMed] [Google Scholar]
- Kock N., Naismith T. V., Boston H. E., Ozelius L. J., Corey D. P., Breakefield X. O., Hanson P. I. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 2006;15:1355–1364. doi: 10.1093/hmg/ddl055. [DOI] [PubMed] [Google Scholar]
- Kustedjo K., Bracey M. H., Cravatt B. F. Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J. Biol. Chem. 2000;275:27933–27939. doi: 10.1074/jbc.M910025199. [DOI] [PubMed] [Google Scholar]
- Kustedjo K., Deechongkit S., Kelly J. W., Cravatt B. F. Recombinant expression, purification, and comparative characterization of torsinA and its torsion dystonia-associated variant Delta E-torsinA. Biochemistry. 2003;42:15333–15341. doi: 10.1021/bi0349569. [DOI] [PubMed] [Google Scholar]
- Liu Q., Pante N., Misteli T., Elsagga M., Crisp M., Hodzic D., Burke B., Roux K. J. Functional association of Sun1 with nuclear pore complexes. J. Cell Biol. 2007;178:785–798. doi: 10.1083/jcb.200704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Zolkiewska A., Zolkiewski M. Characterization of human torsinA and its dystonia-associated mutant form. Biochem. J. 2003;374:117–122. doi: 10.1042/BJ20030258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzlyak E. M., Goedhart J., Shcherbo D., Bulina M. E., Shcheglov A. S., Fradkov A. F., Gaintzeva A., Lukyanov K. A., Lukyanov S., Gadella T. W., Chudakov D. M. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods. 2007;4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- Muchir A., Massart C., van Engelen B. G., Lammens M., Bonne G., Worman H. J. Proteasome-mediated degradation of integral inner nuclear membrane protein emerin in fibroblasts lacking A-type lamins. Biochem. Biophys. Res. Commun. 2006;351:1011–1017. doi: 10.1016/j.bbrc.2006.10.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naismith T. V., Heuser J. E., Breakefield X. O., Hanson P. I. TorsinA in the nuclear envelope. Proc. Natl. Acad. Sci. USA. 2004;101:7612–7617. doi: 10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery F. C., Zeng J., Niland B. P., Hewett J., Farley J., Irimia D., Li Y., Wiche G., Sonnenberg A., Breakefield X. O. TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J. Cell Sci. 2008;121:3476–3486. doi: 10.1242/jcs.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov A. V., Snapp E., Lippincott-Schwartz J., Kreibich G. Active translocon complexes labeled with GFP-Dad1 diffuse slowly as large polysome arrays in the endoplasmic reticulum. J. Cell Biol. 2002;158:497–506. doi: 10.1083/jcb.200201116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius L. J., et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Ozelius L. J. The gene (DYT1) for early-onset torsion dystonia encodes a novel protein related to the Clp protease/heat shock family. Adv. Neurol. 1998;78:93–105. [PubMed] [Google Scholar]
- Rechsteiner M., Rogers S. W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- Siggia E. D., Lippincott-Schwartz J., Bekiranov S. Diffusion in inhomogeneous media: theory and simulations applied to whole cell photobleach recovery. Biophys. J. 2000;79:1761–1770. doi: 10.1016/S0006-3495(00)76428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp E. L., Altan N., Lippincott-Schwartz J. Measuring protein mobility by photobleaching GFP chimeras in living cells. Curr. Protocol. Cell Biol. 2003a doi: 10.1002/0471143030.cb2101s19. Chapter 21, Unit 21–1. [DOI] [PubMed] [Google Scholar]
- Snapp E. L., Hegde R. S., Francolini M., Lombardo F., Colombo S., Pedrazzini E., Borgese N., Lippincott-Schwartz J. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 2003b;163:257–269. doi: 10.1083/jcb.200306020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp E. L., Sharma A., Lippincott-Schwartz J., Hegde R. S. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr D. A. A nuclear-envelope bridge positions nuclei and moves chromosomes. J. Cell Sci. 2009;122:577–586. doi: 10.1242/jcs.037622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart-Hutchinson P. J., Hale C. M., Wirtz D., Hodzic D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 2008;314:1892–1905. doi: 10.1016/j.yexcr.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres G. E., Sweeney A. L., Beaulieu J. M., Shashidharan P., Caron M. G. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc. Natl. Acad. Sci. USA. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K., Litjens S. H., Kuikman I., Tshimbalanga N., Janssen H., van den Bout I., Raymond K., Sonnenberg A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005;171:799–810. doi: 10.1083/jcb.200506083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I., Braun H. P., Schagger H. Blue native PAGE. Nat. Protocol. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Worman H. J., Gundersen G. G. Here come the SUNs: a nucleocytoskeletal missing link. Trends Cell Biol. 2006;16:67–69. doi: 10.1016/j.tcb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Zhu L., Wrabl J. O., Hayashi A. P., Rose L. S., Thomas P. J. The Torsin-family AAA+ protein OOC-5 contains a critical disulfide adjacent to sensor-II that couples redox state to nucleotide binding. Mol. Biol. Cell. 2008;19:3599–3612. doi: 10.1091/mbc.E08-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.