

Abstract
Keratin intermediate filaments (KIFs) form a fibrous polymer network that helps epithelial cells withstand external mechanical forces. Recently, we established a correlation between the structure of the KIF network and its local mechanical properties in alveolar epithelial cells. Shear stress applied across the cell surface resulted in the structural remodeling of KIF and a substantial increase in the elastic modulus of the network. This study examines the mechanosignaling that regulates the structural remodeling of the KIF network. We report that the shear stress–mediated remodeling of the KIF network is facilitated by a twofold increase in the dynamic exchange rate of KIF subunits, which is regulated in a PKC ζ and 14-3-3–dependent manner. PKC ζ phosphorylates K18pSer33, and this is required for the structural reorganization because the KIF network in A549 cells transfected with a dominant negative PKC ζ, or expressing the K18Ser33Ala mutation, is unchanged. Blocking the shear stress–mediated reorganization results in reduced cellular viability and increased apoptotic levels. These data suggest that shear stress mediates the phosphorylation of K18pSer33, which is required for the reorganization of the KIF network, resulting in changes in mechanical properties of the cell that help maintain the integrity of alveolar epithelial cells.
INTRODUCTION
Intermediate filaments (IFs) are the components of the cytoskeleton of eukaryotic cells and are involved in the maintenance of cell shape, locomotion, and intracellular organization (Herrmann et al., 2007). IF proteins comprise a large family, which includes ∼70 different genes (Omary et al., 2006; Kim and Coulombe, 2007; Herrmann et al., 2007; Goldman et al., 2008) are expressed in a cell type and differentiation-dependent manner (Omary et al., 2004; Kim and Coulombe 2007; Goldman et al., 2008). Individual IF proteins consist of a conserved central coiled-coil α-helical rod domain that is flanked by N- (head) and C-terminal (tail) domains (Omary et al., 2006; Kim and Coulombe, 2007; Herrmann et al., 2007; Godsel et al., 2008; Goldman et al., 2008). The N- and C-terminal domains contribute to the structural heterogeneity and are major sites of posttranslational modifications, with phosphorylation being the best characterized one (Omary et al., 2006). This makes them important regulatory domains, because dynamic changes in phosphorylation status are responsible for alterations in IF dynamics, solubility, and organization (Omary et al., 2006).
Epithelial cells express cell type-specific KIF proteins, which assemble into complex networks of 10-nm-diameter fibers made up of heterodimers of acidic type I and neutral–basic type II isoforms (Herrmann and Aebi, 2004). The alveolar epithelial cells used in this study contain polymers of keratin 8 and 18 (K8/K18; Ridge et al., 2005; Jaitovich et al., 2008; Sivaramakrishnan et al., 2008). KIF proteins are important for maintaining the mechanical integrity of epithelial cells (Fuchs and Cleveland, 1998; Herrmann and Aebi, 2004). Several keratin mutations are known to compromise the integrity of the epithelium, resulting in a wide variety of diseases including cirrhosis, hepatitis, skin blistering disease, and inflammatory bowel disease (Omary et al., 2004).
Intermediate filaments are dynamic structures that undergo continuous assembly and disassembly to facilitate structural reorganization of the network in response to mechanical forces (Helmke et al., 2000; Yoon et al., 2001). We recently demonstrated that there is a correlation between the structure of the KIF network and the local mechanical properties in primary alveolar epithelial cells and A549 cells; shear stress applied across the cell surface causes a structural remodeling of KIF network and a substantial increase in the elastic modulus of the network (Sivaramakrishnan et al., 2008). The mechanosignaling that regulates the shear stress–mediated structural reorganization of the KIF network is not known and forms the focus of this study.
The assembly state of KIFs is regulated primarily by posttranslational modifications, in particular, the phosphorylation of specific residues in the head and tail domains of the proteins (Omary et al., 1998). Five phosphorylation sites have been characterized for K8 and K18, namely K8pSer23, K8pSer73, K8pSer431, K18pSer33, and K18pSer52 (Ku and Omary, 1997; Ku et al., 1998, 2002a). Phosphorylation levels of these sites increase during different cellular processes; for instance, K18pSer33 is highly phosphorylated during mitosis, K18pSer52 is the major interphase phosphorylation site, and K8pSer431 is phosphorylated in response to epidermal growth factor stimulation (Ku and Omary, 1994, 1997; Ku et al., 1998). Phosphorylation of K8/K18 is regulated by a variety of protein kinases including PKC and mitogen-activated protein kinase (MAPK; Ku et al., 1998, 2002a, 2004; He et al., 2002; Ridge et al., 2005). Importantly, PKCs and MAPKs are known to be activated by mechanical forces such as shear stress and stretch (Liu et al., 1999; Ridge et al., 2005).
In this study, we report that shear stress activates PKC ζ, which then phosphorylates K18pSer33 and promotes the interaction of 14-3-3 with KIFs. These posttranslational modifications regulate the twofold increase in the dynamic exchange rate of KIF subunits, which facilitates the structural reorganization of the KIF network. Blocking the shear stress–mediated structural changes in the KIF network significantly reduces cell viability and increases the number of apoptotic cells.
MATERIALS AND METHODS
Suppliers
Microcystin-LR and bisindolylamaleimide IV were purchased from EMD Biosciences (La Jolla, CA). Myristoylated-PKC ζ peptide antagonist (N-Myristoyl-Ser-Ile-Tyr-Arg-Arg-Gly-Ala-Arg-Arg-Trp-Arg-Lys-Leu), R18 14-3-3 peptide antagonist, and glutathione S-transferase (GST)-14-3-3ζ recombinant protein were purchased from BIOMOL (Plymouth Meeting, PA). GSH Sepharose beads were from Amersham Biosciences (GE HealthCare Bio-Sciences, Piscataway, NJ). K18 mAb (Ks 18.04) for Western blotting was purchased from Research Diagnostics (Flanders, NJ). K18pSer33 antibody was kindly provided by Dr. Bishr Omary (Stanford University, CA). K8/18 polyclonal antibody for immunoprecipitation was produced in rabbits using keratins from a rat liver preparation as antigen (Toivola et al., 1997). PKC ζ mAb was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies for immunofluorescence, goat anti-mouse and goat anti-rabbit fluorescein, and rhodamine-tagged immunoglobulins were obtained from Molecular Probes (Eugene, OR). Secondary antibodies for immunoblotting were peroxidase-labeled goat anti-mouse or goat anti-rabbit immunoglobulins (Bio-Rad Laboratories, Hercules, CA). Protein A/G agarose beads were purchased from Santa Cruz Biotechnology.
Cell Culture
Cells derived from a human lung adenocarcinoma (A549) were obtained from the American Type Culture Collection (Rockville, MD) and grown in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were incubated in a humidified atmosphere of 5% CO2/95% air at 37°C. A549 cells expressing a dominant negative PKC ζ (Dada et al., 2003) were propagated in complete DMEM supplemented with G418.
Shear Stress
A549 cells were cultured on 25 × 75 × 1-mm glass slides and subjected to continuous laminar flow to generate shear stress (30 dyn/cm2) using the FlexCell Streamer Device (FlexCell Streamer Device; FlexCell International, Hillsborough, NC).
Immunofluorescence
A549 cells grown on glass slides were rinsed three times in PBS and fixed in either methanol (−20°C) for 5 min or 3.7% formaldehyde at room temperature (RT) for 9 min. After formaldehyde fixation, cells were permeabilized with 1% Triton X-100 in PBS for 30 min. Cells were then washed three times with PBS and processed for indirect immunofluorescence as previously described (Ridge et al., 2005). After staining, the glass slides were washed in PBS and mounted in gelvatol containing 100 mg/ml Dabco (1,4-diazabicyclo [2.2.2] octane; Sigma-Aldrich, St. Louis, MO; Prahlad et al., 1998). Images of fixed, stained preparations were taken with a Zeiss LSM 510 microscope (Carl Zeiss, Thornwood, NY; Prahlad et al., 1998). Intensity modulated display (IMD) images were processed using the Ratio Imaging tool in MetaMorph Software (Universal Imaging, Downingtown, PA). Changes in KIF structure were evaluated blind by two different individuals, who compared immunofluorescence images of static control and shear-stressed cells.
Transmission Electron Microscopy
Ultrastructural observations of cytoskeletal preparations were performed as described previously (Sivaramakrishnan et al., 2008). For preservation of IFs, cells on coverslips were extracted with PEM buffer (100 mM Pipes, pH 6.9, 1 mM MgCl2, 1 mM EGTA) containing 1% Triton X-100, 0.4 M NaCl, 4% polyethylene glycol, and 1 mg/ml DNase 1 (Sigma-Aldrich, St. Louis, MO) for 10 min at RT. To remove any residual actin, all preparations were treated with recombinant gelsolin NH2-terminal domain. Cells were then fixed with 2% glutaraldehyde, stained with 0.2% uranyl acetate, and processed by critical point drying followed by rotary shadowing with platinum and carbon (Sivaramakrishnan et al., 2008). Replicas were removed from the coverslips and transferred to copper grids as described (Sivaramakrishnan et al., 2008) for observation by transmission electron microscopy.
Cell Viability Assay
AEC apoptosis was assessed by annexin V staining (Roche Diagnostics, Alameda, CA) and DNA nucleosomal fragmentation ELISA, as previously described. Briefly, A549 cells were exposed to shear stress, and then the cells in the supernatant and attached to the dish were collected for determination of apoptosis. Annexin V–stained cells were assessed under a fluorescence microscope (Eclipse TE200; Nikon, Melville, NY) by two investigators who were blinded to the experimental protocol. Lactate dehydrogenase released from control and shear-stressed cells was measured using a commercially available assay (Cytotoxicity Detection Kit, Roche Pharmaceuticals, Indianapolis, IN).
Cell Cycle Analysis
Cell cycle analysis was determined in A549 cells that were suspended in 100 μl PBS and fixed by adding 900 μl ice-cold 70% ethanol (overnight, 4°C). Cells were centrifuged (200 × g, 5 min) and subsequently resuspended in PBS containing 0.1% Triton X-100 (Sigma Chemicals), 0.2 mg/ml RNase (Qiagen, Chatsworth, CA), and 50 μg/ml propidium iodide (Molecular Probes) and incubated at 37°C for 30 min. Fluorescence-activated cell sorting (FACS) was performed using the MoFlo High-Performance Cell Sorter (Dako North America, Carpinteria, CA) to determine the proportion of cells in G0, G2/M, and S phases.
Biochemical Analysis
Cells were lysed in buffer containing 0.2% Triton X-100 in PBS solution with protease inhibitors (phenyl methyl sulfonate, 100 μg/ml; leupeptin, 2 μg/ml; and N-tosyl-l-phenylal-anine chloromethyl ketone [TPCK], 100 μg/ml) and phosphatase inhibitors (sodium orthovanadate [Na3VO4], 1 mg/ml and 0.126 μg/μl of DNASEI; Sigma Chemicals). Cells were homogenized using a 26-gauge syringe needle (Becton Dickinson, Franklin Lakes, NJ) and were incubated on ice for 45 min to obtain total cell lysates. Protein concentrations were determined using the Standard Bradford Assay (Bio-Rad Laboratories). Samples containing equal amounts of protein were then centrifuged at 10,000 × g for 5 min at 4°C. The pellet constitutes the filamentous keratin fraction, whereas the supernatant is the soluble keratin fraction. All samples were solubilized in boiling Laemmli buffer, loaded on 10% SDS-PAGE, transferred to nitrocellulose membranes, and blotted with primary antibodies as follows: 1) K18pSer33 mAb (1:100 in TBS; gift from Dr. Bishr Omary at Stanford University, CA); 2) K18 mAb (1:100 in TBS), clone Ks 18.04, Research Diagnostics; 3) PKC ζ mAb (1:250 in TBS) from Santa Cruz Biotechnology. Membranes were washed three times with TBS containing 0.1% Tween for 30 min, then incubated with secondary antibodies coupled to horseradish peroxidase (in dilutions recommended by supplier), and visualized using enhanced chemiluminescence (Amersham Biosciences).
Immunoprecipitation
Cells were lysed in immunoprecipitation buffer (IP buffer) containing 0.2% Triton X-100 in PBS with protease and phosphatase inhibitors (see above). Cells were homogenized using a 26-gauge needle and incubated on ice for 45 min to obtain total cell lysates. Samples containing equal amounts of protein were sonicated on ice and mixed with IP buffer. Samples were centrifuged at 10,000 × g for 15 min to pellet insoluble K8/18. The supernatant was then mixed with antibodies to K8/18 and incubated on a rotator overnight at 4°C. Samples were again centrifuged at 10,000 × g for 15 min to remove any filamentous K8/18, mixed with protein A/G agarose beads (Santa Cruz Biotechnology) and incubated for 90 min at 4°C on a rotator. Protein A/G beads were spun down and washed three times with IP buffer followed by three washes with cold PBS containing the phosphatase inhibitor. Bead suspensions were then incubated for 120 min in 1× Laemmli buffer at 37°C to release K8/K18 from beads.
PKC Translocation Assay
After the exposure of A549 cells to shear stress, cells were scraped into a lysis buffer containing 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 5 μg/ml trypsin inhibitor, and 20 μM leupeptin and homogenized for 2 min. Lysates were then centrifuged at 1000 × g for 10 min to obtain nuclear and supernatant fractions (P1). The supernatant fraction was further centrifuged at 100,000 × g for 60 min to obtain the membrane fraction (P2) and cytosol fraction (S). The P2 fraction was suspended in lysis buffer containing 0.1% Triton X-100 for 20 min and centrifuged (16,000 × g, 20 min, 4°C) to separate the detergent-insoluble and -soluble material. Twenty to 50 μg of cytosolic and membrane fractions were then subjected to immunoblotting using isozyme-specific anti-PKC antibodies. Specificity of membrane fractionation was determined by histone H1, a nuclear protein that mainly localizes in the P1 nuclear fraction, and MEK-1, a marker of cytosol protein, present in the S fraction but not in the P1 or P2 fractions (Correa-Meyer et al., 2002; Ridge et al., 2002).
In Vitro Kinase Assay
Cells were lysed and homogenized as described earlier and incubated on ice for 45 min to obtain total cell lysates. Protein concentrations were determined using the Standard Bradford Assay (Bio-Rad Laboratories). Equal protein amounts were then centrifuged at 10,000 × g for 5 min at 4°C. The pellet was solubilized by sonication in lysis buffer (defined earlier) to obtain purified KIF. Either purified KIF or recombinant keratin 18 was added along with recombinant PKC ζ (EMD Biosciences) to reaction buffer containing 10 mM MgCl2, 250 μM EGTA, 400 μM CaCl2, diacylgylcerol 30 μg/μl (Sigma Chemicals), phosphatidylserine 25 μg/μl (Sigma Chemicals), BSA 0.1 μg/μl (Sigma Chemicals), and 100 μM ATP (containing trace 32γ-ATP) in 20 mM Tris HCl, pH 7.4. The reaction mixture was incubated for 30 min at 30°C followed by quenching with 1× Laemmli buffer. Samples were then loaded onto 10% SDS-PAGE and transferred to nitrocellulose membranes. 32P activity associated with the keratin 18 band was measured using a Phosphor Imager (Cyclone, PerkinElmer, Norwalk, CT). Protein loading and phosphorylation was checked with antibodies to K18 and K18pSer33.
Live Cell Imaging
Green fluorescent protein (GFP)-K18 stably expressing A549 cells were cultured to ∼30% confluence on Bioptechs coverslips (Butler, PA) for 48 h before the experiments. Coverslips were placed into cell-imaging medium (DMEM without phenol red, Hanks' F12 medium, and 0.5 M Tris, pH 7.5 combined 6:3:1) for 60 min at 37°C (Note: this medium is used throughout imaging experiment). PKC ζ antagonist (1 μM final concentration, BIOMOL) was then added to the media for 30 min at 37°C. Coverslips were then sealed into a Bioptechs FCS2 chamber with serum-free media containing appropriate PKC ζ inhibitor (Note: PKC ζ inhibitors remain in the media throughout the experiment for both unsheared and shear stress conditions). The Bioptechs FCS2 chamber maintains the cells and media at 37°C. Unsheared (CT) cells were incubated for 120 min at 37°C. Shear stress (SS) cells were subject to flow through the chamber to yield an average laminar shear stress of 15 dyn/cm2 for 120 min at 37°C. Finally, the Bioptechs chamber was mounted onto the stage of an LSM 510Meta confocal microscope (Carl Zeiss) whose stage and objective preequilibrated at 37°C by an air stream stage incubator (model ASI 400; Nevtek, Burnsville, VA). For SS cells flow was continued through the chamber for the entire duration of the experiment. No flow was passed through the chamber for the CT cells. To ensure the health of the cells for the duration of the experiment, samples of media were taken from the Bioptechs chamber at the start and end of the experiment to measure pH, pCO2 and pO2 using Novamed Blood Gas Analyzer. No significant changes in pH, pCO2 or pO2 were observed in the cell imaging medium from the start to the end of experiment.
Fluorescence Recovery After Photobleaching
Fluorescence recovery after photobleaching (FRAP) was carried out with the LSM 510Meta confocal microscope (Carl Zeiss). Phase-contrast images of cells were taken before and immediately after FRAP to ensure that there were no significant changes in cell shape or position. Bar-shaped regions were bleached using the area-scan function at 488 nm (50% power, 1% attenuation), and recovery of fluorescence was monitored (7% power, 10% attenuation) using the time-series function at 30-min intervals for up to 4 h.
Image Analysis
Position, length, and intensity measurements were made on digitized confocal images using Metamorph image analysis software (Universal Imaging). Pixel values were converted to distance using confocal calibration bars. Analyses of the dynamic properties of GFP-K18 fibrils were restricted to cells that showed no obvious shape changes for the duration of the FRAP experiment (3–4 h). FRAP half-times (t1/2) were calculated by monitoring the gray-scale pixel values (intensity measurements) across the bleached GFP-K18 fibrils.
GST-14-3-3 ζ Binding Assay
A549 cells cultured on 25 × 75 × 1-mm glass slides for 3 d were subjected to continuous laminar flow to generate shear stress (30 dyn/cm2) for 60 min using the FlexCell Streamer Device. Unstressed (CT) and SS cells were lysed in 1% NP-40 buffer with protease inhibitors (phenylmethyl sulfonate, 100 μg/ml; leupeptin, 2 μg/ml; and TPCK, 100 μg/ml), phosphatase inhibitors (Na3VO4, 1 mg/ml) and 0.126 μg/μl of DNASEI (Sigma Chemicals). Cells were homogenized using a 26-gauge syringe needle (Becton Dickinson) and were incubated on ice for 45 min to obtain total cell lysates. Protein concentrations were determined using the Standard Bradford Assay (Bio-Rad Laboratories). One hundred micrograms of protein from CT and SS conditions was sonicated (Branson Sonifier, Danbury, CT) to solubilize KIF followed by centrifugation to obtain supernatant fraction (10,000 × g for 5 min). Alternatively recombinant K18 (rK18) was phosphorylated in vitro by recombinant PKC ζ (see below). Supernatant fractions from CT, SS, and rK8 were mixed with 2 μg of recombinant GST-14-3-3ζ and incubated overnight on rotary shaker (4°C). GST-14-3-3ζ was recovered with GSH-Sepharose beads, and washed, and proteins bound to GST-14-3-3ζ were separated on 10% SDS-PAGE, transferred, and immunoblotted with anti-K8/K18 and anti-14-3-3ζ antibodies.
Statistical Analysis
Comparisons were performed using the unpaired Student's t test. One-way ANOVA with Tukey's test was used to analyze the data. p < 0.05 values were considered significant.
RESULTS
Effect of Shear Stress on the KIF Network in A549 Cells
A549 cells were exposed to a fluid shear stress of 30 dyn/cm2 for periods of time ranging from 5 min to 4 h, and the effect of shear stress on the KIF network was assessed by immunofluorescence. In control cells, the KIF network was characterized by thin, long, relatively straight KIFs that surrounded the nucleus and extended out to the periphery of the cell (Figure 1A). This KIF network was unaltered in A549 cells exposed to brief time periods of shear stress (5 and 30 min, data not shown). In contrast, the KIF network was reorganized into thick, wavy, tonofibrils in cells exposed to shear stress for 1 h (Figure 1B). At the ultrastructural level, platinum replica electron microscopic preparations confirmed the immunofluorescence observations (Figure 1, A and B, compared with C and D). In control cells the KIF network was comprised of individual ∼10-nm filaments with no preferred alignment of adjacent filaments (Figure 1C). After shear stress, filaments frequently align parallel to each other, resulting in the formation of tonofibrils comprised of bundles of KIFs (Figure 1D). Additionally, A549 cells were exposed to shear stress (30 dyn/cm2, 1 h) and allowed to recover in media with 10% FBS at 37°C, 5% CO2 for 24 h. Cell were then fixed and processed for immunofluorescent staining with anti-K18pSer33 and anti-K18 antibodies. The remodeling of the keratin IF network and the phosphorylation of K18Ser33 are maintained for up to 24 h after exposure to shear stress (Supplementary Data, Supplementary Figure S1).
Figure 1.

Shear stress results in the structural reorganization of the KIF network. A549 cells exposed to static control and shear stress (30 dyn/cm2; 60 min; arrow in B indicates direction of flow) were fixed and processed for indirect immunofluorescence using anti-K18. In cells exposed shear stress the KIF network was reorganized into thick, wavy tonofibrils; representative photomicrographs are shown in A and B. Cells were grown on glass coverslips and exposed to shear stress (30 dyn/cm2; 4 h). The cells were extracted with a buffer containing 100 mM PIPES, pH 6.9, 1 mM MgCl2, 1 mM EDTA, 0.4 M NaCl, 1% TX-100, 0.5 mg/ml DNase I, and 4% polyethylene glycol for ∼4 min at RT, then treated with the N-terminal domain of gelsolin to remove actin, fixed, dehydrated, and dried by the critical point method. These preparations were also devoid of microtubules. Once dried, the preparations were rotary shadowed with carbon/platinum, and the shadowed replicas were transferred to copper grids for observation by transmission electron microscopy (for details see Helfand et al., 2002) Representative electron micrographs show that the KIF network is comprised of individual ∼10-nm filaments in control cells (Figure 1C); after shear stress the formation of tonofibrils comprised of bundles of KIF was observed (Figure 1D; arrowheads in D indicate formation of keratin “tonofibrils”). White scale bar, 10 μm.
FRAP Analyses of Keratin Tonofibrils in Live A549 Cells Exposed to Shear Stress
We used FRAP analyses of GFP-tagged keratin in order to determine whether shear stress altered the exchange rate between subunits and polymerized keratin IFs in live epithelial cells. Live A549 cells expressing GFP-tagged keratin 18 displayed a typical KIF network (Figure 2, A and C). An identical pattern was observed when the same cell was fixed and examined by immunofluorescence (Figure 2, A–D). FRAP analyses of GFP-tagged K18 in live A549 cells were carried out to determine whether shear stress altered the exchange rate between subunits and polymerized KIF. The results showed an average t1/2 of 120 ± 22 min (n = 5) in unsheared control cells (Figure 2, E–G and K). In contrast, shear stress increased the rate of exchange to an average t1/2 of 55 ± 16 min (n = 5; Figure 2, H–J and K).
Figure 2.

Shear stress increases the dynamic exchange rate of the KIF network. Representative confocal micrographs of live A549 cells expressing GFP-tagged K18 exposed to static control or shear stress (15 dyn/cm2; 120 min) conditions (A and C). The cells in A and C were fixed and processed for indirect immunofluorescence with anti-K18 antibody. An identical pattern of expression of the KIF network is observed (B and D compared with A and C). FRAP analyses of GFP-tagged K18 in live A549 cells was used to determine the exchange rate of the KIF network. Representative confocal micrographs of GFP-tagged K18 in live A549 cells before photobleaching (E and H), immediately after photobleaching (F and I), and after 60 min of fluorescence recovery (G and J). Cells were subject to shear stress (15 dyn/cm2; 120 min) before photobleaching; shear stress was continued throughout fluorescence recovery (H–J). Time-matched static controls (E–G). Boxes in E and H highlight the FRAP zone. Arrows in C and H indicate direction of flow. Relative recovery of fluorescence in the FRAP zones (mean ± SEM of at least four different cells for each time point; K). White scale bar, 10 μm.
Effects of Shear Stress on KIF Network Phosphorylation and Structural Reorganization
KIF assembly dynamics are regulated by the state of phosphorylation of their constituent keratin proteins (Eriksson et al., 1992; Ku et al., 1996; Toivola et al., 1997; Dada et al., 2003). Therefore, we examined the effects of shear stress on keratin phosphorylation. Changes in phosphorylation were assessed by immunoprecipitating K8 or K18 from A549 cells exposed to shear stress, followed by immunoblotting with phosphospecific keratin antibodies (Ku and Omary, 1994, 1997; Ku et al., 1998, 2002a). The results showed that there were no significant changes in the phosphorylation levels of K8Ser73, K8Ser431, and K18Ser52 in response to shear stress (Figure 3A). In contrast, K18pSer33 showed a fivefold increase in phosphorylation after shearing, as demonstrated both in IP assays (Figure 3A) and in total cell lysates (Figure 3B). Immunostaining of KIF networks with K18pSer33 specific antibody revealed that the KIF tonofibrils that form after shear stress showed a ∼10–16-fold increase in fluorescence intensity (Figure 3, C–F). Further, this enhanced K18pSer33 phosphorylation was specific to those cells that displayed significant changes in KIF structure after shear stress (Figure 3G).
Figure 3.

Shear stress causes an increase in K18pSer33 phosphorylation. A549 cells were subjected to shear stress (30 dyn/cm2; 60 min; SS) and compared with static controls (CT). (A) Keratin 18 was immunoprecipitated by dilution of whole cell extracts with 1% NP-40 buffer using a polyclonal anti-K18 and recovered with protein A/G Sepharose. Proteins were separated on 10% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with phospho-specific antibodies to K18pSer52, K18pSer33, K8pSer73, and K8pSer431 along with K18 or K8 antibody as loading control. (B) Equal amounts of protein from cell lysates were separated by 10% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with phospho-specific antibodies to anti-K18pSer33 and anti-K18 antibodies. Microcystin (MC) was used as a positive control for K18pSer33 phosphorylation. Graph represents relative intensity of K18pSer33 to K18 (mean ± SD, n = 8, p < 0.01). (C–F) Cells were fixed and processed with anti-K18 and anti-K18pSer33 antibodies for indirect immunofluorescence; representative confocal micrographs show immunolocalization of K18pSer33 (C and E). Ratio imaging was used to assess relative levels of K18Ser33 phosphorylation to K18; blue indicates low levels and red indicates high levels of K18pSer33 phosphorylation using an intensity-modulated display (D and F). (G) Percentage of cells with K18pSer33 staining associated with keratin tonofibril bundles under static control and shear stress conditions (mean ± SD, ∼750 cells each from three separate experiments counted by two different blinded individuals).
K18pSer33 is the predominant K18 phosphorylation site in mitotic cells (Ku et al., 1998). However, the increase in K18pSer33 phosphorylation in shear-stressed cells did not result from an increase in the number of mitotic cells after shear stress as assessed by propidium iodide and FACS (Table 1). Additionally, the increased K18pSer33 phosphorylation did not result from an arrest of cells in G2/M or S, because arresting cells in G2/M (colchicine) or late S (aphidicolin) followed by release into G1 (4 h) did not prevent the shear stress–mediated increase in K18pSer33 phosphorylation (data not shown).
Table 1.
Cell cycle distribution
| G1 | G2/M | S | |
|---|---|---|---|
| CT | 58 ± 6 | 13 ± 4 | 29 ± 10 |
| SS | 56 ± 5 | 8 ± 2 | 37 ± 5 |
To determine whether K18pSer33 phosphorylation is required for the shear stress–mediated structural changes in KIF network, A549 cells were stably transfected with either wild-type GFP-K18 or the GFP-K18Ser33A mutant and subjected to shear stress (30 dyn/cm2; 60 min). Both GFP-K18 and GFP-K18Ser33A become properly incorporated into the endogenous KIF network, as assessed by the direct observation of live A549 cells and corroborated by fixing and processing the same cell for double immunofluorescence (Figure 4, compare A with B and E with F). On exposure to shear stress, wild-type GFP-K18–expressing cells displayed the characteristic tonofibril formation in both the endogenous KIF network and GFP-K18 network (Figure 4, C and D). In contrast, the exogenous network expressing GFP-K18Ser33A is partially disassembled in shear-stressed cells (Figure 4H), whereas the endogenous network, in which K18Ser33 is phosphorylated, is intact (Figure 4G).
Figure 4.

Shear stress–mediated structural reorganization of the KIF network requires K18pSer33-A549 cells were transiently transfected with GFP-tagged K18 wild-type (A–D) or GFP-tagged K18 S33A mutant (E–H). The wild-type and mutant cells were exposed to shear stress (30 dyn/cm2; 60 min) fixed, and processed for indirect immunofluorescence using anti-K18 and direct immunofluorescence for GFP. In GFP-K18S33A and GFP-K18 cells exposed to shear stress for 60 min, the endogenous keratin IF network (red channel) was thick and wavy and formed tonofibrils (C and G). In the GFP-K18S33A cells the exogenously expressed keratin IF network (green channel) was disassembled/degraded after exposure to shear stress (H). In the GFP-K18 cells the exogenously expressed GFP-keratin IF network was intact (D).
We studied the reorganization of the keratin IF network in both low-density culture conditions (e.g., single isolated cells; Figures 1 and 4) and high-density culture conditions (e.g., cells within a monolayer; Figure 2 and Supplemental Figure 2); the percentage of cells that undergo a structural reorganization of the keratin IF network after shear stress was analyzed by two blinded individuals. We show that the nature and extent of the change in IF organization are similar both for isolated cells, cell clusters, and cells within the monolayer. From these results we infer that the cell culture density does not appear to affect the response of the cells to shear stress (Supplementary data; Supplementary Figure S2).
Role of PKC ζ in K18pSer33 Phosphorylation under Shear Stress
PKC is activated in mechanically stimulated epithelial cells (Liu et al., 1999; Ridge et al., 2002, 2005). To determine whether K18pSer33 was phosphorylated by PKC, A549 cells were pretreated with 10 μM bisindolylmaleimide, a PKC inhibitor, and then exposed to shear stress. Bisindolylmaleimide prevented the shear stress–mediated phosphorylation of K18pSer33 (Figure 5A). A549 cells express several isozymes of PKC, including PKC α, βI, βII, δ, ε, θ, and ζ (Ridge et al., 2002). PKC isozymes become activated by translocating from the cytosol to new subcellular sites, including the plasma membrane (Shoji et al., 1986), nucleus (Soh et al., 1999), and cytoskeletal elements (Prekeris et al., 1996). To determine which PKC isozymes were activated, A549 cells were exposed to shear stress and then subfractionated into cytosol (C), membrane (M), and cytoskeletal (T) fractions. The classical PKCs, PKC α and β, were not activated in response to shear stress. The novel and atypical PKCs, PKC δ, PKC ε, and PKC ζ, were activated in shear stressed A549 cells, with PKC δ and ζ being translocated to the cytoskeletal fraction after exposure to shear stress (Figure 5B).
Figure 5.
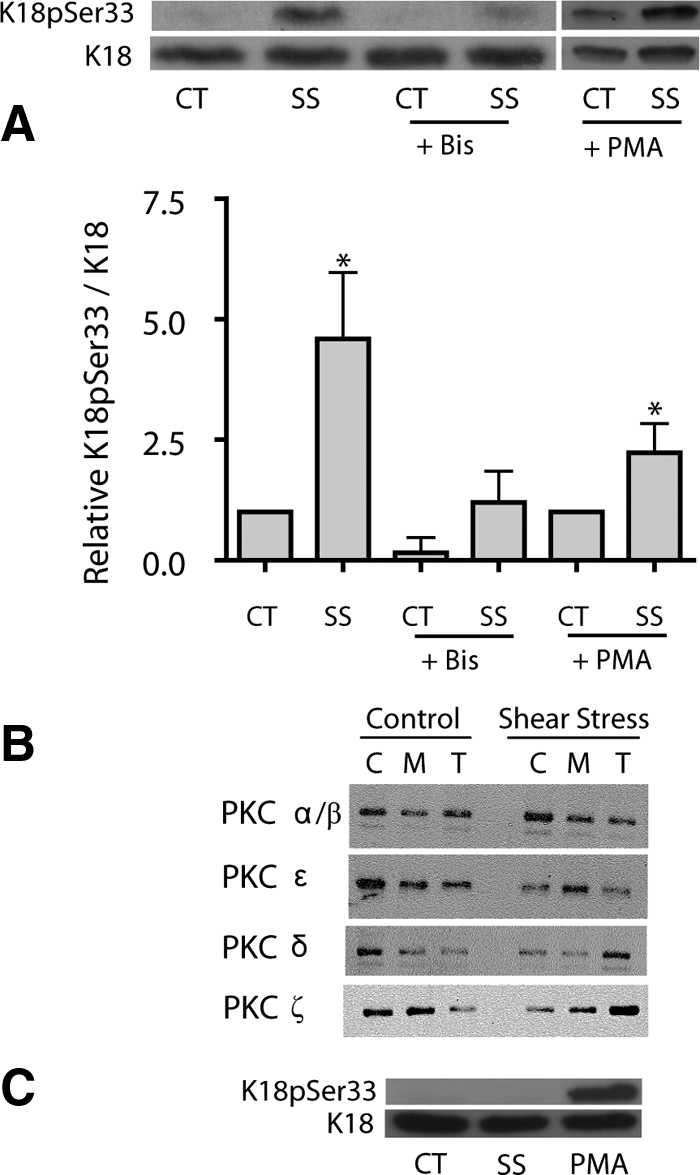
PKC is required K18pSer33 phosphorylation. (A) A549 cells were pretreated with either bisindolylameide (10 μM, 30 min, +Bis) or PMA (1 μM, 16 h, +PMA) before subjecting cells to shear stress (30 dyn/cm2; 60 min). Equal amounts of protein from cell lysates were separated by 10% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-K18pSer33 and anti-K18 antibodies. Graph represents relative intensity of K18pSer33 to K18 bands (mean ± SD, n = 4; *p ≤ 0.01). (B) A549 cells under static control conditions (Control) or shear stress (30 dyn/cm2; 60 min) were subject to subcellular fractionation to obtain cytoplasmic (C), membrane (M), and Triton X-100–insoluble (T) cytoskeletal fractions. Equivalent protein amounts of each fraction were separated by 10% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with anti-PKC α/β, ε, δ, and ζ antibodies. (C) A549 cells treated with 10 μM phorbol ester (PMA) for 15 min or 1 μM microcytsin-LR (MC) for 30 min. Equal amounts of protein from cell lysates were separated by 10% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-K18pSer33 and anti-K18 antibodies.
Short-term treatment with the phorbol ester, phorbol 12-myristate 13-acetate (PMA; 1 μM, 15 min) is known to activate classical and novel PKCs, but not atypical PKCs (e.g., PKC δ, but not PKC ζ). Therefore, we exposed A459 cells to PMA (1 μM, 15 min) or to microcystin-LR (MC, 1 μM, 30 min; as a positive control for phosphorylation of K18pSer33) and observed that activation of classical and novel PKCs did not affect the phosphorylation state of K18Ser33 (Figure 5C). Further, classical and novel PKCs (e.g., PKC δ, but not PKC ζ) can be down-regulated by long-term treatment with PMA. Therefore, A549 cells were treated with PMA (1 μM, 20 h) and then exposed to shear stress or static, control conditions. The down-regulation of classical and novel PKCs did not alter the shear stress–mediated phosphorylation of K18pSer33 (Figure 5A, right panel).
To demonstrate that the atypical PKC ζ regulated the shear stress–mediated phosphorylation of K18pSer33, we used three different approaches. First, A549 cells were stably transfected with an enzymatically inactive DN-PKCζ kinase. Control cells exposed to shear stress had a fivefold increase in K18pSer33/K18 (see Figures 5A and 6A). In the presence of DN-PKC ζ the relative expression of K18pSer33/K18 in 0.6 ± 0.3 and 0.3 ± 0.3 in control and shear-stressed cells, respectively. Thus, these data demonstrate that the shear stress–mediated phosphorylation of K18 Ser33 was completely prevented in the presence of DN-PKC ζ (Figure 6A). Next, we performed a back phosphorylation assay in lysates prepared from control and shear-stressed A549 cells to determine whether K18pSer33 was phosphorylated by PKC ζ. Keratin 18 was immunoprecipitated and subjected to an in vitro phosphorylation reaction with purified PKC ζ and [γ-32P]ATP. Proteins that were phosphorylated in the intact cell should not incorporate 32P, because they cannot be furthered phosphorylated in vitro. Conversely, proteins that were not phosphorylated in the intact cell can then be phosphorylated in the in vitro reaction. As shown in Figure 6B, 31% less 32P-labeled phosphate was incorporated into K18 (identified by immunoblotting) during the in vitro phosphorylation in shear-stressed A549 cells than in static control cells. Finally, we conducted an in vitro phosphorylation assay, in which purified PKC ζ phosphorylates recombinant K18 as determined by incorporation of [γ-32P]ATP uptake and specifically on K18pSer33 using K18pSer33-specific antibody (Figure 6C).
Figure 6.
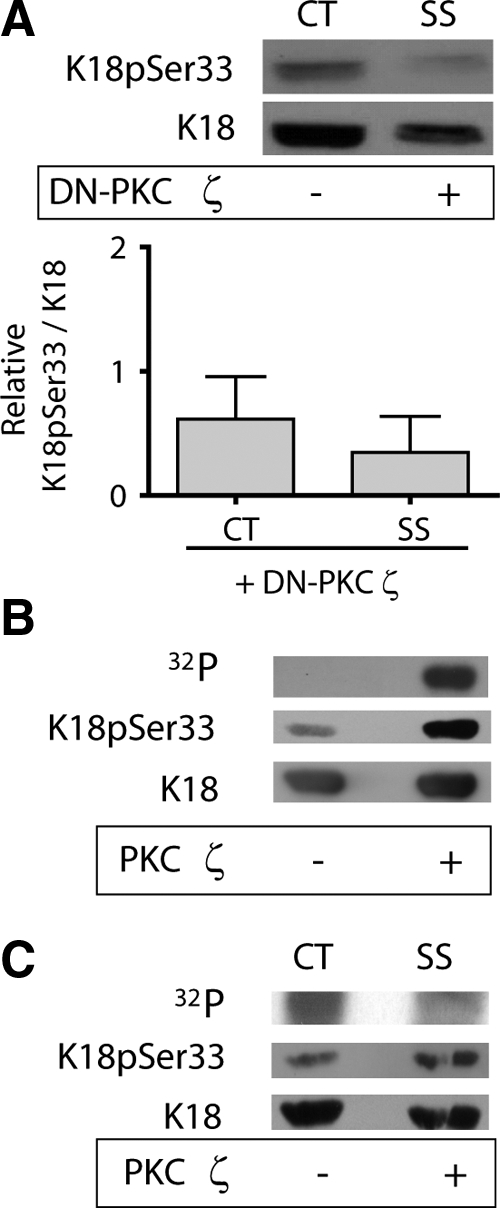
PKC isoform ζ is necessary and sufficient for K18pSer33 phosphorylation. (A) A549 cells stably transfected with dominant negative PKC ζ construct were subject to either static control conditions (CT) or shear stress (30 dyn/cm2; 60 min; SS). Equal amounts of protein from cell lysates were separated by 10% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with anti-K18pSer33 and anti-K18 antibodies. Graph represents relative intensity of K18pSer33 to K18 bands (mean ± SD, n = 3). (B) In vitro phosphorylation reaction of purified recombinant K18 with recombinant PKC ζ and [γ-32P]ATP. Samples were separated by 10% SDS-PAGE, transferred to nitrocellulose, autoradiographed (32P), and immunoblotted with anti-K18pSer33 and anti-K18 antibodies. (C) In vitro “back” phosphorylation reaction of KIF purified from static control (CT) and shear stressed (SS) cells using recombinant PKC ζ and [γ-32P]ATP.
Role of PKC ζ in Structural Reorganization of KIF Network and Dynamics of KIF
Because K18pSer33 phosphorylation is required for structural changes caused by shear stress (Figure 4), we examined the role of PKC ζ in both the structural reorganization and dynamics of KIF. PKC ζ activity was transiently inhibited with a myristoylated PKC ζ peptide antagonist (1 μM). This PKC ζ-specific peptide had no effect on the KIF network in static control cells compared with cells treated with a scrambled peptide (Figure 7, A and C). After shear stress the KIF network did not form extensive tonofibrils in cells treated with the PKC ζ peptide antagonist compared with cells treated with the scrambled peptide (Figure 7, B and D). Instead, the KIF network undergoes a partial disassembly of keratin filaments similar to that observed for K18pSer33A mutant cells (Figure 4H). The PKC ζ peptide antagonist also significantly reduces fluorescence recovery of GFP-K18 filaments in FRAP experiments on static control and shear-stressed cells (Figure 8, A–F and M). This result is specific to the PKC ζ peptide as a scrambled control peptide, used at the same concentration (1 μM), does not alter FRAP recovery (Figure 8, G–L and M).
Figure 7.

PKC ζ is required for structural reorganization of the KIF network. Cells were pretreated with either a myristoylated scrambled peptide (1 μM, 60 min; A and B) or a myristoylated PKC ζ peptide antagonist (1 μM, 60 min; C and D) and exposed to static control (A and C) or shear stress (30 dyn/cm2; 60 min; B and D; arrow in B indicates direction of flow) conditions. Cells were then fixed and processed for indirect immunofluorescence using anti-K18; representative confocal micrographs are shown. White scale bar, 10 μm.
Figure 8.

Dynamic exchange of the KIF network requires PKC ζ. Cells were pretreated with myristoylated PKC ζ peptide antagonist (1 μM, 60 min; A–F) or with a scrambled myristoylated peptide control (1 μM, 60 min; G–L). Cells were subject to either static control or shear stress (15 dyn/cm2; 120 min) conditions. Representative confocal micrographs of live A549 cells expressing GFP-tagged before photobleaching (A, D, G, and J), immediately after photobleaching (B, E, H, and K) and after 60 min of fluorescence recovery (C, F, I, and L). (A–F). Boxes in A, D, G, and J highlight the FRAP zone. Arrow in D indicates direction of flow. Relative recovery of fluorescence in the FRAP zones (mean ± SEM of at least four different cells for each time point; M). White scale bar, 10 um.
To understand the importance of the structural reorganization of KIF after shear stress on cellular integrity, we examined cell viability and apoptosis levels in GFP-K18Ser33A cells and in A549 cells that were stably transfected with an enzymatically inactive DN-PKCζ kinase under static control and shear stress conditions. Under static control conditions neither the GFP-K18Ser33A mutation nor the DN-PKCζ A549 cells had any affect cell viability or apoptotic levels (Figure 9, A–C) as assessed by LDH, annexin V, and DNA fragmentation assays. However, in both the GFP-K18Ser33A mutant cells and in the DN-PKCζ A549 cells exposed to shear stress, there is a significant increase the number of annexin V–positive cells and a significant increase in DNA fragmentation levels (Figure 9, B and C), which correlated with increased cell death (Figure 9A).
Figure 9.

Phosphorylation of K18Ser33 is cytoprotective in shear-stressed cells. A549 cells transiently transfected with GFP-tagged K18 wild-type or GFP-tagged K18 S33A mutant and A549 cells stably transfected with enzymatically inactive DN-PKCζ kinase were exposed to shear stress (30 dyn/cm2; 1 or 4 h). Cell viability was assessed by (A) lactate dehydrogenase (LDH) release and (B) cell apoptosis detected by annexin V staining (∼1000 cells/condition were evaluated by two different blinded individuals). (C) DNA fragmentation detected by ELISA death assay. Bars, mean ± SD, n = 3. Doxorubicin (0.5 μM, Dox) was used as a positive control for death assays.
Binding of 14-3-3 ζ to KIF and its Role in Structural Reorganization
Using recombinant GST-14-3-3 ζ mixed with K8/K18 extracts from static control and shear-stressed cells (30 dyn/cm2; 60 min), we showed that there is an increase in 14-3-3 ζ binding to K18 isolated from SS cells when compared with static CT cells (Figure 10A, lanes 1 and 2 from left). 14-3-3 ζ appeared to selectively bind phosphorylated K18, as GST-14-3-3 ζ binds specifically to K18 after its phosphorylation in vitro by PKC ζ (Figure 10A, lanes 3 and 4 from left). To study the role of 14-3-3 ζ on KIF structure and dynamics, we blocked 14-3-3 ζ binding to KIF in vivo using a dominant negative R18 peptide. R18 binds to 14-3-3 proteins with very high affinity (Qi and Martinez, 2003) and prevents their interaction with other proteins. R18 peptide did not affect KIF structure in static control cells (Figure 10B) but resulted in partial disassembly of the KIF network in shear-stressed cells (Figure 10C) and significantly retarded FRAP recovery in both static control and shear-stressed cells (Figure 10D).
Figure 10.

14-3-3 ζ preferentially binds to phosphorylated K18 and is required to KIF network remodeling. (A) Cell lysates from static control (CT) and shear stress (30 dyn/cm2; 60 min, SS) cells were incubated with GST-14-3-3ζ (lanes 1 and 2). Alternatively, recombinant K18 (rK18) was phosphorylated in vitro by recombinant PKC ζ (rPKC ζ) and incubated with GST-14-3-3 ζ (lanes 3 and 4). Proteins bound to GST-14-3-3ζ were recovered using GSH-Sepharose, separated on 10% SDS-PAGE, transferred and immunoblotted with anti-K8/K18 and anti-GST antibodies. Lane 5 is negative control without GST-14-3-3 ζ, whereas lane 6 is GST control without 14-3-3 ζ. (B and C) Confocal micrographs showing immunofluorescence staining of A549 cells pretreated with R18 peptide under static control (B) and shear stress (30 dyn/cm2; 60 min) conditions (C), processed with anti-K18 antibody. Arrow in B indicates direction of flow. (D) Recovery of fluorescence in the FRAP zones of GFP-K18 stably transfected A549 cells pretreated with R18 peptide for 60 min before shear stress (SS) or time matched controls (CT). Cells were subject to shear stress (15 dyn/cm2; 120 min) before photobleaching; shear stress was continued throughout fluorescence recovery. Each data point is mean ± SEM of at least four different cells. White scale bar, 10 μm.
DISCUSSION
Mechanical ventilation is often required to manage patients with acute respiratory failure of different origins. However, mechanical ventilation itself can also cause or exacerbate lung injury, resulting in ventilator induced lung injury (VILI; Lecuona et al., 1999; Sznajder et al., 1998). The mechanisms associated with VILI have not been fully elucidated, but it is believed that the opening of a fluid-filled, collapsed airway generates a shear force across the epithelium (Bilek et al., 2003). These harmful mechano-forces within the lung are borne by the lung epithelial cells (Ridge et al., 2005; Sivaramakrishnan et al., 2008).
The current paradigm of the regulation of the assembly and structure of KIF networks suggests that in general increased phosphorylation drives their disassembly (Omary et al., 2006). This is supported by the finding that treatment of cells with a phosphatase inhibitor causes the rapid disassembly of the KIF network (Ku et al., 1996; Toivola et al., 1997). Furthermore, the Ser73 to Ala mutation in K8 blocks disassembly of the KIF network by the phosphatase inhibitor okadaic acid (Ku et al., 2002a) and prolonged shear stress (30 dyn/cm2; 24 h) results in increased phosphorylation of K8Ser73, leading to the disassembly and degradation of the KIF network (Ridge et al., 2005; Jaitovich et al., 2008). However, in this study, we find that brief periods of shear stress (30 dyn/cm2; 60 min) induced an increase in K18Ser33 phosphorylation, without alteration of K8pSer73, K8pSer52, and K8pSer431 phosphorylation levels. The phosphorylation of K18Ser33 is required for the reorganization of the KIF network and was accompanied by a decrease in solubility of the KIF network, a significant increase in the exchange rate of keratin subunits and the formation of tonofibrils comprised of bundles of KIF.
We find that initially shear stress activates a protective signaling cascade involving PKC ζ that results in K18Ser33 phosphorylation. K18Ser33 phosphorylation is required for the incorporation of detergent-soluble subunits into the filamentous network of KIF. Coincident with this loss of the soluble fraction there is a structural reorganization of the KIF network that appears to involve “tonofibril” formation. Furthermore, in cells exposed to brief periods of shear stress, the mesh size of the KIF network was reduced. This reduction in mesh size was associated with a ∼40% increase in the average stiffness of A549 cells, enabling them to withstand harmful mechanical forces without injury to the alveolar epithelium (Sivaramakrishnan et al., 2008). This conclusion is borne out in the current study as cells expressing the GFP-K18Ser33A mutation were unable to reorganize their KIF networks and had increased rates of cell death after exposure to shear stress. In contrast, wild-type cells exposed to shear stress under the same conditions showed no changes in cell viability. Partial disassembly of the KIF network was observed in shear-stressed cells pretreated with a myristoylated PKC ζ peptide antagonist or an R18 peptide antagonist, which inhibits 14-3-3 activity. 14-3-3 is known to bind to target proteins in a phosphorylation-dependent manner (Dougherty and Morrison, 2004). In epithelial cells, 14-3-3 ζ proteins bind to phosphorylated keratin 18, a type I IF protein, during the cell cycle, and act as solubility cofactors to modulate keratin filaments and hepatocyte mitotic progression (Liao and Omary, 1996; Ku et al., 1998, 2002b). Although we are in agreement with existing data in the literature that 14-3-3 binding requires the phosphorylation of K18Ser33; our findings appear to differ from those obtained in mitotic cell extracts (Liao and Omary, 1996) because we did not observe an increase in the solubility of KIF. Shear stress did not alter the fraction of mitotic cells (Table 1); hence the role of 14-3-3, PKC ζ, and K18pSer33 in modulating the structure of KIF in response to shear stress are likely to be different from those in mitotic extracts. We reason that these proteins play important roles in reorganizing the KIF network, which may then modulate a number of different signal transduction pathways by acting as scaffolds for and regulators of key signaling proteins.
It is noteworthy that although five specific phosphorylation sites have been characterized for K8/K18, both the head and tail domains have upward of 40 serine/threonine residues that are candidate phosphorylation sites for a variety of kinases (Omary et al., 1998). Hence a comparative map of the extent of phosphorylation of all potential sites on the head and tail domains of KIF under different levels of shear stress and physiological conditions, such as mitosis, is necessary to determine the role of different phosphorylation sites in the assembly state of KIF. A complete phosphorylation map of vimentin has recently been completed (Eriksson et al., 2004), and we are poised to undertake such an effort for K8/K18 as well, but this is beyond the scope of this study.
The specific phosphorylation state/site and subsequent structural reorganization of the KIF network maybe a key determinant in defining whether or not lung epithelial cells can withstand external mechanical forces. Presently, we show that PKC ζ is required, but not sufficient, for the regulation of KIF assembly and reorganization in response to mechanical stimuli. Although the mechanisms underlying the activation of PKC isozymes during the cellular responses to mechanical forces remain unclear, it is evident that PKCs are temporally activated in response to shear stress. Thus, in the initial response to shear stress the cells respond in a cytoprotective mechanism by activating PKC ζ, phosphorylating K18Ser33, and recruiting 14-3-3. These signaling mechanisms coincide with a decrease in mesh size of the KIF network and an increase in storage modulus or stiffness of the KIF network. This initial response to mechanical stimuli may enable the cells to withstand mechanical forces with minimal deformation. The physiological significance of KIF networks becomes evident in disease conditions, such as acute respiratory distress syndrome, where the maintenance of the mechanical integrity of the lung epithelium may prevent alveolar flooding, improve gas exchange, and decrease patient mortality.
These initial changes in the assembly state of the KIF network after exposure to shear stress could have a significant impact on alternative signaling pathways that may affect the cell's response to changes in its environment. In other words, the initially protective signaling mechanisms that lead to the reorganization and increased stiffness of the KIF network may also lead to the activation of additional shear stress–mediated signal transduction pathways that could be deleterious to the integrity of the KIF network and the viability of the cell. For example, when cells are exposed to prolonged periods of shear stress, we find that PKC δ phosphorylated at K8Ser73 drives the disassembly and ubiquitin-mediated degradation of the KIF network (Ridge et al., 2005; Jaitovich et al., 2008). It is important to note that mechanical strain induces a temporal sequence of changes involving the reorganization of keratin IF: After 1–4 h of shear stress, thin arrays of keratin give way to tonofibril formation/keratin bundling; after 12–16 h of shear stress we observed the formation of Mallory-like body formation, and finally the KIF network is disassembled and degraded after 24–36 h of exposure to shear stress (Ridge et al., 2005; Jaitovich et al., 2008). There is also an increase level of apoptosis in cells that cannot reorganize their KIF networks to reduced mesh size and increase stiffness in response to mechanical stimuli. These results emphasize the importance of KIF network reorganization in maintaining the structural integrity of epithelial cells exposed to external mechanical forces. Importantly, these results are supported by numerous reports in which keratin mutations cause blistering diseases of the skin and alterations in liver function (Fuchs and Cleveland, 1998; Omary et al., 2004).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health (PO1-HL71643 (R.G.D. and KM.R.), RO1-HL079190 (K.M.R.), the American Heart Association Grant AHA 0415476Z to S.S.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-10-1028) on April 8, 2009.
REFERENCES
- Bilek A. M., Dee K. C., Gaver D. P. Mechanism of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J. Appl. Physiol. 2003;94:770–783. doi: 10.1152/japplphysiol.00764.2002. [DOI] [PubMed] [Google Scholar]
- Correa-Meyer E., Pesce L., Guerrero C., Sznajder J. Cyclic stretch activates ERK1/2 via G proteins and EGFR in alveolar epithelial cells. Am. J. Physiol. 2002;282:L883–L891. doi: 10.1152/ajplung.00203.2001. [DOI] [PubMed] [Google Scholar]
- Dada L. A., Chandel N. S., Ridge K. M., Pedemonte C., Bertorello A. M., Sznajder J. I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J. Clin. Invest. 2003;111:1057–1064. doi: 10.1172/JCI16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty M. K., Morrison D. K. Unlocking the code of 14-3-3. J. Cell Sci. 2004;117:1875–1884. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- Eriksson J. E., He T., Trejo-Skalli A. V., Harmala-Brasken A. S., Hellman J., Chou Y. H., Goldman R. D. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 2004;117:919–932. doi: 10.1242/jcs.00906. [DOI] [PubMed] [Google Scholar]
- Eriksson J. E., Opal P., Goldman R. D. Intermediate filament dynamics. Curr. Opin. Cell Biol. 1992;4:99–104. doi: 10.1016/0955-0674(92)90065-k. [DOI] [PubMed] [Google Scholar]
- Fuchs E., Cleveland D. W. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:574–579. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Godsel L. M., Hobbs R. P., Green K. J. Intermediate filament assembly: dynamics to disease. Trends Cell Biol. 2008;18:28–37. doi: 10.1016/j.tcb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Goldman R. D., Grin B., Mendez M. G., Kuczmarski E. R. Intermediate filaments: versatile building blocks of cell structure. Curr. Opin. Cell Biol. 2008;20:28–34. doi: 10.1016/j.ceb.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T., Stepulak A., Holmstrom T., Omary M., Eriksson J. The intermediate filament protein keratin 8 is a novel cytoplasmic substrate for c-Jun N-terminal kinase. J. Biol. Chem. 2002;277:10767–10774. doi: 10.1074/jbc.M111436200. [DOI] [PubMed] [Google Scholar]
- Helfand B. T., Mikami A., Vallee R. B., Goldman R. D. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J. Cell Biol. 2002;157:795–806. doi: 10.1083/jcb.200202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmke B. P., Goldman R. D., Davies P. F. Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ. Res. 2000;86:745–752. doi: 10.1161/01.res.86.7.745. [DOI] [PubMed] [Google Scholar]
- Herrmann H., Aebi U. Intermediate filaments: molecular structure, assembly mechanism and integration into functionally distinct intracellular scaffolds. Annu. Rev. Biochem. 2004;73:749–789. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- Herrmann H., Bär H., Kreplak L., Strelkov S. V., Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat. Rev. Mol. Cell Biol. 2007;8:562–573. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- Jaitovich A., Mehta S., Na N., Ciechanover A., Goldman R. D., Ridge K. M. Ubiquitin-proteasome-mediated degradation of keratin intermediate filaments in mechanically stimulated A549 cells. J. Biol. Chem. 2008;283:25348–25355. doi: 10.1074/jbc.M801635200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Coulombe P. A. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21:1581–1597. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- Ku N., Azhar S., Omary M. Keratin 8 phosphorylation by p38 kinase regulates cellular keratin filament reorganization: modulation by a keratin 1-like disease causing mutation. J. Biol. Chem. 2002a;277:10775–10782. doi: 10.1074/jbc.M107623200. [DOI] [PubMed] [Google Scholar]
- Ku N., Fu H., Omary M. Raf-1 activation disrupts its binding to keratins during cell stress. J. Cell Biol. 2004;166:479–485. doi: 10.1083/jcb.200402051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N., Liao J., Chou C., Omary M. Implications of intermediate filament protein phosphorylation. Cancer Metastasis Rev. 1996;15:429–444. doi: 10.1007/BF00054011. [DOI] [PubMed] [Google Scholar]
- Ku N., Omary M. Identification of the major physiologic phosphorylation site of human keratin 18, potential kinases and a role in filament reorganization. J. Cell Biol. 1994;127:161–171. doi: 10.1083/jcb.127.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Liao J., Omary M. B. Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J. 1998;17:1892–1906. doi: 10.1093/emboj/17.7.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Michie S., Resurreccion E. Z., Broome R. L., Omary M. B. Keratin binding to 14-3-3 proteins modulates keratin filaments and hepatocyte mitotic progression. Proc. Natl. Acad. Sci. USA. 2002b;99:4373–4378. doi: 10.1073/pnas.072624299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N. O., Omary M. B. Phosphorylation of human keratin 8 in vivo at conserved head domain serine 23 and at epidermal growth factor-stimulated tail domain serine 431. J. Biol. Chem. 1997;272:7556–7564. doi: 10.1074/jbc.272.11.7556. [DOI] [PubMed] [Google Scholar]
- Lecuona E., Saldías F., Comellas A., Ridge K., Guerrero C., Sznajder J. I. Ventilator-associated lung injury decreases lung ability to clear edema and downregulates alveolar epithelial cell Na,K-adenosine triphosphatase function. Chest. 1999;116:29S–30S. doi: 10.1378/chest.116.suppl_1.29s. [DOI] [PubMed] [Google Scholar]
- Liao J., Omary M. B. 14-3-3 proteins associate with phosphorylated simple epithelial keratins during cell cycle progression and act as a solubility cofactor. J. Cell Biol. 1996;133:345–357. doi: 10.1083/jcb.133.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Tanswell A. K., Post M. Mechanical force-induced signal transduction in lung cells. Am. J. Physiol. 1999;277:L667–L683. doi: 10.1152/ajplung.1999.277.4.L667. [DOI] [PubMed] [Google Scholar]
- Omary M., Coulombe P., McLean I. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004;351:2087–2100. doi: 10.1056/NEJMra040319. [DOI] [PubMed] [Google Scholar]
- Omary M. B., Ku N. O., Tao G. Z., Toivola D. M., Liao J. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem. Sci. 2006;31:383–394. doi: 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Omary M.B., Ku O.-N., Price D. Keratin modifications and solubility properties in epithelial cells and in vitro. In: Harris H. A., editor. Subcellular Biochemistry: Intermediate Filaments. Vol. 31. New York: Plenum Press; 1998. pp. 105–140. [PubMed] [Google Scholar]
- Prahlad V., Yoon M., Moir R., Vale R., Goldman R. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris R., Mayhew M. W., Cooper J. B., Terrian D. M. Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. J. Cell Biol. 1996;132:77–90. doi: 10.1083/jcb.132.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W., Martinez J. Reduction of 14-3-3 proteins correlates with increased sensitivity to killing of human lung cancer cells by ionizing radiation. Radiat. Res. 2003;160:217–223. doi: 10.1667/rr3038. [DOI] [PubMed] [Google Scholar]
- Ridge K., Linz L., Flitney F., Kuczmarski E., Chou Y., Omary M., Sznajder J., Goldman R. Keratin 8 phosphorylation by protein kinase C δ regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J. Biol. Chem. 2005;280:30400–30405. doi: 10.1074/jbc.M504239200. [DOI] [PubMed] [Google Scholar]
- Ridge K. M., Dada L., Lecuona E., Bertorello A. M., Katz A. I., Mochly-Rosen D., Sznajder J. I. Dopamine-induced exocytosis of Na,K-ATPase is dependent on activation of protein kinase C-epsilon and -delta. Mol. Biol. Cell. 2002;13:1381–1389. doi: 10.1091/mbc.01-07-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M., Girard P. R., Mazzei G. J., Vogler W. R., Kuo J. F. Immunocytochemical evidence for phorbol ester-induced protein kinase C translocation in HL60 cells. Biochem. Biophys. Res. Commun. 1986;135:1144–1149. doi: 10.1016/0006-291x(86)91047-8. [DOI] [PubMed] [Google Scholar]
- Sivaramakrishnan S., DeGuilio J., Lorand L., Goldman R., Ridge K. Micromechanical Properties of Keratin Intermediate Filament Networks. Proc. Natl. Acad. Sci. 2008;105:889–894. doi: 10.1073/pnas.0710728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh J. W., Lee E. H., Prywes R., Weinstein I. B. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol. Cell. Biol. 1999;19:1313–1324. doi: 10.1128/mcb.19.2.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajder J. I., Ridge K. M., Saumon G., Dreyfuss D. Lung injury induced by mechanical ventilation. In: Matthay M., Ingbar D., editors. Pulmonary Edema. Vol. 13. New York: Marcel Dekker; 1998. pp. 413–430. [Google Scholar]
- Toivola D., Goldman R., Garrod D., Eriksson J. Protein phosphatases maintain the organization and structural interactions of hepatic keratin intermediate filaments. J. Cell Sci. 1997;110:23–33. doi: 10.1242/jcs.110.1.23. [DOI] [PubMed] [Google Scholar]
- Yoon K. H., Yoon M., Moir R. D., Khuon S., Flitney F. W., Goldman R. D. Insights into the dynamic properties of keratin intermediate filaments in living epithelial cells. J. Cell Biol. 2001;153:503–516. doi: 10.1083/jcb.153.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.