

Abstract
Background
Studies on the relation between thrombin activatable fibrinolysis inhibitor (TAFI) and arterial thrombosis have produced conflicting results.TAFI regulates fibrinolysis, but other roles of this inhibitor, including anti-inflammatory properties, have also been demonstrated.
Design and Methods
We investigated the association between TAFI activity and the risk of myocardial infarction. Additionally, we studied the association of common single nucleotide polymorphisms in the TAFI gene with levels of the TAFI protein and risk of myocardial infarction.We included 554 men under 70 years old with a first myocardial infarction and 643 controls participating in the Study of Myocardial Infarctions Leiden (SMILE), a case-control study.
Results
We found odds ratios (95% confidence intervals) of a first myocardial infarction of 2.4 (1.6–3.6), 3.2 (2.1–4.7) and 3.4 (2.3–5.1) for subjects whose TAFI levels were in the third, second and first quartiles (lowest TAFI levels), respectively, compared with the fourth quartile, after adjusting for arterial disease risk factors. The rare -438A and 1040T alleles were associated with lower, and the rare 505G allele with higher TAFI levels than the common alleles. Carriers of the -438A allele had an increased risk of myocardial infarction (odds ratio 1.6 (1.0–2.5) for AA; odds ratio 1.2 (0.9–1.5) for AG compared with GG). The other single nucleotide polymorphisms were not associated with myocardial infarction.
Conclusions
Low TAFI activity levels are associated with increased risk of a first myocardial infarction in men. The results on the association between TAFI single nucleotide polymorphisms and myocardial infarction were inconsistent.
Keywords: TAFI, myocardial infarction, genetics, epidemiology
Introduction
Thrombin activatable fibrinolysis inhibitor (TAFI) forms the molecular link between the coagulation and fibrinolytic systems.1 Various animal models have demonstrated that TAFI regulates both venous and arterial fibrinolysis in vivo.2–5 Furthermore, epidemiological studies have shown elevated TAFI levels to be a mild risk factor for the development of a first or recurrent venous thrombosis.6–8 However, studies on the association between single nucleotide polymorphisms (SNP) that are associated with TAFI levels and the risk of venous thrombosis have given conflicting results.9–11
Epidemiological data on the association between TAFI levels and the risk of arterial disease are inconsistent. Several case-control studies have shown high TAFI levels to be associated with the risk of coronary artery disease.12–14 However, elevated TAFI levels, or polymorphisms in the TAFI gene associated with increased TAFI levels, have been related to a decreased risk of myocardial infarction.15 In one case-control study, high TAFI antigen decreased but high TAFI activity increased the risk of myocardial infarction in young patients.16 Furthermore, in a prospective study, decreased TAFI levels were associated with refractiveness to medical treatment in patients with unstable angina pectoris.17
Recently, biological functions of TAFI distinct from fibrinolysis regulation have been described, including regulation of inflammation, blood pressure, cell migration, and wound healing.18 These functions may depend on substrates of TAFI other than fibrin, for instance bradykinin, the anaphylatoxins C3a and C5a, annexin II, and osteopontin.19,20
The contradictory results from epidemiological studies may be explained by two opposite effects of TAFI on the development of arterial thrombosis. While the association between high levels of TAFI and arterial thrombosis may be the result of a hypofibrinolytic state, the association between low TAFI levels and arterial thrombosis may be explained by a defective regulation of inflammation.
Another explanation of the contradictory results could be the use of different methods for measuring TAFI. Several different antigen assays as well as distinct activity assays have been used in epidemiological studies. With the discovery that some enzyme linked immunosorbent assays of TAFI antigen are dependent on the 1040C/T polymorphism, revealing a lack of reactivity with the 1040T variant,21 even more caution is required in the interpretation of results.
The -438G/A SNP in the promoter region of the TAFI gene,22 the 505G/A SNP (which encodes an Ala to Thr substitution at position 147),22 and the 1040C/T SNP (encoding a Thr to Ile substitution at position 325)23 are known to be associated with TAFI plasma antigen levels. In Caucasians, three main haplotypes can be identified (www.hapmap.org). The -438G/A and 1040C/T are generally located in the same haplotype, but are not in complete linkage disequilibrium.24 The 505A/G is typical to a second haplotype. A third haplotype is characterized by a fourth SNP in intron 4 (i4 + 164 A/C). In the Study of Myocardial Infarctions Leiden (SMILE), a case-control study of men with a first myocardial infarction, we investigated determinants of the level of TAFI. In addition we studied the effect of plasma TAFI levels, as determined by a functional assay, and the -438G/A, 505A/G, 1040C/T, and i4 + 164A/C SNP in the TAFI gene on the risk of a first myocardial infarction.
Design and Methods
Selection of participants
The design of SMILE has been described previously.25 In brief, the recruited patients were men who had a first myocardial infarction between January 1990 and January 1996, who were below the age of 70. Control subjects were frequency matched on 10-year age groups to the patients and were men without a history of myocardial infarction, without renal disease or severe (neuro)psychiatric problems, with a life expectancy of more than 1 year, and who had not taken any oral anticoagulants in the 6-month period prior to participation in the study. All participants completed a questionnaire and an interview took place prior to collection of blood samples. Questions referred to present and former smoking habits and alcohol use, diabetes, and current use of medications. Blood pressure was measured after a rest of at least 10 min with the person sitting in an upright position. In addition, for patients, information on diabetes and medication use prior to myocardial infarction was retrieved from discharge letters. A person was classified as hypertensive or hyper-cholesterolemic when he was taking prescription drugs for these conditions. All participants signed an informed consent form. Approval for this study was obtained from the the Medical Ethics Committee of the Leiden University Medical Center, Leiden, The Netherlands.
Blood collection and laboratory analysis
Fasting blood samples were drawn from the antecubital vein into Sarstedt Monovette tubes (Sarstedt, Nümbrecht, Germany) and were obtained between July 1994 and February 1997. Most blood samples were collected in the morning (at a median of 9:35 am, with 95% before 11:00 am). The time between myocardial infarction and blood sampling ranged from 88 days to 5.8 years with a median of 2.6 years.
Total cholesterol, HDL-cholesterol, triglyceride, and C-reactive protein (CRP) concentrations were measured as described previously.25
Plasma TAFI activity levels were determined in citrated plasma with a chromogenic assay (Pefakit TAFI, Pentapharm Ltd., Basel, Switzerland) by converting TAFI into its active form using a reagent containing thrombinthrombomodulin and subsequently measuring carboxypeptidase activity. Measurements were run on a BCS coagulation analyzer (Dade Behring Inc., Marburg, Germany). TAFI activity was expressed in percentages using a calibration curve of pooled normal plasma (pool of samples from more than 200 healthy volunteers). The inter-assay coefficients of variation were 5.8% (n=32) at ~100% TAFI concentration and 8.0% (n=31) at ~28% TAFI concentration. For six patients and three control subjects, plasma was unavailable or reliable results were not obtained; therefore 554 patients and 643 control subjects were included in the analysis.
The -438G/A (rs2146881), i4 + 164 A/C (rs3818477), 505G/A (rs3742264), and 1040C/T (rs1926447) polymorphisms were determined by a 5’nuclease assay (Taqman; Applied Biosystems, Foster City, CA) using a standard polymerase chain reaction (PCR) mix (Eurogentec, Seraing, Belgium) and allele-specific fluorescent probes equipped with a minor groove binding moiety (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
Mean TAFI levels were calculated with 5th and 95th percentiles for categories of arterial risk factors. Quartiles of continuous variables (blood pressure, total cholesterol, HDL-cholesterol, triglyceride, and CRP) were defined based on the distribution among control subjects. A multiple linear regression analysis was performed among the control subjects to determine the independent effect of arterial risk factors on TAFI levels. As the association between lipid levels or CRP and TAFI is not linear, these variables were entered in the model using quartiles.
The association between TAFI levels and the risk of a first myocardial infarction was investigated by calculating odds ratios (OR), as an approximation of relative risks, and 95% confidence intervals (CI). TAFI levels were grouped into quartiles based on the distribution among the control subjects. The highest quartile (highest TAFI levels) was taken as the reference group. OR adjusted for age and other potential confounders were calculated using multivariate unconditional logistic regression. Age, levels of triglycerides, cholesterol, CRP, and body mass index (BMI) were used as continuous variables in the model. Levels of triglycerides and CRP were entered into the model after 10log transformation because these variables were not normally distributed. Genotypic frequencies for each of the four loci were determined from the observed genotypic counts, and χ2 analysis was used to check for departures from the Hardy-Weinberg equilibrium. The association between the four TAFI SNP and risk of myocardial infarction was also assessed by means of unconditional logistic regression. The homozygous genotype of the common allele was taken as the reference group. SPSS 14.0 (SPSS, Chicago, IL, USA) was used for all statistical analyses.
Results
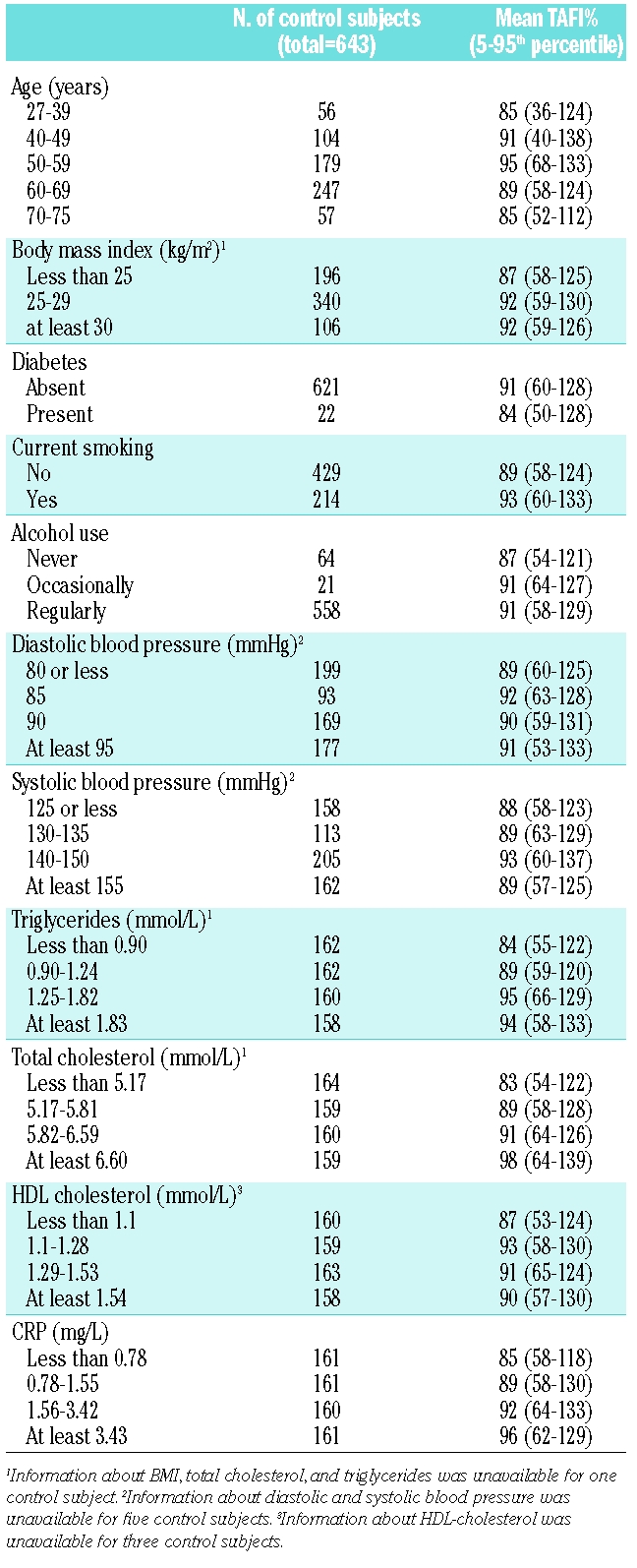
The mean age of the 554 patients with myocardial infarction was 56.4 years (5th–95th percentiles, 40.4–68.8 years) and that of the 643 control subjects was 57.3 (5th–95th percentiles, 34.7–72.1 years). Arterial risk factors such as smoking, obesity, diabetes, hypertension, and hypercholesterolemia were more prevalent in patients than in control subjects (Table 1). The mean TAFI level in patients was 84% (5th–95th, percentile 56–112%) and 90% in controls (5th–95th, percentile 58–128%). Age, BMI, presence of diabetes, smoking, or alcohol use had no or little effect on TAFI levels (Table 2). Cholesterol and triglyceride levels were positively associated with TAFI levels. TAFI levels were 83% in subjects with low cholesterol levels (first quartile) and 98% in those with high cholesterol levels (4th quartile). Similarly, TAFI levels increased from 84 to 94% when comparing individuals with low and high triglyceride levels. TAFI levels also increased with CRP levels (85% for those with low and 96% for individuals with high CRP). No apparent association was present with HDL cholesterol or blood pressure. Multiple linear regression was performed to determine the independent effect of CRP, total cholesterol, and triglycerides on TAFI levels. With each increase in quartile of CRP, TAFI increased 2.8% (β =2.8%/quartile of CRP; 95% CI 1.4–4.4). For total cholesterol this was 3.8%/quartile (95% CI 2.3–5.3) and 1.7% (95% CI 0.2–3.2) for triglycerides. Adding smoking to the model did not change the coefficients (data not shown).
Table 1.
Characteristics of men with a first myocardial infarction and control subjects.
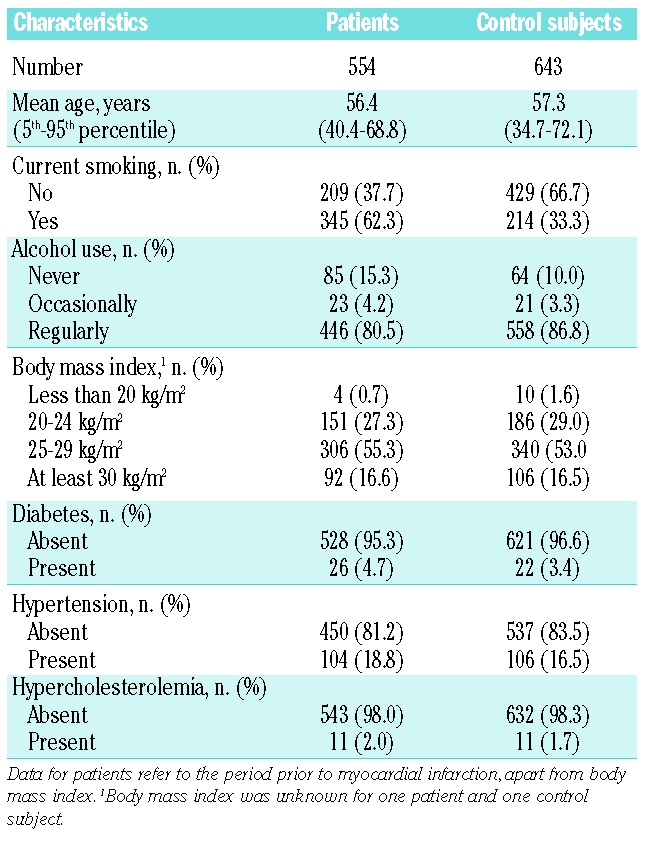
Table 2.
Arterial risk factors among control subjects and the association with TAFI levels.
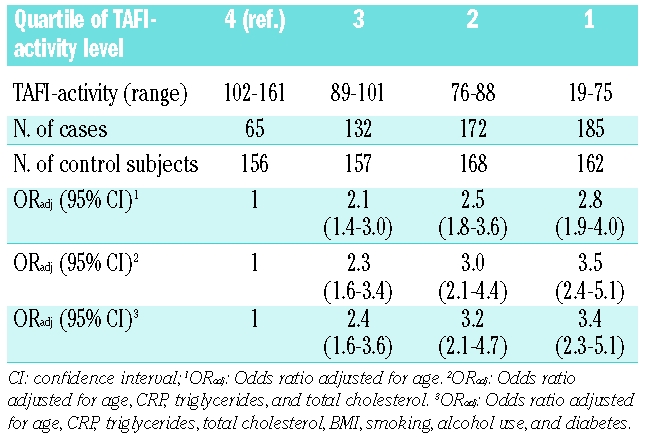
The association between TAFI levels (in quartiles) and the risk of myocardial infarction is shown in Table 3. The fourth quartile (highest TAFI levels) was taken as the reference group. The risk of myocardial infarction increased with each decreasing quartile. Men with the lowest TAFI levels (first quartile) had a 2.8-fold age-adjusted increased risk of myocardial infarction compared with men with TAFI levels in the fourth quartile (OR 2.8; 95% CI 1.9–4.0). Adjusting for other factors found to be associated with TAFI in the control subjects (CRP, triglycerides, and cholesterol), resulted in similar or slightly higher odds ratios (OR 3.4; 95% CI 2.3–5.1). Further adjustment for BMI, smoking, alcohol use, and diabetes (full model) did not substantially change the estimates.
Table 3.
Risk of myocardial infarction with decreasing quartile of TAFI level.
Excluding individuals who were using aspirin at the time of blood sampling (127 patients and 35 control subjects) did not change the results, nor did excluding the 132 patients who used oral anticoagulants at the time of blood collection (data not shown).
We also investigated the effect of the combination of low TAFI levels and smoking or increased CRP levels on the risk of myocardial infarction. Table 4 shows the risk of myocardial infarction for quartiles of TAFI in smokers and non-smokers. The reference group was formed of the non-smokers with the highest levels (4th quartile) of TAFI, who had the lowest risk of myocardial infarction. Within the non-smokers, decreased TAFI levels (1st quartile) were associated with an almost two-fold increased risk of myocardial infarction (OR 1.9; 95% CI 1.1–3.2). In men who smoked and had high TAFI levels (4th quartile) an OR of 2.1 was found (95% CI 1.1–3.7). Smokers with TAFI levels in the 1st quartile had a risk of myocardial infarction that was increased 9.4-fold (95% CI 5.4–16.4). The odds ratios were 2.8 (95% CI 1.8–4.4) for men with low TAFI levels and a CRP level below the 75th percentile, 1.9 (95% CI 1.0–3.5) for those with CRP levels above the 75th percentile and high TAFI levels, and the risk of myocardial infarction was increased 8.2-fold (95% CI 4.4–15.2) in men with high CRP levels and low TAFI levels, all compared with men with low CRP and high TAFI levels.
Table 4.
Combined risk of myocardial infarction by quartiles of TAFI and smoking status.
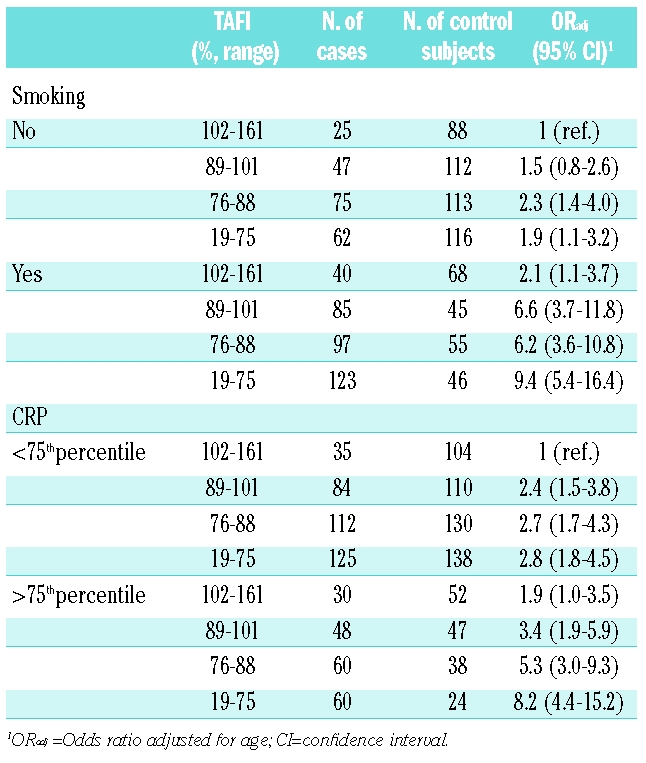
Table 5 shows the risk of myocardial infarction associated with four SNP in the TAFI gene as well as mean TAFI level for each genotype. The observed frequency distribution of the TAFI genotypes in the control group did not deviate from that predicted for the four SNP under the Hardy-Weinberg equilibrium. The SNP at position -438, 505, and 1040 were strongly associated with TAFI levels. The mean TAFI level in those subjects carrying the -438GG genotype was 94%, while the mean TAFI level in carriers of the -438AA genotype was 76%. Mean TAFI level was 82% for carriers of the 505GG genotype and 107% for those with the 505AA genotype. Individuals with the 1040CC genotype had a mean TAFI level of 94%, which was higher than that of subjects with the 1040TT genotype (mean TAFI level 75%). The association between the i4 + 164A/C SNP and TAFI levels was less clear. Carriers of the -438AA genotype, who had low TAFI levels, had an increased risk of myocardial infarction (OR 1.6; 95% CI 1.0–2.5) compared with carriers of the -438GG genotype. The odds ratio for carriers of the -438AG genotype was 1.2 (0.9–1.5). The other SNP appeared not to be associated with risk.
Table 5.
Effect of TAFI SNP on functional TAFI levels and on risk of myocardial infarction.
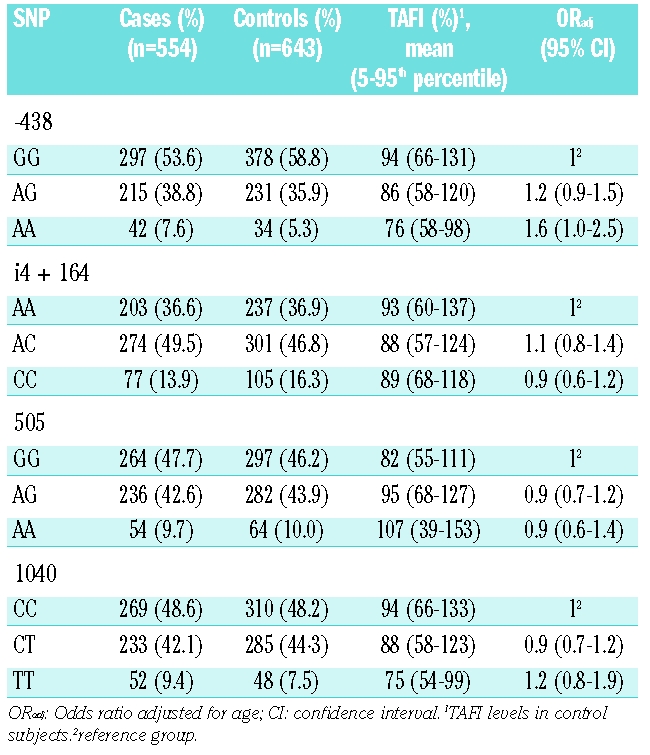
Results of several studies on the association between levels of TAFI and risk of myocardial infarction were, in retrospect, considered not reliable as assays depended on the 1040C/T genotype. To verify whether the association between low TAFI activity and myocardial infarction was independent of the 1040T allele, the association of TAFI levels and myocardial infarction was determined only in those men who carried the 1040CC genotype. Among men carrying the 1040CC genotype, the age-adjusted odds ratio for myocardial infarction among those with TAFI levels in the first quartile was 2.0 (95% CI 1.2–3.3), compared with those with TAFI levels in the fourth quartile. Using the full model, the odds ratio was 2.2 (95% CI 1.2–3.9).
Discussion
This case-control study shows that individuals with low TAFI levels, as measured with a functional assay, had an increased risk of a first myocardial infarction compared with those with high TAFI levels. In accordance with these results, individuals carrying the A allele of the -438A/G SNP in the promoter region of the TAFI gene, who have low TAFI levels, had an increased risk of myocardial infarction, compared with individuals carrying the GG genotype. The 505G/A and 1040C/T polymorphisms, which are also associated with levels of TAFI activity, were not associated with the risk of myocardial infarction.
The increased risk of myocardial infarction in men with lower TAFI levels remained after adjusting for several arterial risk factors, some of which (i.e., triglycerides and total cholesterol) were shown to be strongly associated with TAFI levels. The increased risk was found to be independent of the 1040T allele. A dose-response association was found between TAFI levels and myocardial infarction, but the risk difference between the third and fourth quartile was most pronounced while the odds ratios for the first, second and third quartiles were similar. In other words, there might be a threshold, above which levels of TAFI protect against myocardial infarction.
The results of our study partly contrast those of previous studies and are not in line with what would be expected based on the role of TAFI in the regulation of fibrinolysis. Several case-control studies have found increased TAFI levels to increase the risk of coronary artery disease or myocardial infarction13,14,16 or did not find any association between TAFI and arterial thrombosis.26 Explanations for these differences could be different outcome measures,13,14,26 inclusion of men and women,13,14,16,26 restriction to a relatively young population,16 or blood sampling during the acute phase of the disease.14 In some studies comparisons were only made between mean TAFI levels of patients and controls and thus extreme values were not studied and adjustments for possible confounders that could have influenced the results were not performed.14,16 In one case-control study TAFI antigen levels were decreased in patients with myocardial infarction but activity levels of TAFI were increased, a difference that could not be explained by the 1040C/T SNP.16 In the PRIME-study, a large prospective cohort study set up to investigate potential reasons for the difference in incidence of coronary heart disease in France and Northern Ireland, no clear association was found between baseline TAFI levels and angina pectoris or myocardial infarction and coronary death. However, in this study TAFI was measured with an antigen and not activity assay. Surprisingly, the 505A allele, which is associated with increased TAFI levels, was associated with an increased risk of coronary heart disease in France but with a decreased risk in Northern Ireland.27 The HIFMECH study, a European multicenter case-control study, found elevated TAFI levels to be associated with a decreased risk of myocardial infarction, confirmed by a higher frequency of the 505A allele in patients than in controls.15 The TAFI antigen assay used in the HIFMECH study was later found to be dependent on the 1040C/T genotype.
Although TAFI has been shown to inhibit fibrinolysis in vivo, recent studies suggest that TAFI plays an important role in regulating complement-mediated vascular inflammation in vivo. TAFI inhibits inflammation in vivo by inactivating the inflammatory mediators bradykinin and the anaphylatoxins C3a and C5a.19 Furthermore, TAFI−/− plasminogen+/− mice expressed increased leukocyte migration compared with their wild-type counterparts in a model of peritoneal inflammation.28 In another study, TAFI-deficient mice, primed by lipopolysaccharide, showed increased mortality after infusion of cobra venom factor, which is a potent non-specific activator of complement proteins.29 Based on these results, one might expect that TAFI-deficient mice would have a more rapid progression of atherosclerosis, in which inflammation plays a pivotal role. However, to our knowledge, no data on atherosclerosis progression in TAFI knockout mice are available. As inflammation plays an important role in the development of atherosclerosis and arterial thrombosis,30 it is not implausible that increased TAFI levels decrease the risk of myocardial infarction by decreasing the inflammatory response. These two counteracting effects may also explain the conflicting study results on TAFI and arterial thrombosis, and, because inflammation is less important in venous disease, the consistent relation between high TAFI levels and venous thrombosis risk. There is also evidence that TAFI binds to collagen resulting in reduced platelet adhesion to collagen under flow conditions,31 which could also contribute to the protective effect of TAFI.
The high risk of myocardial infarction found in smokers with low TAFI levels is also in line with an excessive inflammatory response. It is known that smoking leads to an inflammatory state,32 a fact also supported by increased CRP levels in smokers compared to non-smokers in this study.25 It is possible that the effect of smoking on risk of myocardial infarction is most pronounced in those individuals with decreased TAFI levels, who have reduced potential to neutralize this inflammatory response. This hypothesis is also supported by the 8.2-fold increased risk in men with high levels of CRP, indicating elevated inflammatory state, and low TAFI levels. In addition to smoking and increased CRP levels, hypercholesterolemia is also associated with an increased inflammatory state.33 A large number of patients, however, were using lipid-lowering drugs at the time of blood sampling, making an analysis on the combination of cholesterol and TAFI levels unreliable. The data on hypercholesterolemia based on the use of lipid-lowering drugs prior to myocardial infarction (Table 1) were insufficient for such an analysis.
We found a higher risk in individuals carrying the -438A allele than in those carrying the -438G allele, which supports our finding that decreased TAFI levels increase the risk of myocardial infarction. No clear increase in risk was found for those carrying the 1040T allele compared with the 1040C, while both the -438A and 1040T alleles appeared to decrease TAFI levels. It is known that the 1040 T for C substitution results in a TAFIa species with a prolonged half-life at 37°C.34 Possibly, this increased stability of TAFIa compensates for the decreased levels of the protein.
Surprisingly, the 505G/A SNP (or Ala147Thr), which has the largest effect on TAFI levels, was not associated with the risk of myocardial infarction. It could be that this SNP has an unknown function that counteracts the protective effect of the increased levels. The TAFI crystal structure shows that the amino acid at position 147 is solvent exposed.35 Thus, theoretically, this residue, which is located close to the Arg145 that is known to be one of the essential residues for substrate peptide anchoring,36 might contribute to ligand binding and the 505G/A polymorphism might affect this process. Although the 505G/A polymorphism does not affect TAFI fibrinolytic activity,34 the effect of the polymorphism on the catalytic activity towards other molecular substrates such as bradykinin and anaphylatoxins has, to our knowledge, not yet been tested. A decreased activity of the A variant towards TAFI substrates involved in inflammation could plausibly explain why no association is found between the 505G/A SNP and the risk of myocardial infarction. Another possibility is that it is not the 505G/A SNP itself that is the functional variant but that this SNP is in strong linkage disequilibrium with another functional SNP. It has been suggested that the 505G/A SNP is not directly related to TAFI levels but that the 1583T>A SNP, which is in strong linkage disequilibrium with the 505G/A SNP, is actually the functional variant.24 Similarly, the -438G/A might itself not be functional.37 However, the linkage between SNP in the TAFI gene is so strong that the results of the SNP measured in our study are reliable indicators of the effects of the putative functional SNP.
It is possible that reduced TAFI levels are not causally linked to an increased risk of myocardial infarction, but that TAFI levels are reduced by another causal factor preceding the myocardial infarction. We are not aware of any risk factor for myocardial infarction that would reduce TAFI levels. The only disease state that could lead to low TAFI levels would be liver disease, which was only present in two cases and three controls, who had normal TAFI levels. Furthermore, TAFI levels were found to increase with risk factors for myocardial infarction, such as lipid levels and CRP, rather than decrease. As TAFI is also suggested to be an acute phase protein38 it is not likely that decreased TAFI levels are a result of some other underlying factor. Samples were frozen and stored for approximately 10 years. This could have influenced the levels of TAFI. However, if TAFI activity decreases during storage this effect would be equal in patients and controls so no bias of the results is expected, or at most an attenuation of the observed effects.
In conclusion, our study provides evidence that decreased TAFI levels are associated with an increased risk of a first myocardial infarction in men. The results on the association between TAFI SNP and myocardial infarction were inconsistent.
Acknowledgments
the authors thank the cardiologists at Leiden University Medical Center and Diaconessenhuis Leiden, FJM van der Meer, T Visser, JJ Schreijer, I de Jonge, JHM Souverijn, WF Kopatz, PJ Noordijk, MA Weijne, and EJ Hoenderdos-Suijdendorp for their assistance.
Footnotes
Authorship and Disclosures
MEM designed the study, analyzed and interpreted the data, and drafted the manuscript; CJMD designed the overall study, collected and interpreted the data, and critically reviewed the analyses and the manuscript; PGdG interpreted the data and critically reviewed the analyses and the manuscript; JCMM designed the present study, was responsible for performing the assay, interpreted the data and critically reviewed the analyses and the manuscript; FRR designed the overall study, interpreted the data, and critically reviewed the analyses and the manuscript; TL designed the study, interpreted the data, critically reviewed the analyses and participated in writing the manuscript.
The authors declare that they have no conflicts of interest.
Funding; this work was supported by grants from the Netherlands Heart Foundation (Grant n. 2005B060 and 92.345).
References
- 1.Mosnier LO, Bouma BN. Regulation of fibrinolysis by thrombin activatable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathways of coagulation and fibrinolysis. Arterioscler Thromb Vasc Biol. 2006;26:2445–53. doi: 10.1161/01.ATV.0000244680.14653.9a. [DOI] [PubMed] [Google Scholar]
- 2.Minnema MC, Friederich PW, Levi M, von dem Borne PA, Mosnier LO, Meijers JCM, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101:10–4. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redlitz A, Nicolini FA, Malycky JL, Topol EJ, Plow EF. Inducible car-boxypeptidase activity. A role in clot lysis in vivo. Circulation. 1996;93:1328–30. doi: 10.1161/01.cir.93.7.1328. [DOI] [PubMed] [Google Scholar]
- 4.Klement P, Liao P, Bajzar L. A novel approach to arterial thrombolysis. Blood. 1999;94:2735–43. [PubMed] [Google Scholar]
- 5.Wang YX, da Cunha V, Vincelette J, Zhao L, Nagashima M, Kawai K, et al. A novel inhibitor of activated thrombin activatable fibrinolysis inhibitor (TAFIa) - part II: enhancement of both exogenous and endogenous fibrinolysis in animal models of thrombosis. Thromb Haemost. 2007;97:54–61. [PubMed] [Google Scholar]
- 6.van Tilburg NH, Rosendaal FR, Bertina RM. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood. 2000;95:2855–9. [PubMed] [Google Scholar]
- 7.Eichinger S, Schönauer V, Weltermann A, Minar E, Bialonczyk C, Hirschl M, et al. Thrombin-activatable fibrinolysis inhibitor and the risk for recurrent venous throm-boembolism. Blood. 2004;103:3773–6. doi: 10.1182/blood-2003-10-3422. [DOI] [PubMed] [Google Scholar]
- 8.Libourel EJ, Bank I, Meinardi JR, Baljé-Volkers CP, Hamulyak K, Middeldorp S, et al. Co-segregation of thrombophilic disorders in factor V Leiden carriers; the contributions of factor VIII, factor XI, thrombin activatable fibrinolysis inhibitor and lipoprotein(a) to the absolute risk of venous thromboembolism. Haematologica. 2002;87:1068–73. [PubMed] [Google Scholar]
- 9.Franco RF, Fagundes MG, Meijers JCM, Reitsma PH, Lourenço D, Morelli V, et al. Identification of polymorphisms in the 5'-untranslated region of the TAFI gene: relationship with plasma TAFI levels and risk of venous thrombosis. Haematologica. 2001;86:510–7. [PubMed] [Google Scholar]
- 10.Morange PE, Aillaud MF, Nicaud V, Henry M, Juhan-Vague I. Ala147Thr and C+1542G polymorphisms in the TAFI gene are not asssociated with a higher risk of venous thrombosis in FV Leiden carriers. Thromb Haemost. 2001;86:1583–4. [PubMed] [Google Scholar]
- 11.Martini CH, Brandts A, de Bruijne EL, van Hylckama Vlieg A, Leebeek FWG, Lisman T, Rosendaal FR. The effect of genetic variants in the thrombin activatable fibrinolysis inhibitor (TAFI) gene on TAFI-antigen levels, clot lysis time and the risk of venous thrombosis. Br J Haematol. 2006;134:92–4. doi: 10.1111/j.1365-2141.2006.06117.x. [DOI] [PubMed] [Google Scholar]
- 12.Silveira A, Schatteman K, Goossens F, Moor E, Scharpé S, Strömqvist M, et al. Plasma procarboxypeptidase U in men with symptomatic coronary artery disease. Thromb Haemost. 2000;84:364–8. [PubMed] [Google Scholar]
- 13.Santamaría A, Martínez-Rubio A, Borrell M, Mateo J, Ortín R, Fontcuberta J. Risk of acute coronary artery disease associated with functional thrombin activatable fibrinolysis inhibitor plasma level. Haematologica. 2004;89:880–1. [PubMed] [Google Scholar]
- 14.Schroeder V, Wilmer M, Buehler B, Kohler HP. TAFI activity in coronary artery disease: a contribution to the current discussion on TAFI assays. Thromb Haemost. 2006;96:236–7. [PubMed] [Google Scholar]
- 15.Juhan-Vague I, Morange PE, Aubert H, Henry M, Aillaud MF, Alessi MC, et al. Plasma thrombin-activatable fibrinolysis inhibitor antigen concentration and genotype in relation to myocardial infarction in the north and south of Europe. Arterioscler Thromb Vasc Biol. 2002;22:867–73. doi: 10.1161/01.atv.0000015445.22243.f4. [DOI] [PubMed] [Google Scholar]
- 16.Zorio E, Castelló R, Falcó C, España F, Osa A, Almenar L, et al. Thrombin-activatable fibrinolysis inhibitor in young patients with myocardial infarction and its relationship with the fibrinolytic function and the protein C system. Br J Haematol. 2003;122:958–65. doi: 10.1046/j.1365-2141.2003.04549.x. [DOI] [PubMed] [Google Scholar]
- 17.Brouwers GJ, Leebeek FWG, Tanck MW, Wouter Jukema J, Kluft C, de Maat MP. Association between thrombin-activatable fibrinolysis inhibitor (TAFI) and clinical outcome in patients with unstable angina pectoris. Thromb Haemost. 2003;90:92–100. [PubMed] [Google Scholar]
- 18.Boffa MB, Koschinsky ML. Curiouser and curiouser: recent advances in measurement of thrombin-activatable fibrinolysis inhibitor (TAFI) and in understanding its molecular genetics, gene regulation, and biological roles. Clin Biochem. 2007;40:431–42. doi: 10.1016/j.clinbiochem.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ, et al. Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem. 2003;278:51059–67. doi: 10.1074/jbc.M306977200. [DOI] [PubMed] [Google Scholar]
- 20.Fogg DK, Bridges DE, Cheung KK, Kassam G, Filipenko NR, Choi KS, et al. The p11 subunit of annexin II heterotetramer is regulated by basic carboxypeptidase. Biochemistry. 2002;41:4953–61. doi: 10.1021/bi012045y. [DOI] [PubMed] [Google Scholar]
- 21.Gils A, Alessi MC, Brouwers E, Peeters M, Marx P, Leurs J, et al. Development of a genotype 325-specific proCPU/TAFI ELISA. Arterioscler Thromb Vasc Biol. 2003;23:1122–7. doi: 10.1161/01.ATV.0000074145.58172.BD. [DOI] [PubMed] [Google Scholar]
- 22.Henry M, Aubert H, Morange PE, Nanni I, Alessi MC, Tiret L, Juhan-Vague I. Identification of polymorphisms in the promoter and the 3' region of the TAFI gene: evidence that plasma TAFI antigen levels are strongly genetically controlled. Blood. 2001;97:2053–8. doi: 10.1182/blood.v97.7.2053. [DOI] [PubMed] [Google Scholar]
- 23.Brouwers GJ, Vos HL, Leebeek FWG, Bulk S, Schneider M, Boffa M, et al. A novel, possibly functional, single nucleotide polymorphism in the coding region of the thrombin-activatable fibrinolysis inhibitor (TAFI) gene is also associated with TAFI levels. Blood. 2001;98:1992–3. doi: 10.1182/blood.v98.6.1992. [DOI] [PubMed] [Google Scholar]
- 24.Frère C, Tregouet DA, Morange PE, Saut N, Kouassi D, Juhan-Vague I, et al. Fine mapping of quantitative trait nucleotides underlying thrombin-activatable fibrinolysis inhibitor antigen levels by a transethnic study. Blood. 2006;108:1562–8. doi: 10.1182/blood-2006-01-008094. [DOI] [PubMed] [Google Scholar]
- 25.Doggen CJM, Berckmans RJ, Sturk A, Manger Cats V, Rosendaal FR. C-reactive protein, cardiovascular risk factors and the association with myocardial infarction in men. J Intern Med. 2000;248:406–14. doi: 10.1046/j.1365-2796.2000.00754.x. [DOI] [PubMed] [Google Scholar]
- 26.Folkeringa N, Coppens M, Veeger NJ, Bom VJ, Middeldorp S, Hamulyak K, et al. Absolute risk of venous and arterial thromboembolism in thrombophilic families is not increased by high thrombin-activatable fibrinolysis inhibitor (TAFI) levels. Thromb Haemost. 2008;100:38–44. doi: 10.1160/TH07-11-0659. [DOI] [PubMed] [Google Scholar]
- 27.Morange PE, Tregouet DA, Frere C, Luc G, Arveiler D, Ferrieres J, et al. TAFI gene haplotypes, TAFI plasma levels and future risk of coronary heart disease: the PRIME Study. J Thromb Haemost. 2005;3:1503–10. doi: 10.1111/j.1538-7836.2005.01486.x. [DOI] [PubMed] [Google Scholar]
- 28.Swaisgood CM, Schmitt D, Eaton D, Plow EF. In vivo regulation of plasminogen function by plasma carboxypeptidase B. J Clin Invest. 2002;110:1275–82. doi: 10.1172/JCI15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asai S, Sato T, Tada T, Miyamoto T, Kimbara N, Motoyama N, et al. Absence of procarboxypeptidase R induces complement-mediated lethal inflammation in lipopolysaccharide-primed mice. J Immunol. 2004;173:4669–74. doi: 10.4049/jimmunol.173.7.4669. [DOI] [PubMed] [Google Scholar]
- 30.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 31.Marx PF, Lisman T, Adelmeijer J, Marquart J, Meijers JCM. Binding of TAFI to collagen: a role for TAFI in the regulation of platelet adhesion? J Thromb Haemost. 2006;4(Suppl 1):208. [Google Scholar]
- 32.Bazzano LA, He J, Muntner P, Vupputuri S, Whelton PK. Relationship between cigarette smoking and novel risk factors for cardiovascular disease in the United States. Ann Intern Med. 2003;138:891–7. doi: 10.7326/0003-4819-138-11-200306030-00010. [DOI] [PubMed] [Google Scholar]
- 33.Stokes KY, Cooper D, Tailor A, Granger DN. Hypercholesterolemia promotes inflammation and micro-vascular dysfunction: role of nitric oxide and superoxide. Free Radic Biol Med. 2002;33:1026–36. doi: 10.1016/s0891-5849(02)01015-8. [DOI] [PubMed] [Google Scholar]
- 34.Schneider M, Boffa M, Stewart R, Rahman M, Koschinsky M, Nesheim M. Two naturally occurring variants of TAFI (Thr-325 and Ile-325) differ substantially with respect to thermal stability and antifibrinolytic activity of the enzyme. J Biol Chem. 2002;277:1021–30. doi: 10.1074/jbc.M104444200. [DOI] [PubMed] [Google Scholar]
- 35.Marx PF, Brondijk THC, Plug T, Romijn RA, Hemrika W, Meijers JCM, Huizinga EG. Crystal structures of TAFI elucidate the inactivation mechanism of activated TAFI: a novel mechanism for enzyme autoregulation. Blood. 2008;112:2803–9. doi: 10.1182/blood-2008-03-146001. [DOI] [PubMed] [Google Scholar]
- 36.Avilés FX, Vendrell J, Guasch A, Coll M, Huber R. Advances in metallo-procarboxypeptidases. Emerging details on the inhibition mechanism and on the activation process. Eur J Biochem. 1993;211:381–9. doi: 10.1111/j.1432-1033.1993.tb17561.x. [DOI] [PubMed] [Google Scholar]
- 37.Boffa MB, Maret D, Hamill JD, Bastajian N, Crainich P, Jenny NS, et al. Effect of single nucleotide polymorphisms on expression of the gene encoding thrombin-activatable fibrinolysis inhibitor: a functional analysis. Blood. 2008;111:183–9. doi: 10.1182/blood-2007-03-078543. [DOI] [PubMed] [Google Scholar]
- 38.Sato T, Miwa T, Akatsu H, Matsukawa N, Obata K, Okada N, et al. Pro-carboxypeptidase R is an acute phase protein in the mouse, whereas carboxypeptidase N is not. J Immunol. 2000;165:1053–8. doi: 10.4049/jimmunol.165.2.1053. [DOI] [PubMed] [Google Scholar]