

Abstract
We describe a 16-year-old girl and her 41-year-old father who both had a bleeding tendency, dramatic prolongation of all standard clotting assays, undetectable levels of plasma protein C activity, and low or borderline levels of factors X, XI and XII. Plasma and serum electrophoresis revealed a minor peak following the main α1 globulin peak, of which the proportion was increased. Platelet aggregation by thrombin (final concentration 1 U/mL) was absent in both patients, but this inhibition can be overcome by increasing the concentration of thrombin (4 U/mL). The molecular defect responsible for these coagulation abnormalities was identified by genomic sequencing. Both patients are heterozygous for α1-antitrypsin Met 358 to Arg (α1-antitrypsin Pittsburgh). Seven other members of this pedigree had normal coagulation tests and do not carry the same genetic mutation. This unique family with α1-antitrypsin Pittsburgh sheds some light on the study of this extremely rare mutation and its inheritance.
Keywords: α1-antitrypsin Pittsburgh, bleeding, inheritance, mutation
Introduction
α1-antitrypsin Pittsburgh (α1-AT-P), initially designated antithrombin Pittsburgh,1 was characterized as Met 358 to Arg substitution at the reactive Met-Ser site of α1-antitrypsin (α1-AT) in 1983 in the plasma of a boy who had died at the age of 14 of a severe bleeding disorder.2 This variant of α1-AT allows the protein to function as a potent thrombin inhibitor, thus displaying strong functional analogy with the physiological clotting inhibitor antithrombin III (AT-III), whose reactive site consists of an Arg-Ser bond. Hence the mutation explained the index patient’s recurrent and ultimately fatal hemorrhagic diathesis. To date, only 2 individuals with α1-AT-P have been described.1–3 Both patients had the same genetic mutation, even though the hemorrhagic manifestations were different. The second case also had a protein C deficiency (13%), which may contribute to the in vivo hemostatic balance.3 His bleeding tendency was mild only manifesting at the age of 17 after an increase in the level of the mutant enzyme, which is an acute phase reactant. Neither of these cases had a family history of spontaneous bleeding.
We suspected the presence of a similar abnormality in a 16-year-old girl whose routine test before and after surgery revealed markedly impaired coagulation and profoundly decreased plasma protein C activity. Her father showed similar results on his coagulation profile. We were able to confirm the presence in heterozygosity of the Met 358 to Arg mutation, α1-AT-P, by genomic DNA analysis both in the girl and her father.
Design and Methods
Case reports
The hemostatic abnormality was discovered during a routine pre-operative laboratory investigation of a 16-year-old Chinese girl (Figure 1 III-2) suffering from rupture of a left ovarian corpus luteum, and presenting with prolonged activated partial thromboplastin time (APTT), prothrombin time (PT), and thrombin time (TT). Repair of the ovary was performed, and two hematomas developed in her pelvic cavity 36 h after the operation. Her hemoglobulin dropped to 66 g/L, and she was transferred to intensive care. Whole blood, fresh frozen plasma or cryoprecipitate partially corrected the coagulation tests temporarily; 30 mg of protamine had no effect. Her condition was stabilized after six weeks of intermittent treatment. She received regular ultrasound examination, which revealed the development of a hematoma in her right ovary. Initially measuring 3.5×2.4 cm, it reduced to 1.1×1.2 cm within 20 days. To date, the hematomas have not been completely re-absorbed. The patient had no previous history of soft tissue hematoma, hematuria, melena or menorrhagia. Laboratory evaluation included platelet count, antinuclear antibodies (ANA), double strands DNA (ds-DNA), extracted nuclear antigens (ENA), anticardiolipin antibodies (ACA), all of which were normal.
Figure 1.
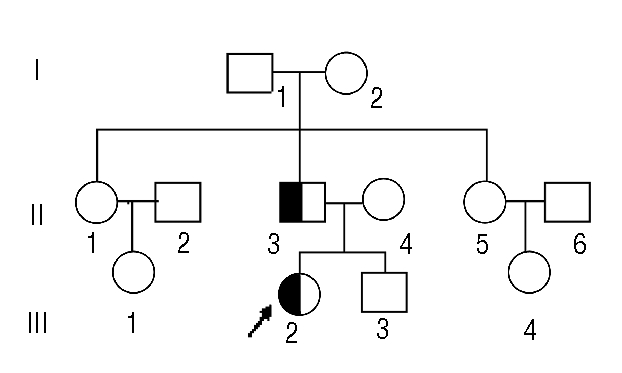
Pedigree of the family with α-1-antitrypsin Pittsburgh. White square and circle, healthy male and female; half darkened square and circle, affected male and female. Arrow: proband.
Her father, a 41-year-old Chinese (Figure 1 II–3), has a positive bleeding history and shares a similar coagulation abnormality with his daughter. At the age of 14 he developed a hematoma in his buttock after a severe fall onto hard ground.
Subsequently, and in response to trauma, aged 15 and 24 respectively, he developed a hematoma in his forearm and in his calf. These bleeding episodes were controlled by whole blood transfusion. He had undergone a blood evaluation and was found to have low levels of factor VIII and IX activity. Three years ago he experienced melena after heavy alcohol consumption but recovered after blood transfusion. He had no history of joint bleeding. Bleeding from minor wounds usually ceased after compression without need for further management. We suspected a hereditary disorder in this family resulting in the presence of a strong coagulation inhibitor in plasma, as the coagulation abnormalities were not corrected by mixing studies. After acquiring appropriate informed consent, peripheral venous blood was drawn from a total of 9 members of this pedigree (Figure 1). None of the other family members, including the girl’s mother and younger brother, had bleeding histories. Three of them had no excessive bleeding after moderate or major surgery.
Coagulation assays
All plasma clotting assays were performed using commercial reagents from Diagnostica Stago (Asnières, France) for Factors II, V, VII, VIII, IX, X, XI, XII, and for PT, APTT, TT, Fibrinogen, and reptilase time (RT).
AT-III and protein C activities were measured using the synthetic chromogenic substrate method (SACHROM® AT III and PROTEIN C). Protein S activity was evaluated by a clotting assay (STACLOT® PROTEIN S).
Platelet aggregation was evaluated by aggregometry (CHRONO-LOG, USA).
Protein studies
Serum and plasma electrophoresis was performed using capillary system (SEBIA, France).
Amplification of α1-AT Pittsburgh by polymerase chain reaction
Genomic DNA was isolated from peripheral blood leukocyte using QIAamp DNA Mini Kit (QIAGEN, Germany), and the target sequence from position 9706 to 10261 in the numbering system of Long et al. was amplified.4
PCR amplification was performed in reaction mixtures of 100 μL final volume: 4 μL of the DNA template preparation (0.1 μg/μL), 8 μL dNTP mixture (2.5 mmol/L), 4 μL each primer (10 pmol/L), and I U Ex Taq polymerase. The sense primer (AT-F) had the sequence 5'-GCTCTTCCCTGTTCTGAGTTGT-3', and the anti-sense primer (AT-R) 5’-ATGGGAGGGATTTACAGT-CACA-3'. Initial denaturation was performed at 95°C for 5 min, followed by 35 cycles of 95°C for 30s, 60°C for 30 s and 72°C for 40 s. The final extension was performed at 72°C for 7 min and the reaction was terminated at 4°C.
DNA sequencing
The 556bp PCR products were purified and both strands were sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit® (Applied Biosystems, Foster City, CA, USA), in an ABI 3730 DNA Sequencer (Applied Biosystems).
Results and Discussion
Coagulation assays
Laboratory findings are summarized in Table 1. The results of coagulation tests suggested the presence of a potent coagulation inhibitor in the plasma samples of the girl and her father, because they showed no correction by 50:50 mixing with normal pool plasma. Both the father and the daughter had similar and parallel results in coagulation assays. Factors II, V and VII were normal. The activity of factors VIII, IX, X, XI, XII was less than normal. However, the results of Factor VIII and IX activity were within the normal range when performed on further dilution of samples. AT III activities were increased in both affected cases. Even though the protein S activities were low, they increased to the normal range when further dilutions were made.
Table 1.
Laboratory findings.

Protein C activity was undetectable in both patients and there was no change on further dilution of test plasma.
Platelet aggregation
Platelet aggregation by thrombin (final concentration 1 U/mL) was absent in both patients when measured using aggregometry. Their platelet rich plasma (PRP) could inhibit thrombin aggregation of normal PRP. But this inhibition could be overcome by increasing the concentration of thrombin. In both cases PRP had a good aggregation response to adenosine diphosphate (ADP, final concentration 10 μM). Other members of this pedigree had normal coagulation test results and platelet aggregation results.
Protein studies
Serum and plasma electrophoresis on capillary showed that both father and daughter had a higher proportion of α1-globulin (6.0% and 5.7% for the father, 5.6% and 6.8% for daughter, normal 2.2–4.8%). Electrophoresis pictures showed a minor peak following the main α1-globulin peak. The girl’s mother and younger brother had normal proportion and electrophoretic traces of α1-globulin. All of them had normal levels of prealbumin and complement C3.
Genetic studies
T to G substitution at nucleotide 10038 in the α1-AT gene encoding for the reactive site of the serpin was identified by direct sequencing of amplified DNA from the girl and her father. They are both heterozygous for this substitution, which generates an AGG codon instead of an ATG codon at position 358 of the mature protein and is pathognomonic for α1-AT-P. This confirmed our speculation that all the coagulation abnormalities resulted from the presence of α1-AT-P with marked inhibition of thrombin, FXa and protein C.
Furthermore, all the coding regions of the α1-AT gene, as well as the intron/exon junctions, were sequenced. No other mutation was found in these patients. The other 7 family members did not carry any genetic mutation in their α1-AT genes.
α1-antitrypsin-Pittsburgh is a spontaneously occurring point mutation of α1-antitrypsin in which methionine-358, the reactive center of the molecule, is substituted by an arginine residue, causing a loss of antielastase activity and a marked increase in the antithrombin properties of the inhibitor. The P1 arginine residue is required for the maximal thrombin inhibition of α1-proteinase inhibitor.5 This mutation was first observed in a patient from Pittsburgh.1,2
Previous to this paper, only two individuals with α1-AT-P have been described.1–3 Neither had a family history of a bleeding diathesis, suggesting that a new mutation occurred either during gamete formation in one of their progenitors, or during the first steps of embryogenesis.3 Our patients are the first family group with the condition. Both the girl and her father are heterozygous for the causative T to G mutation. The girl’s mother is normal. Hence the defective allele in the girl must be inherited from her father, which proves that α1-AT-P is inherited in an autosomal dominant manner.
Comparison of our cases with the 2 previously recorded cases of α1-AT-P reveals a strong similarity with the second case, where the patient displayed a mild bleeding tendency with a first episode at the age of 17. Protein C deficiency was observed both in the second case and our patients, which might explain the maintenance of in vivo hemostatic balance as was postulated by Brennan et al.2 This concept was further supported by biochemical studies in which α1-AT was described as a physiological inhibitor of activated protein C (APC).6 Furthermore, it was reported that the substitution of Met358 by Arg in the reactive center of α1-AT resulted in an increase of over 4,400-fold in the association rate constant for activated APC, and at least 4,000-fold for thrombin.7
Using DGGE screening, Emmerich et al. failed to identify an abnormality on the second patient’s protein C gene, but found that the strong affinity of mutant α1-AT for protein C leads to an increased turnover and thus to a low circulating level.8
A protease responsible for the intracellular processing of precursor polypeptides is also inhibited by the mutated α1-AT.9 Its coexpression with complement C3 and albumin in transfected cells leads to the inhibition of propeptide cleavage.10 But the serum levels of C3 and prealbumin in our patients proved to be normal when tested by chemical methods. Other coagulation factors except factors X, XI, and XII were found to have markedly reduced activities in clotting assays performed with the patients’ plasma, most likely due to the presence of the mutant inhibitor. Factor X, XI, and XII are also inhibited by α1-AT-P.3,11 The minor peak following the main α1-globulin peak in our electrophoresis pictures may be the complexes of α1-AT-P and its target proteins, such as thrombin and/or APC. APC/α1- AT complexes were found in the plasma of the previously reported second patient.8 Bleeding manifestations in index cases and our patients occurred after trauma, which induced an increase in the level of the mutant enzyme that is an acute phase reactant. In the second patient, levels of the abnormal coagulation inhibitor rose to more than twice baseline values during the acute episode.8 This would be a particularly strong challenge for our young female patient, as she ovulates regularly, and has a high risk of bleeding in each cycle. Indeed, after the presenting episode, we observed that a new hematoma had developed in her right ovary.
In conclusion, the cases of α1-AT-P we describe here provide new insights into the consequences of this single amino acid substitution, and the manner in which this bleeding disorder is inherited. It also supports the hypothesis that the mutation results in in vivo consequences which contribute to maintain a normal hemostatic balanced out with episodes of trauma, follicle rupture or surgery. It will be mandatory to prescribe an estroprogestative pill to avoid further follicle ruptures and associated life threatening bleeding for the girl.
Acknowledgments
we thank professor Yaping Zhai, Henan Provincial Hospital, for recommending patients to us, and Wei Su and Qinyi Cheng, Clinical Laboratory Department of PUMCH, for performing protein studies.
Footnotes
BH and LF contributed equally to this work.
Authorship and Disclosures
BLH designed research, performed research, analyzed data, and wrote the paper; YL performed gene research and analyzed data; LKF performed coagulation assays, and analyzed data; YQZ designed research, analyzed data, and wrote the paper; EGDT designed research and revised the manuscript. The authors declare no conflicts of interest.
References
- 1.Lewis JH, Iammarino RM, Spero JA, Hasiba U. Antithrombin Pittsburgh: an α1-antitrypsin variant causing hemorrhagic disease. Blood. 1978;51:129–37. [PubMed] [Google Scholar]
- 2.Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. α1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N Engl J Med. 1983;309:694–8. doi: 10.1056/NEJM198309223091203. [DOI] [PubMed] [Google Scholar]
- 3.Vidaud D, Emmerich J, Alhenc-Gelas M, Yvart J, Flessinger JN, Aiach M. Met 358 to Arg mutation of α1-antitrypsin associated with protein C deficiency in a patient with mild bleeding tendency. J Clin Invest. 1992;89:1537–43. doi: 10.1172/JCI115746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long GL, Chandra T, Woo SL, Davie EW, Kurachi K. Complete sequence of the cDNA for human α1-antitrypsin and the gene for the S variant. Biochemistry. 1984;23:4828–37. doi: 10.1021/bi00316a003. [DOI] [PubMed] [Google Scholar]
- 5.Filion ML, Bhakta V, Nguyen LH, Liaw PS, Sheffield WP. Full or partial substitution of the reactive center loop of α1-proteinase inhibitor by that of heparin cofactor II: P1 Arg is required for maximal thrombin inhibition. Biochemistry. 2004;43:14864–72. doi: 10.1021/bi048833f. [DOI] [PubMed] [Google Scholar]
- 6.Heeb MJ, Griffin JH. Physiologic inhibition of human activated protein C by α1-antitrypsin. J Biol Chem. 1988;263:11613–6. [PubMed] [Google Scholar]
- 7.Heeb MJ, Bischoff R, Courtney M, Griffin JH. Inhibition of activated protein C by recombinant α1-antitrypsin variants with substitution of arginine or leucine for methionine 358. J Biol Chem. 1990;265:2365–9. [PubMed] [Google Scholar]
- 8.Emmerich J, Alhenc-Gelas M, Gandrille S, Guichet C, Fiessinger JN, Aiach M. Mechanism of protein C deficiency in a patient with arginine 358 α1-antitrypsin (Pittsburgh mutation): role in the maintenance of hemostatic balance. J Lab Clin Med. 1995;125:531–9. [PubMed] [Google Scholar]
- 9.Brennan SO, Peach RJ. Calcium-dependent KEX2-like protease found in hepatic secretory vesicles converts proalbumin to albumin. FEBS Lett. 1988;229:167–70. doi: 10.1016/0014-5793(88)80819-6. [DOI] [PubMed] [Google Scholar]
- 10.Misumi Y, Ohkubo K, Sohda M, Takami N, Oda K, Ikehara Y. Intracellular processing of complement pro-C3 and proalbumin is inhibited by rat α1-protease inhibitor variant (Met352 Arg) in transfected cells. Biochem Biophys Res Commun. 1990;171:236–42. doi: 10.1016/0006-291x(90)91382-3. [DOI] [PubMed] [Google Scholar]
- 11.Scott CF, Carrell RW, Glaser CB, Kueppers F, Lewis JH, Colman RW. α1-antitrypsin-Pittsburgh. A potent inhibitor of human plasma factor XIa, kallikrein, and factor XIIf. J Clin Invest. 1986;77:631–4. doi: 10.1172/JCI112346. [DOI] [PMC free article] [PubMed] [Google Scholar]