

Abstract
There is a persistent need for small-molecule fluorescent labels optimized for single-molecule imaging in the cellular environment. Application of these labels comes with a set of strict requirements: strong absorption, efficient and stable emission, water solubility and membrane permeability, low background emission, and red-shifted absorption to avoid cell autofluorescence. We have designed and characterized several fluorophores, termed “DCDHF” fluorophores, for use in live-cell imaging based on the push–pull design: an amine donor group and a 2-dicyanomethylene-3-cyano-2,5-dihydrofuran (DCDHF) acceptor group, separated by a π-rich conjugated network. In general, the DCDHF fluorophores are comparatively photostable, sensitive to local environment, and their chemistries and photophysics are tunable to optimize absorption wavelength, membrane affinity, and solubility. Especially valuable are fluorophores with sophisticated photophysics for applications requiring additional facets of control, such as photoactivation. For example, we have reengineered a red-emitting DCDHF fluorophore so that it is dark until photoactivated with a short burst of low-intensity violet light. This molecule and its relatives provide a new class of bright photoactivatable small-molecule fluorophores, which are needed for super-resolution imaging schemes that require active control (here turning-on) of single-molecule emission.
Keywords: biosensors, fluorescent probes, imaging agents, photochemistry, single-molecule studies
1. Introduction
1.1. Nonlinear Optical Uses for DCDHFs
In the late 1980s and 1990s, the nonlinear optical properties of push-pull chromophores—structures containing electron donor and electron acceptor units separated by a π-electron-rich conjugated linker—were well documented.[1-3] The 2-dicyanomethylene-3-cyano-2,5-dihydrofuran (DCDHF) unit is a useful π-accepting unit, and continues to be used in nonlinear applications, especially in electro-optic media.[4] Chromophores containing a DCDHF acceptor and an amine donor, separated by a π-conjugated system (Figure 1), were also optimized for photorefractive polymer applications.[5-7] In these materials, the linear polarizability anisotropy is very important and the push-pull character of the chromophore increases both the hyperpolarizability β and the ground state dipole moment μG via the charge-transfer absorption and the asymmetric charge distribution, respectively. Increases in donor and acceptor strength also red-shift the absorption, lowering the energy required to produce an intramolecular charge-transfer upon photoexcitation.[8]
Figure 1.
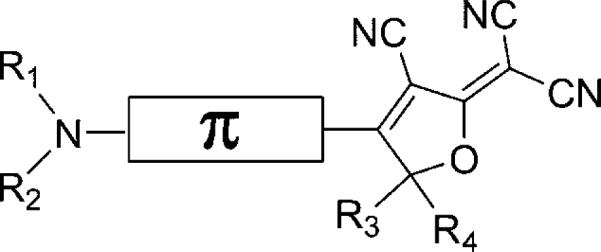
Schematic structure of the DCDHF fluorophores. The amine donor and DCDHF acceptor are connected by a π-conjugated linker. The R1–R4 groups can be modified (usually without affecting the photophysics) in order to add reactive functionality or increased solubility. The naming scheme used in this paper specifies the π system: “DCDHF-(π unit closest to acceptor)-...-(π unit closest to donor)” with the π units denoted P = phenylene, V = vinyl, T = thiophene, N = naphthalene, A = anthracene; the amine donor is not specified because it is present in all structures (see Table 1 for structure drawings.)
1.2. Discovery of DCDHF Fluorescence Suitable For Single-Molecule Imaging
Most nonlinear optical chromophores are not known to be efficient fluorescent emitters, especially because very strong charge transfer can produce large excited-state structure distortions. In the case of photorefractive polymer materials, the dominant figure of merit is not fluorescence but μ2(Δα),[9-11] where Δα is the polarizability anisotropy (the tensor that reports how easily the electron distribution of the molecule distorts in an applied electric field). Chromophores containing a carbocyanine structure were found to be optimal for such nonlinear optical applications.[12]
In 2003, we discovered[13] that some chromophores designed for photorefractive polymers,[14] the DCDHFs, have high fluorescence quantum yields in glassy polymer films and that they emit millions of photons before photobleaching. This means that when the local environment inhibits certain intramolecular twists, the excited-state distortion is not so large as to prevent fluorescence. Since then, we have designed, synthesized, and characterized hundreds of members of the DCDHF class for applications as fluorophores for single-molecule imaging. This paper concentrates on molecules specifically optimized for potential use as cellular labels[13,15-24] and the DCDHF class should be regarded as equally useful for single-molecule experiments as other dyes in the well-known rhodamine, cyanine, rylene, etc. classes.
2. General Characteristics and Photophysics
2.1. Environment-Sensing
DCDHF fluorophores exhibit two types of sensitivity to the local environment: solvatochromism (as a result of the charge-transfer character of the excitation) and viscosity dependence (due to suppression of bond twists that permit nonradiative pathways).[15] A simple consequence of the strong solvatochromism that DCDHFs demonstrate is that the fluorescence is substantially red-shifted with respect to the excitation in biological media, making it easy to filter out background due to scatter and autofluorescence, which typically has a smaller Stokes shift.[25] Solvatochromism in other fluorophores has been harnessed to report local hydrophobicity, because changes in polarity cause changes in the color or intensity of the emission signal.[26] Moreover, the viscosity-dependent brightness that DCDHFs exhibit has a more direct advantage in background suppression: copies of DCDHF or DCDHF-labeled biomolecules not in a rigid region of interest are dim, and contribute little to the fluorescence background. Microscopy in total-internal-reflection (TIR) mode is not always a viable option (e.g. if one needs to image away from the glass interface such as at the upper membrane of a cell or if the optical polarization of TIR cannot be tolerated), and fluorescence readout only in certain regions of a cell permits low-background epifluorescence imaging of those regions.[18,27]
Solvatochromism is an expected outcome of the push–pull character of donor–π–acceptor chromophores. Conventionally, the Lippert–Mataga equation [Eq. (1)] may be used to probe the influence of the solvent polarity on the Stokes shift.[28] In most push–pull chromophores, the charge separation is greater in the excited-state manifold. When such a charge-transfer fluorophore is excited from the ground to excited state, the nearby solvent dipoles can reorient around the larger excited-state dipole moment, thus stabilizing the system and lowering the energy of the excited state (and simultaneously destabilizing the ground state). This effect becomes more pronounced as the solvent polarity is increased, and the Stokes shift increases in more polar solvents. In the Lippert–Mataga approximation, the orientation polarizability Δf is used as a parameter to represent the degree of molecular rearrangement around a dipole in a continuous medium, thus leading to the observed dependence of the Stokes shift on the square of the dipole moment change:
| (1) |
where and and are the wavenumbers of the absorption and emission, μG and μE are the ground- and excited-state dipole moments, a is the Onsager cavity radius (assumed to be 5 Å), n is the refractive index of the solvent, εr is the relative dielectric constant of the solvent, h is Planck's constant, and c is the speed of light. The solvent parameter Δf takes into account only the low-frequency polarizability (i.e. the slow rearrangement of nuclear coordinates), and removes the high-frequency polarizability component (from electron redistribution, which occurs on the same timescale as photoinduced charge transfer in the fluorophore) from the total polarizability.[28,29]
Figure 2 demonstrates the influence that the π structure has on solvatochromism for a series of DCDHF fluorophores with different acene π units. Increasing the conjugation length (from a single phenyl group P to larger naphthalene N and anthracene A) between the longitudinally situated donor and acceptor increases the sensitivity of Stokes shift to solvent polarity. This trend can be explained by the fact that the charge redistribution in the excited state occurs over a greater distance in the cases of naphthalene and anthracene, thus resulting in a larger change in the dipole moment between the ground and excited state. (This of course assumes that the magnitude of charge displaced does not change. In push–pull chromophores, the dipole moment eventually plateaus as the π-system is made very large.)
Figure 2.

Solvatochromism exhibited in a Lippert–Mataga plot (see text for details). The slopes of the fits for DCDHF-A, DCDHF-N, and DCDHF-P (structures in Figure 3) are 7757, 6921, and 1588 cm−1, respectively; these slopes correspond to the change in dipole (μE–μG) values of 9.7, 9.4, and 4.4 D. As expected, increasing the conjugation length between the donor and acceptor increases the sensitivity to solvent polarity, because photoinduced charge transfer across a greater distance results in a larger change in the dipole moment. (Data from refs. [15] and [20].)
The fluorescence quantum yield (ΦF) of DCDHFs increases in rigid environments as demonstrated in Figure 3 for the three acene-based fluorophores. A strong dependence of fluorescence quantum yield on viscosity of the solvent was also observed.[15] The working model for this dependence is a twisted intramolecular charge transfer (TICT) state that opens a nonradiative relaxation channel.[30] Quantum-mechanical calculations have indicated that the excited-stated structure with the lowest energy involves an approximately 90° rotation of the dicyano group around the methylene bond (labeled δ in Figure 4).[15] In viscous media, such structural isomerizations are hindered, decreasing the probability of entering the TICT state (the pathway on the right in Figure 4). Because the radiative decay rate remains relatively unchanged, the nonradiative pathway becomes kinetically less accessible and the fluorescence quantum yield increases. It is worth noting that this mechanism relies on local effects, and the strong connection between fluorescence and viscosity observed for methanolglycerol mixtures may arise from networks of hydrogen bonds than can form around the acceptor portion of the molecule.
Figure 3.

A) Three acene DCDHF structures with increasing conjugation, and thus increasing absorption wavelength. B) Demonstrating the quantum-yield dependence on viscosity: in rigidified frozen samples, the fluorescence emission increases drastically. The “6” in the names refer to the six carbon tails on the amine. (Adapted with permission from J. Phys. Chem. A 2007, 111, 8934−8941. Copyright 2007 American Chemical Society.)
Figure 4.

Proposed mechanism accounting for the interplay between radiative and nonradiative pathways of DCDHF fluorophores. DCDHF-P in its excited state S1 can relax either via channel I to S1ϕ or via channel III to S1δ.In channel III, a twist around the methylene bond δ produces a stable TICT state (S1δ), from which the molecule can relax back nonradiatively to ground state Γ0. In viscous media, this twist is hindered and relaxation to the relatively planar S1ϕ state via channel I becomes more competitive and fluorescence emission can compete with the nonradiative pathway; thus, the fluorescence quantum yield increases. (Adapted with permission from J. Phys. Chem. B 2004, 108, 10 465−10 473. Copyright 2004 American Chemical Society.)
Other such “molecular rotors” have been reported in the literature.[31-33] Figure 5 demonstrates the utility of obtaining strong fluorescence only from viscous local environments: when studying the plasma membrane of living cells, the observed emission signal is brightest from fluorophores located in the lipid bilayer.[18,27] Because molecules that are not in the membrane are dim, they do not contribute to the fluorescence background. Therefore, there is less need for washing away un-bound fluorophores or for background-reducing microscopy configurations (e.g. TIR or confocal[34]). This study, discussed in more detail below, required a DCDHF derivative with reactive maleimide functionality;[35] N-hydroxysuccinimide variants have also been prepared (see the supporting material of ref. [21]). In most cases, adding reactive functionality to DCDHFs via single-bonded alkyl attachments had no affect on the photophysics, because the added groups do not significantly interact electronically with the fluorophore's charge-transfer absorption.
Figure 5.

Octaarginine cell-penetrating peptides labeled with DCDHF-V-P in the plasma membrane of living CHO cells. At low enough concentrations of labeled peptides (right), single molecules can be observed and tracked as they interact with the membrane. (Reproduced with permission from J. Am. Chem. Soc. 2008, 130, 9364−9370. Copyright 2008 American Chemical Society.)
2.2. Photostablility
Inherent photostability is one of the most important parameters of a single-molecule fluorophore: the longer each molecule survives before it photobleaches, the more it reports on its location, environment, orientation, diffusion kinetics, or whatever parameter the experiment aims to measure. Singlet molecular oxygen, produced when triplet oxygen interacts with a fluorophore in its triplet state, is often blamed as the culprit in many photodestruction reactions.[36-38] Therefore, various oxygen-scavenging systems are often used in single-molecule biophysics experiments to extend the survival time of the fluorophore.[39,40] However, molecular oxygen is a potent triplet quencher,[41] because the molecular oxygen triplet ground state helps couple the fluorophore triplet state back to the ground singlet state (thus increasing the intersystem-crossing rate). Therefore, removing oxygen from solution can result in dim, intermittent fluorescence because emitters become trapped in long-lived dark states. Careful experiments have demonstrated that removing oxygen can even reduce the total number of cycles to the excited state while simultaneously impeding the emission rate.[41-44] Because oxygen removal can generate inconsistent behavior from structure to structure, fluorophores with inherently robust photostability are always desirable.
Two fundamental measures of fluorophore photostability are the total number of photons emitted for a single molecule before it permanently photobleaches (Ntot,e) and the photobleaching quantum yield (ΦB) which is the probability of photobleaching for each photon absorbed. These two parameters scale inversely to each other (Ntot,e = ΦF/ΦB), but can also be regarded as independent useful tests, measured using different experiments and assumptions. For the Ntot,e measurement, single-molecule time traces from movies are used to extract the total number of detected photons before photobleaching molecule-by-molecule, where all the photons (minus background) contributing to a single-molecule spot are spatially and temporally integrated. Results from hundreds of single molecules are histogrammed[20] or plotted versus a probability distribution,[23] and an average number of photons detected is extracted from an exponential fit. To convert to the photons emitted, the electron-multiplying and conversion gains of the camera as well as collection efficiency D of the microscopy setup must be measured: D = ηQFcollFoptFfilter, which is the product of the camera quantum efficiency ηQ, the angular collection factor Fcoll determined by the objective NA, the transmission factor through the objective and microscope optics Fopt, and the transmission factor through the various filters Ffilter, respectively.[34] Typical values of D are approximately 10 % for epifluorescence. True photon-counting detectors (e.g. avalanche photodiodes) provide an alternative detection scheme which reduces error introduced by inaccuracies in camera gain values, but imaging with such detectors requires confocal scanning and measuring molecules one at a time. Note that the Ntot,e calculation does not depend on irradiance, wavelength, or extinction coefficient (unless the molecule is excited so strongly as to experience excitation from the first excited state to higher excited or triplet states that lead to bleaching, and this regime is to be avoided).
On the other hand, the photobleaching quantum yield can be measured from a bulk sample if the laser irradiance (Ilzλ) and wavelength (λ), and absorption cross-section (σλ = 2303ελ/NA) are known, from which the ratio of the bleaching rate (RB)to the absorption rate (Rabs) can be calculated using Equation (2):
| (2) |
where τB is the time constant in an exponential fit of the decay of bleaching fluorescence, h is Planck's constant, and c is the speed of light. If measured under similar conditions to a single-molecule imaging experiment but at approximately an order of magnitude higher concentration, ΦB is an independent check of the photostability. Because of experimental variability (standard errors of the mean for Ntot,e or ΦB can be as high as 30 %) and differences in fluorescence quantum yields among fluorophores, the two photostability measures may not be exactly inverse; however, trends in each measure should be similar among fluorophores.
In Table 1, values for Ntot,e and ΦB are reported for many DCDHFs, as well as for the standard fluorophores rhodamine 6G and fluorescein. Many DCDHFs exhibit comparable or superior photostability to rhodamine 6G, which is a demonstrably good single-molecule emitter; moreover, all DCDHFs measured are at least an order of magnitude more photostable than fluorescein. Table 1 also contains extinction coefficients, absorption and fluorescence emission maxima, and ΦF values in solution and in polymer films.
Table 1.
Names, structures, and photophysical parameters of various DCDHFs and standard fluorophores. The structures are generalized: the methyl groups on the amine and dihydofuran ring may vary in length and composition; the data presented is representative of chromophores with varying R1–R4 groups, because varying the length of the alkyl chains has little effect on the photophysics. See text for details on photophysical parameters. (Data from refs. [19,20], and [24].)
| Compound | Structure | εmax [m−1 cm−1][a] | λabs [nm][a] | λem [nm][a] | ΦF in toluene {PMMA} | ΦB in gelatin {PMMA} (× 10−6) | Ntot,e in PMMA (× 106) |
|---|---|---|---|---|---|---|---|
| DCDHF-P | 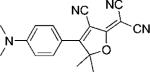 |
71 000 | 486 | 505 | 0.044 {0.92} | 6.6 | 2.4 |
| DCDHF-P with 1 constraining ring | 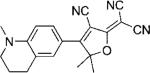 |
95 900[b] | 495 | 515 | 0.10 | ||
| DCDHF-P with 2 constraining rings | 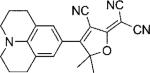 |
90 400[b] | 503 | 527 | 0.21 | 1.1 | |
| DCDHF-V-P | 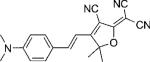 |
45 500 | 562 | 603 | 0.02 {0.39} | 2.8 | ∼1.9 |
| DCDHF-T | 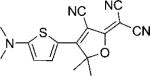 |
100 000 | 514 | 528 | 0.11 | ∼0.91 | |
| DCDHF-V-T | 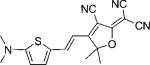 |
114 000 | 614 | 646 | 0.02 | ∼0.23 | |
| DCDHF-N | 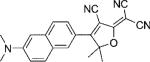 |
42 000 | 526 | 579 | 0.85 {0.98} | 3.4 | 1.1 |
| DCDHF-V-N | 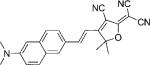 |
58 100[b] | 574 | 671 | 0.01 | ||
| DCDHF-V-(1,4)N | 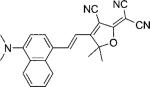 |
22 200[b] | 538 | 661 | 0.01 | ||
| DCDHF-A | 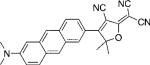 |
35 000 | 585 | 689 | 0.54 | 1.7 | 2.2 |
| DCDHF-P-P | 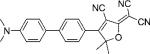 |
31 000 | 506 | 623 | 0.82 | 4.5 | |
| DCDHF-T-T | 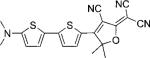 |
71 800 | 634 | 679 | 0.50 | {2.1} | |
| DCDHF-V-T-T | 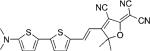 |
49 800 | 708 | 779 | 0.13 | {3.4} | |
| DCDHF-P-T | 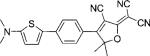 |
44 000 | 591 | 663 | 0.21 | 6.4 | |
| DCDHF-T-P | 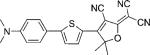 |
22 000 | 575 | 631 | 0.74 | 2.9 | |
| DCDHF-V-T-P | 47 300 | 611 | 723 | 0.07 | {0.13} | ||
| DCDHF-P-T-P | 28 000 | 541 | 709 | 0.34 | 0.91 | ||
| Rhodamine 6G | 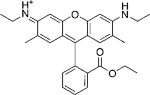 |
105 000[c] | 530[c] | 556[c] | 0.95[c] | 3.5 | 1.4 |
| Fluorescein | 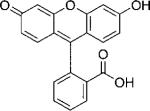 |
92 300[c] | 483[c] | 515[c] | 0.79[c] | 64 |
In toluene.
In dichloromethane.
In ethanol.
3. Structure-Property Design
3.1. Long-Wavelength Absorption
One of the more straightforward structure-property relationships is the red-shift that accompanies an increase in the length of the conjugated linker between the donor and acceptor. Assuming a particle-in-a-box model, increasing the length of the fluorophore should lower the energy and spacing between the ground and excited state, and thus produce red-shifted absorption. This can be seen, for example, in going from DCDHF-P to DCDHF-V-P: there is a 75 nm absorption red-shift (in toluene) with the addition of a vinyl group.
Because cis–trans isomerism and bond twists involving the vinyl group can reduce the fluorescence quantum yield, it may be preferable to extend the conjugation using more rigid groups. For instance, replacing the phenyl group with a naphthalene or anthracene increases the absorption wavelength in toluene 40 nm for DCDHF-N and nearly 100 nm for DCDHF-A (see Figure 3). Table 1 includes various other red DCDHF fluorophores with combinations of vinyl, phenyl, and thiophene linkers (see Figure 1 for nomeclature.)
3.2. Constraining the Amine Twist
Although quantum-mechanical modeling revealed methylene-bond twists as one major source of viscosity sensing in DCDHFs (see Figure 4), the various other twists in the molecule may also significantly contribute to lowering the fluorescence quantum yield in solution. Moreover, the methylene bond does not have synthetic “handles” with which to constrain rotations, because modifying the cyano groups would also drastically alter the photophysics.
Therefore, in order to experimentally test the influence of other bond twists on the fluorescence quantum yield, a series of fluorophores were synthesized with increasing constraint on the amine rotation (angle α in Figure 4) produced by tetrahydroquinoline rings.[19] Table 2 demonstrates that restraining the amine not only increases the fluorescence quantum yield but also slightly red-shifts the absorption and emission. However, the quantum yields of molecules in solution do not approach the values measured in the rigid polymer film, which indicates that the amine bond alone is not responsible for the viscosity sensing.
Table 2.
Influence of constraining the amine from twisting on photophysical properties. (Data from ref. [19].)
| Parent Compound | Number of constraining rings | εmax [m−1cm−1][a] | λabs [nm][b] | λem [nm][b] | ΦF in toluene {PMMA} |
|---|---|---|---|---|---|
| DCDHF-P | 0 | 486 | 505 | 0.044 {0.92} | |
| 1 | 95 900 | 495 | 515 | 0.10 | |
| 2 | 90 400 | 503 | 527 | 0.21 | |
| DCDHF-V-P | 0 | 562 | 603 | 0.02 {0.39} | |
| 1 | 17 400 | 581 | 618 | 0.028 | |
| 2 | 80 900 | 594 | 628 | 0.053 | |
| DCDHF-N | 0 | 40 900 | 527 | 576 | 0.85 {0.98} |
| 1 | 545 | 607 | 0.64 | ||
| DCDHF-V-N | 0 | 58 100 | 574 | 671 | 0.0049 |
| 1 | 44 200 | 600 | 699 | 0.30 | |
| DCDHF-V-(1,4)N | 0 | 22 200 | 538 | 661 | 0.0067 |
| 1 | 37 500 | 571 | 677 | 0.0096 |
In dichloromethane.
In toluene.
These experimental results do not entirely match the calculations,[15] and suggest that the TICT mechanism in DCDHFs is more complex than just one bond rotation. Further computational studies or ultrafast measurements might help clarify the exact mechanism of viscosity sensing of DCDHF fluorophores.
3.3. Water Solubility
Biophysics and cell-imaging experiments require fluorophores that can be used in aqueous environments, and most of the molecules described so far require relatively nonpolar solvents. Full water-solubility is not always necessary or desirable (e.g. fluorescent lipid analogs must have some hydrophobicity); however, many applications do require fluorophores that are soluble and photostable in water.
The original work on molecules in the DCDHF class was carried out in organic solvents, in which the fluorophores are very soluble. For single-molecule cell experiments, stock fluorophore solutions in DMSO or ethanol were diluted into the aqueous buffer;[18] because of the very low dye concentrations, aggregation in water was not a problem. Nevertheless, in order to introduce true water solubility, synthetic efforts were undertaken to add alcohol, carboxylic acid, and sulfonic acid groups to two DCDHFs.[22] As expected, sulfonic acid groups imparted the most solubility (up to 104 ppm). Significant water solubility was achieved without compromising desirable photophysical properties of the DCDHF class of fluorophores.
3.4. Photoactivation
Sophisticated photophysics (e.g. photoswitching, fluorogenic reactions, multicolor emission) are required for more complex imaging modalities now becoming popular. For instance, super-resolution imaging schemes (PALM, F-PALM, STORM)[45-47] require active control over fluorescence in order to “turn on” only a few emitters in each imaging cycle.[48,49] The DCDHFs are synthetically tailorable, and designing in such sophisticated qualities are possible. Disrupting the push–pull character of the fluorophore corresponds to significant changes in the absorption and emission wavelengths and fluorescence quantum yield, rendering such modified DCDHFs promising candidates for fluorogenic probes.[23,50]
Because amines are strong electron-donating substituents and azides are weakly electron-withdrawing,[51] replacing the amine on the DCDHF fluorophore with an azide should disrupt the donor–π–acceptor character of the molecule and blue-shift the absorption and fluorescence. Moreover, aryl azides are known to be photolabile, often losing dinitrogen and rearranging to seven-membered azepine heterocycles.[52] However, Platz et al.[53] demonstrated that electron-withdrawing substituents on the benzene can stabilize the nitrene intermediate and promote the formation of amines and azo groups versus rearrangement to the azepine. Because the DCDHF is a very strong electron-withdrawing substituent, an azido DCDHF should photoconvert to a fluorescent amino DCDHF upon irradiation with resonant light.[23]
Scheme 1 displays several possible photoconversion pathways. With the loss of N2, the azide on 1a can be photoconverted to a reactive nitrene. The nitrene can then convert into an amine (1b) or a nitro group (1c); both these structures were actually isolated and characterized.[23] Structure 1d is hypothetical, but has literature precedent: nitrenes are reactive enough to insert into carbon-carbon bonds of nearby molecules. This approach, called photoaffinity labeling, can be used to bioconjugate aryl azides to biological targets of interest.[54] The azido DCDHF could act as a fluorogenic photoaffinity label: photoactivation should both induce fluorescence and form a covalent bond between the fluorophore and a neighboring biomolecule.
Scheme 1.

Photoactivation reactions of the azido DCDHF fluorogen. Aryl azides are known to be photolabile; the loss of dinitrogen leaves a reactive nitrene intermediate, which can rearrange to form a seven-membered azepine heterocycle (not shown), an amine (1b), or a nitro (1c) group. Compounds 1a and 1c are not fluorescent when pumped at long wavelengths, but photoproducts 1b and 1d are fluorescent. Compound 1d is hypothetical and the result of nitrene inserting into C–C bonds of a nearby biomolecule. (Adapted with permission from J. Am. Chem. Soc. 2008, 130, 9204−9205. Copyright 2008 American Chemical Society.)
Figure 6 demonstrates the utility of the azido DCDHF as a fluorogen; before photoactivation, cells incubated with 1a are dark; after photoactivation, the cells light up as 1a converts to 1b. The intensity of activating light required to generate fluorescent DCDHF molecules is very low, at least three orders of magnitude lower than the intensity of the imaging light. This helps ensure that the high-energy blue or UV activating light does not kill the cells of interest or alter their morphology. Furthermore, photobleaching is three orders of magnitude less likely than photoconverting, so the activated fluorophore emits millions of photons before photobleaching (see Table 3).
Figure 6.

Azido DCDHFs are photoactivatable. In contrast to the amine group, the azide group is not electron donating; therefore, charge-transfer band of the chromophore is disrupted and the absorption is significantly blue-shifted, so it is no longer resonant with the imaging laser. Irradiance with low-intensity 407 nm light converts the azide to an amine, which repairs the donor-acceptor character of the fluorophore and returns the absorption and emission to longer wavelengths. See Scheme 1. A) Three CHO cells incubated with azido fluorogen are dark before activation. B) The fluorophore lights up in the cells after activation with a 10 s flash of diffuse, low-irradiance (0.4 W cm−1) 407 nm light. The white-light transmission image is merged with the fluorescence images (white), excited at 594 nm (∼ 1 kW cm−1). Scalebar: 20 μm. C) Single molecules of the activated fluorophore in a cell under higher magnification. Scalebar: 800 nm. D) Absorption curves in ethanol showing photoactivation of fluorogen 1a (λabs = 424 nm) over time to fluorescent product 1b (λabs = 570 nm). Different curves represent time points between 0 and 1320 s of illumination by 3.1 mW cm−1 of diffuse 407 nm light (trends denoted by arrows). Dashed line is the absorbance of pure, synthesized 1b. (Inset) Dotted line is weak pre-activation fluorescence of 1a excited at 550 nm; solid line is strong post-activation fluorescence resulting from exciting 1b at 550 nm, showing an increase in the fluorescence emission. (Adapted with permission from J. Am. Chem. Soc. 2008, 130, 9204−9205. Copyright 2008 American Chemical Society.)
Table 3.
Photophysical properties of the photoactivatable DCDHF in Scheme 1 and other photoswitchable fluorophores.[a]
| λabs [nm] | λfl [nm] | εmax [m−1 cm−1] | ΦF | ΦP[b] | ΦB in gelatin | Ntot,e in gelatin | |
|---|---|---|---|---|---|---|---|
| 1a | 424 | 552 | 29100 | n/a | good (0.0059) | n/a | n/a |
| 1b | 570 | 613 | 54100 | 0.025−0.39 | n/a | 4.1 × 10−6 | 2.3 × 106 |
| Dronpa[c] | 503 | 518 | 95000 | 0.85 | very good | ∼ 3.2 × 10−5 | – |
| PA-GFP[d] | 504 | 517 | 17400 | 0.79 | moderate | ∼ 6.9 × 10−5 | ∼ 140,000 |
| EYFP[e] | 514 | 527 | 84000 | 0.61 | moderate | 5.5 × 10−5 | ∼ 140,000 |
| Cy3/Cy5[f] | 647 | 662 | 200000 | 0.18 | very good | – | ∼ 670,000 |
| PC-RhB[g] | 552 | 580 | 110000 | 0.65 | moderate | – | ∼ 600,000 |
Values for 1a and 1b reported in ethanol unless otherwise stated. In the other systems, we calculated estimations from available information (see discussion in the supporting material of reference [23]).
Quantum yield of photoconversion; for the DCDHF system, from azide with 407 nm illumination. Quantitative values for other systems have not been reported, so we include qualitative comparisons.
Aqueous photophysical values for the reversibly photoswitchable GFP called Dronpa from refs. [57] and [67].
Aqueous and in-cell photophysical values for the irreversibly photoswitchable GFP called PA-GFP as reported in refs. [25,56], and [68].
The photophysics of this azido DCDHF fluorogen are favorable compared to other popular photoswitchable or photoactivatable molecules: it is red-shifted, emits many photons, and requires only low doses of blue light (see Table 3). However, the disadvantages of the azido DCDHF system are: it is not genetically targeted, as fluorescent proteins are; DCDHFs with a primary amine exhibit more blinking than those with secondary or tertiary amines (see Figure 7 and reference [13]); and the photoactivation is irreversible (once the fluorophore photobleaches, it cannot be reactivated, which is possible in some photoswitches[55-60]).
Figure 7.

Top: The trajectory of a single copy of the amino DCDHF fluorophore diffusing in a CHO cell after photoactivation. Gray lines indicate frames when the fluorophore was dark and not tracked (i.e. blinking or out of focus). Bottom: A background-subtracted intensity time-trace of the molecule in the trajectory on the left. Gray lines indicate when the fluorophore was dark (i.e. initially blinking events, then finally bleaching). (Adapted with permission from J. Am. Chem. Soc. 2008, 130, 9204−9205. Copyright 2008 American Chemical Society.)
In some imaging scenarios, irreversibility is acceptable; because the millions of photons emitted are not spread over many activating cycles, the localization precision can be high for each molecule. Blinking, however, is not particularly desirable, but can sometimes be used to reduce the emitter concentrations in super-resolution microscopy methods.[49] This issue may be addressed in future work by finding a DCDHF version that does not blink as a primary amine, or via fluorogenic photoaffinity labeling that produces the secondary amine (because DCDHFs with a secondary amine are less prone to blink). When combined with a targeting moiety that brings the molecule close to the location of interest, fluorogenic photoaffinity makes such targeting permanent,[54,61,62] and thus could resolve two disadvantages of the azido DCDHF system simultaneously.
4. Cell and Biological Imaging
4.1. Screening DCDHFs in Cell-like Media
Imaging single molecules in living cells has strict criteria; not only must the emitter be bright and photostable, but also it must be red-shifted enough to avoid background from cellular autofluorescence from flavins and other endogenous fluorophores pumped at wavelengths shorter than approximately 500 nm,[25] be water-soluble and membrane-permeable, be washable after incubation, and be specifically targetable.
Quantifying photophysical parameters of single molecules in living cells is not always possible. A more practical approach is to screen fluorophores in cell-like media such as protein gels, agarose gels, or poly(vinyl alcohol) aqueous films. For instance, Figure 8 (left) shows single copies of DCDHF-N-6 fluorophores embedded in gelatin, an aqueous protein gel. Photostability parameters (Ntot,e or ΦB) measured in gelatin offer a better measure of how well the fluorophore will perform in cells than testing only in organic solvents and polymer films; Table 1 includes several values for ΦB in gelatin.
Figure 8.

Surface plot of emission from (left) four distinct DCDHF-N-6 single molecules in a gelatin layer and (right) two distinct DCDHF-A-6 single molecules in a CHO cell membrane. The ability to see single emitters in cells is a strong qualitative test of their utility. In gelatin, photostability and other important photophysical parameters canbe directly quantified. (Adapted with permission from J. Phys. Chem. A 2007, 111, 8934−8941. Copyright 2007 American Chemical Society.)
Nevertheless, the ultimate test of a fluorophore is whether it is visible on the single-molecule level in a living cell. Figure 8 shows two DCDHF-A-6 molecules in the plasma membrane of a living Chinese hamster ovary (CHO) cell. Figure 6 demonstrates that photoactivating the azido DCDHF fluorogen is possible in living cells; moreover, single copies of the photoactivated fluorophore were visible and trackable as they diffused in the cell (Figure 7). Although these are only qualitative measures, they are evidence for the utility of these fluorophores as cellular probes.
4.2. DCDHFs as Lipid Analogs
Quantifying the brightness and photostability of emitters in cells, while difficult, is ultimately necessary to confirm their efficacy. For instance, we measured the diffusion behavior of single emitters of seven DCDHF structures in the cell membrane,[18] and found by measuring signal-to-background levels and total photons detected that DCDHF-N-6 performed comparably to TRITC, a rhodamine derivative. For this experiment, DCDHF versions with long alkyl chains on the amine donor were synthesized in order to mimic the polar head and nonpolar tail of membrane lipids. These DCDHF lipid analogs insert well into the lipid bilayer, where higher viscosity brightens the fluorophore to the point that it is visible and trackable, even with electron-multiplying camera integration times as short as 30 ms. Charged DCDHF versions exhibited slower diffusion kinetics than the neutral versions, possibly indicating that they partitioned into different lipid environments, or possibly resulting from stronger association of neighboring lipid molecules around the additional charges.
4.3. Self-Quenched DCDHF Molecular Beacons
DCDHF emitters can be used for other types of photophysical readouts of molecular associations. We attached the amine-reactive NHS ester of DCDHF-V-P to the two ends of a DNA hairpin,[21] thereby generating a self-quenched dimer molecular beacon which fluoresces upon hybridization.[63] In this study, identical DCDHFs were covalently attached to the ends of a single-stranded DNA molecular beacon in an H-dimer configuration (i.e. transition dipoles oriented parallel).[64] This interaction results in splitting of the electronic excited state into a higher and a lower energy level. Excitation to the lower energy level is dipole-forbidden, as evidenced by the resulting blue-shifted absorption peak. Following photoexcitation from the ground state to the higher energy level of the electronic excited state, rapid internal conversion to the lower energy level occurs, from which fluorescence is a forbidden transition; therefore, fluorescence from the DCDHFs is quenched in the closed molecular beacon. Upon addition of target DNA, which binds to the loop region of the beacon and causes it to open, the H-dimer configuration is broken and the DCDHFs spatially separate and become bright.
Figure 9 displays the beacon scheme and the sensitivity to the addition of target sequence in single-molecule imaging. In addition to achieving a high fluorescence turn-on ratio, this system affords other advantages, including a one-pot synthesis of the molecular beacons, non-fluorescent colorimetric detection of DNA, a two-fold “on” signal (i.e. emission from two fluorophores instead of only one, as in a traditional fluorophorequencher molecular beacon), and the ability to use two-step photobleaching (Figure 9 c) of the molecular beacon in single-molecule studies to reduce false positives resulting from stray fluorescent impurities or singly labeled beacons. Although these were in vitro tests of the beacon, sensing single oligonucleotide target sequences in living cells may be possible.
Figure 9.

Molecular beacons using DCDHF-V-P as a self-quenchable fluorophore. Single-molecule fluorescence images of surface-immobilized beacons a) before and b) after adding target oligonucleotide (scale bars: 5 μm). c) Single-molecule time traces of opened beacons display two-step photobleaching, indicating that two DCDHF fluorophores emit from each bright spot. d) Closed and open conformations of the surface-immobilized molecular beacon: the two DCDHFs (indicated by arrows) are arranged in an H-dimer configuration and the beacon is dark (self-quenched); upon binding of the target oligonucleotide sequence, the two fluorophores become separated and both become bright (no longer self-quenched). (Adapted with permission from J. Phys. Chem. B 2007, 111, 7929−7931. Copyright 2007 American Chemical Society.)
4.4. DCDHFs Labeling Cell-Penetrating Peptides
DCDHFs have also been used to label specific biomolecules of interest to enable studies of the molecules in conjunction with living systems. For example, to shed light on the debated cell-entry mechanisms of cell-penetrating peptides, we labeled a cell-penetrating peptide of the guanidinium-rich oligoarginine type[65] using a reactive maleimide group on a DCDHF-V-P fluorophore.[27] The DCDHF-labeled oligoarginines were then added to living cells and observed as they interacted with the plasma membrane. This experiment was performed at normal oxygen concentration as the use of DCDHF-V-P affords high-contrast imaging without the use of oxygen-scavenger systems.
Figure 5 shows the labeled oligoarginines on a cell at different concentrations; single copies are easily visible and trackable on the surface of the cell at nanomolar concentration of labeled oligoarginines, with low background from fluorophores in the surrounding solution. In this case, since the free upper membrane of the cell was of interest, a TIR imaging configuration was not a feasible option. The use of viscosity-sensing DCDHFs maintained a low enough background to enable single-molecule tracking in epifluorescence mode on the top surface of the cell, which does not interact with the coverslip.
Quantitative parameters such as diffusion coefficients and residence times of the DCDHF-labeled oligoarginines in the plasma membrane were extracted from single-molecule traces. These values were used to demonstrate that oligoarginines interact with the plasma membrane either via a multiple-entry mechanism or via a mechanism distinct from passive diffusion or endocytosis.
5. Conclusions and Outlook
The DCDHF organic fluorophores are attractive as single-molecule emitters, with their high photostability, bright emission, sensitivity to local environment, solubility, and range of colors and reactive chemistries. By harnessing their structure-property relationships, we have designed and optimized DCDHFs for cell-labeling applications by red-shifting the absorption wavelength, adding lipid-like tails, increasing water solubility, and increasing the brightness by constraining bond rotations.
Moreover, the push–pull character of the fluorophores offers a distinctive handle for new fluorogenic reactions. Because each component of the donor–π–acceptor structure is required for effective charge transfer interaction, disrupting any one part of the fluorophore can render it dark; chemically or photochemically regenerating that portion of the molecule restores fluorescence. This property of DCDHFs can be used to design probes that become fluorescent only after a specific bioconjugation reaction or irradiation with photoactivating light. Additionally, this approach could be applied to other push–pull fluorophores. With photoswitching and fluorogenic probes becoming more important as complex and sophisticated microscopy techniques[49,66] are applied to cell imaging, DCDHF fluorophores will be a valuable tool.
Acknowledgements
We warmly thank our additional collaborators for their contributions: S. Bunge, P. J. Schuck, J. Bertke, P. R. Callis, and P. A. Wender. We also thank M. A. Thompson for conversations about relative photoactivation quantum yields. This work was supported in part by the National Institutes of Health through the NIH Roadmap for Medical Research, Grant No. P20-HG003638-04.
Footnotes
DCDHF: 2-dicyanomethylene-3-cyano-2,5-dihydrofuran
References
- 1.Design and Synthesis of Organic Molecular Compounds for Efficient Second Harmonic Generation. Nicoud JF, Twieg RJ. In: Nonlinear Optical Properties of Organic Molecules and Crystals. Chemla DS, Zyss J, editors. Academic Press; New York: 1987. pp. 227–296. [Google Scholar]
- 2.Prasad PN, Williams DJ. Introduction to Nonlinear Optical Effects in Molecules and Polymers. Wiley; New York: 1991. [Google Scholar]
- 3.Zyss J. Molecular Nonlinear Optics. Academic Press; New York: 1994. [Google Scholar]
- 4.Davies JA, Elangovan A, Sullivan PA, Olbricht BC, Bale DH, Ewy TR, Isborn CM, Eichinger BE, Robinson BH, Reid PJ, Li X, Dalton LR. J. Am. Chem. Soc. 2008;130:10565–10575. doi: 10.1021/ja8007424. [DOI] [PubMed] [Google Scholar]
- 5.Gubler U, He M, Wright D, Roh Y, Twieg RJ, Moerner WE. Adv. Mater. 2002;14:313–317. [Google Scholar]
- 6.Ostroverkhova O, Wright D, Gubler U, Moerner WE, He M, Sastre-Santos A, Twieg RJ. Adv. Funct. Mater. 2002;12:621–629. [Google Scholar]
- 7.Ostroverkhova O, Moerner WE. Chem. Rev. 2004;104:3267–3314. doi: 10.1021/cr960055c. [DOI] [PubMed] [Google Scholar]
- 8.Stevenson PE. J. Mol. Spectrosc. 1965;15:220–256. [Google Scholar]
- 9.Kippelen B, Meyers F, Peyghambarian N, Marder SR. J. Am. Chem. Soc. 1997;119:4559–4560. [Google Scholar]
- 10.Grunnet-Jepsen A, Thompson CL, Moerner WE. J. Opt. Soc. Am. B. 1998;15:905–913. [Google Scholar]
- 11.Würthner F, Wortmann R, Meerholz K. ChemPhysChem. 2002;3:17–31. doi: 10.1002/1439-7641(20020118)3:1<17::AID-CPHC17>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 12.Wortmann R, Poga C, Twieg RJ, Geletneky C, Moylan CR, Lundquist PM, DeVoe RG, Cotts PM, Horn H, Rice JE, Burland DM. J. Chem. Phys. 1996;105:10637–10647. [Google Scholar]
- 13.Willets KA, Ostroverkhova O, He M, Twieg RJ, Moerner WE. J. Am. Chem. Soc. 2003;125:1174–1175. doi: 10.1021/ja029100q. [DOI] [PubMed] [Google Scholar]
- 14.Wright D, Gubler U, Roh Y, Moerner WE, He M, Twieg RJ. Appl. Phys. Lett. 2001;79:4274–4276. [Google Scholar]
- 15.Willets KA, Callis PR, Moerner WE. J. Phys. Chem. B. 2004;108:10465–10473. [Google Scholar]
- 16.Schuck PJ, Willets KA, Fromm DP, Twieg RJ, Moerner WE. Chem. Phys. 2005;318:7–11. [Google Scholar]
- 17.Willets KA, Nishimura SY, Schuck PJ, Twieg RJ, Moerner WE. Acc. Chem. Res. 2005;38:549–556. doi: 10.1021/ar0401294. [DOI] [PubMed] [Google Scholar]
- 18.Nishimura SY, Lord SJ, Klein LO, Willets KA, He M, Lu ZK, Twieg RJ, Moerner WE. J. Phys. Chem. B. 2006;110:8151–8157. doi: 10.1021/jp0574145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Lu Z, Lord S, Willets K, Bertke J, Bunge S, Moerner W, Twieg R. Tetrahedron. 2007;63:103–114. [Google Scholar]
- 20.Lord SJ, Lu Z, Wang H, Willets KA, Schuck PJ, Lee HD, Nishimura SY, Twieg RJ, Moerner WE. J. Phys. Chem. A. 2007;111:8934–8941. doi: 10.1021/jp0712598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conley NR, Pomerantz AK, Wang H, Twieg RJ, Moerner WE. J. Phys. Chem. B. 2007;111:7929–7931. doi: 10.1021/jp073310d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Lu Z, Lord SJ, Moerner WE, Twieg RJ. Tetrahedron Lett. 2007;48:3471–3474. doi: 10.1016/j.tetlet.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lord SJ, Conley NR, Lee HD, Samuel R, Liu N, Twieg RJ, Moerner WE. J. Am. Chem. Soc. 2008;130:9204–9205. doi: 10.1021/ja802883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Z, Liu N, Lord SJ, Bunge SD, Moerner WE, Twieg RJ. unpublished results.
- 25.Harms GS, Cognet L, Lommerse PHM, Blab GA, Schmidt T. Biophys. J. 2001;80:2396–2408. doi: 10.1016/S0006-3495(01)76209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SY, Semyonov AN, Twieg RJ, Horwich AL, Frydman J, Moerner WE. J. Phys. Chem. B. 2005;109:24517–24525. doi: 10.1021/jp0534232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HD, Dubikovskaya EA, Hwang H, Semyonov AN, Wang H, Jones LR, Twieg RJ, Moerner WE, Wender PA. J. Am. Chem. Soc. 2008;130:9364–9370. doi: 10.1021/ja710798b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; New York: 2006. p. 954. [Google Scholar]
- 29.Suppan P. J. Photochem. Photobiol. A. 1990;50:293–330. [Google Scholar]
- 30.Grabowski ZR, Rotkiewicz K, Rettig W. Chem. Rev. 2003;103:3899–4031. doi: 10.1021/cr940745l. [DOI] [PubMed] [Google Scholar]
- 31.Sawada S, Iio T, Hayashi Y, Takahashi S. Anal. Biochem. 1992;204:110–117. doi: 10.1016/0003-2697(92)90148-z. [DOI] [PubMed] [Google Scholar]
- 32.Iwaki T, Torigoe C, Noji M, Nakanishi M. Biochemistry. 1993;32:7589–7592. doi: 10.1021/bi00080a034. [DOI] [PubMed] [Google Scholar]
- 33.Kuimova MK, Yahioglu G, Levitt JA, Suhling K. J. Am. Chem. Soc. 2008;130:6672–6673. doi: 10.1021/ja800570d. [DOI] [PubMed] [Google Scholar]
- 34.Moerner WE, Fromm DP. Rev. Sci. Instrum. 2003;74:3597–3619. [Google Scholar]
- 35.Lu Z, Weber R, Twieg RJ. Tetrahedron Lett. 2006;47:7213–7217. doi: 10.1016/j.tetlet.2006.07.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kautsky H. Trans. Faraday Soc. 1939;35:216–219. [Google Scholar]
- 37.Turro NJ. Modern Molecular Photochemistry. University Science Books; Sausalito: 1991. p. 628. [Google Scholar]
- 38.Toutchkine A, Nguyen D, Hahn KM. Org. Lett. 2007;9:2775–2777. doi: 10.1021/ol070780h. [DOI] [PubMed] [Google Scholar]
- 39.Rasnik I, McKinney SA, Ha T. Nat. Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 40.Aitken CE, Marshall RA, Puglisi JD. Biophys. J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Widengren J, Mets U, Rigler R. J. Phys. Chem. 1995;99:13368–13379. [Google Scholar]
- 42.Hübner CG, Renn A, Renge I, Wild UP. J. Chem. Phys. 2001;115:9619–9622. [Google Scholar]
- 43.Renn A, Seelig J, Sandoghdar V. Mol. Phys. 2006;104:409. [Google Scholar]
- 44.Widengren J, Chmyrov A, Eggeling C, Lofdahl P, Seidel C. J. Phys. Chem. A. 2007;111:429–440. doi: 10.1021/jp0646325. [DOI] [PubMed] [Google Scholar]
- 45.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 46.Hess ST, Girirajan TPK, Mason MD. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rust MJ, Bates M. Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moerner WE. Nat. Methods. 2006;3:781–782. doi: 10.1038/nmeth1006-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moerner WE. Proc. Natl. Acad. Sci. USA. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouffard J, Kim Y, Swager TM, Weissleder R, Hilderbrand SA. Org. Lett. 2008;10:37–40. doi: 10.1021/ol702539v. [DOI] [PubMed] [Google Scholar]
- 51.Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 52.Schriven EFV. Azides and Nitrenes: Reactivity and Utility. Academic Press; Orlando: 1984. p. 542. [Google Scholar]
- 53.Soundararajan N, Platz MS. J. Org. Chem. 1990;55:2034–2044. [Google Scholar]
- 54.Kotzyba-Hibert F, Kapfer I, Goeldner M. Angew. Chem. Angew. Chem. Int. Ed. 1995;1995;10734:1391–1408. 1296–1312. [Google Scholar]
- 55.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 56.Patterson GH, Lippincott-Schwartz J. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 57.Ando R, Mizuno H, Miyawaki A. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- 58.Bates M, Blosser TR, Zhuang X. Phys. Rev. Lett. 2005;94:108101-1–108101-4. doi: 10.1103/PhysRevLett.94.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fölling J, Belov V, Kunetsky R, Medda R, Schönle A, Egner A, Eggeling C, Bossi M, Hell SW. Angew. Chem. Angew. Chem. Int. Ed. 2007;2007;11946:6382–6386. 6266–6270. doi: 10.1002/anie.200702167. [DOI] [PubMed] [Google Scholar]
- 60.Conley NR, Biteen JS, Moerner WE. J. Phys. Chem. B. 2008;112:11878–11880. doi: 10.1021/jp806698p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dreyfuss G, Schwartz K, Blout ER, Barrio JR, Liu F, Leonard NJ. Proc. Natl. Acad. Sci. USA. 1978;75:1199–1203. doi: 10.1073/pnas.75.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dockter ME. J. Biol. Chem. 1979;254:2161–2164. [PubMed] [Google Scholar]
- 63.Tyagi S, Kramers HA. Nat. Biotechnol. 1996;14:303. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 64.Kasha M, Rawls HR, El-Bayoumi MA. Pure Appl. Chem. 1965;11:371–392. [Google Scholar]
- 65.Rothbard JB, Jessop TC, Wender PA. Adv. Drug Delivery Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 66.Hell SW. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 67.Habuchi S, Ando R, Dedecker P, Verheijen W, Mizuno H, Miyawaki A. J. Hofkens, Proc. Natl. Acad. Sci. USA. 2005;102:9511–9516. doi: 10.1073/pnas.0500489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt T, Kubitscheck U, Rohler D, Nienhaus U. Single Mol. 2002;3:327. [Google Scholar]
- 69.Patterson G, Day RN, Piston D. J. Cell Sci. 2001;114:837–838. doi: 10.1242/jcs.114.5.837. [DOI] [PubMed] [Google Scholar]
- 70.Soper SA, Nutter HL, Keller RA, Davis LM, Shera EB. Photochem. Photobiol. 1993;57:972–977. [Google Scholar]