

Abstract
The transcription factor REST/NSRF (RE1-Silencing Transcription Factor) is a master repressor of neuronal gene expression and neuronal programs in non-neuronal lineages1−3. Recently, REST was identified as a human tumor suppressor in epithelial tissues4, suggesting that REST regulation may have important physiologic and pathologic consequences. However, the pathways controlling REST have yet to be elucidated. Here, we demonstrate that REST is regulated by ubiquitin-mediated proteolysis, and use an RNAi screen to identify SCFβTRCP as an E3 ubiquitin ligase responsible for REST degradation. βTRCP binds and ubiquitinates REST and controls its stability through a conserved phosphodegron. During neural differentiation REST is degraded in a βTRCP-dependent manner. βTRCP is required for proper neural differentiation only in the presence of REST, indicating that βTRCP facilitates this process through degradation of REST. Conversely, failure to degrade REST attenuates differentiation. Furthermore, we find that βTRCP overexpression, which is common in human epithelial cancers, causes oncogenic transformation of human mammary epithelial cells and this pathogenic function requires REST degradation. Thus, REST is a key target in βTRCP-driven transformation and the βTRCP-REST axis is a new regulatory pathway controlling neurogenesis.
REST levels decline during differentiation of embryonic stem cells to neural stem and progenitor cells5, consistent with a role for REST in restraining neuronal gene expression programs. This decrease results from a 3-fold reduction in REST half-life (Fig. 1a), suggesting that a regulatory pathway controls REST degradation during early neural differentiation. To determine whether ubiquitination is involved, REST was evaluated for ubiquitin-modification in vivo. Immunoprecipitation of HA-ubiquitin revealed slower migrating species of REST suggestive of polyubiquitination (Fig. 1b, lane 3). REST also precipitated with an HA-ubiquitin mutant lacking all lysines except K48 (Fig. 1b, lane 4), suggesting REST is K48 polyubiquitinated which promotes degradation.
Figure 1. Identification of βTRCP and FBW4 ubiquitin ligases as regulators of REST stability.
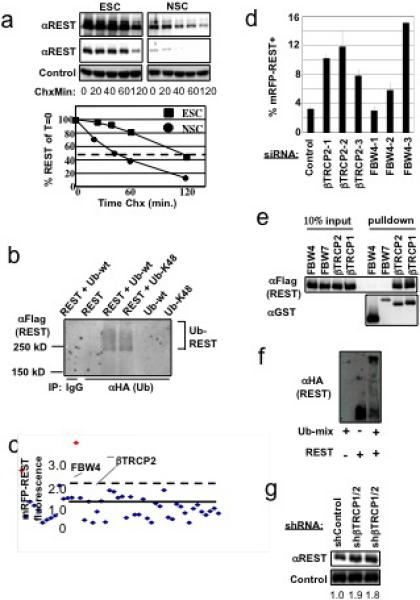
a, Embryonic stem cells (ESC) or Neural Stem Cells (NSC) were examined for REST protein half-life in a cycloheximide (Chx) timecourse. Quantitation of relative REST levels in lower panel (red line denotes half-life). b, 293T cells were transfected with plasmids expressing Flag-REST, HA-ubiquitin, and/or HA-ubiquitin-K48 as indicated, immunoprecipitated with HA-specific antibodies or control IgG, and analyzed by αFlag immunoblot. c, siRNA screen for regulators of mRFP-REST (see Supp. Legend 1c for details). d, siRNAs targeting βTRCP2 or FBW4 sequences independent from library-derived siRNAs were tested for effects on mRFP-REST fluorescence (n=3, error bars +/− s.d.). e, Coimmunoprecipitation of GST-F-box fusion proteins and Flag-REST from mammalian cells. f, in vitro ubiquitination of HA-REST by SCFβTRCP (see Supp. Legend 1f for details). g, Human mammary epithelial cells expressing control shRNA or shRNA-targeting human βTRCP1 and βTRCP2 were analyzed by αREST immunoblot. Two-independent infections with βTRCP-shRNA are shown. Quantitation of relative REST levels is shown above each lane.
To search for the E3 ubiquitin ligase for REST, we began with the SCF superfamily of ligases6. Each SCF family contains a common Cullin scaffold that is required for ligase function. Notably, coexpression of a dominant negative Cullin-1 (Cul1) mutant resulted in a dramatic increase (11-fold) in REST levels (Supp. Fig. 1b), indicating that one or more Cul1-containing ligases negatively regulate REST abundance.
F-box proteins act as substrate receptors for the SCF7,8. To determine which F-box proteins are required for REST turnover, we established a system for monitoring REST abundance in a high-throughput manner using an mRFP-REST fusion protein. Similar to endogenous REST, mRFP-REST was unstable, and its abundance increased upon inhibition of Cul-1 (Supp. Fig. 2a). To identify the F-box proteins regulating REST, individual siRNAs targeting each F-box protein (4 siRNAs/gene) were cotransfected with a plasmid encoding mRFP-REST, and changes in cellular fluorescence were monitored by flow cytometry (Supp. Fig. 2b). siRNAs that increased fluorescence >2 standard deviations from the mean were retested in triplicate for their effects on both mRFP and mRFP-REST to identify siRNAs that specifically alter REST stability (Fig. 1c). This approach identified FBW4 and βTRCP2. Notably, multiple siRNAs targeting additional sequences within FBW4 and βTRCP2 also increased mRFP-REST abundance (Fig. 1d), confirming the specificity of the siRNAs. Supporting this conclusion, coexpression of a dominant negative βTRCP mutant (lacking the F-box) also increased REST levels (Supp. Fig. 4a-b, respectively).
βTRCP2 and FBW4 may control REST abundance by direct ubiquitination of REST or by modulating upstream regulators of REST. βTRCP2, but not FBW4, was capable of binding REST in cells (Fig. 1e), suggesting that FBW4-mediated regulation is indirect. The highly homologous βTRCP1 also interacted with REST (Fig. 1e and Supp. Fig. 3a), consistent with previous reports that βTRCP1 and βTRCP2 have similar substrate specificities and frequently function redundantly9,10. Importantly, endogenous βTRCP and REST interact in cells (Supp. Fig. 3b), and REST was polyubiquitinated by SCFβTRCP1 in vitro (Fig. 1f), suggesting that SCFβTRCP regulates REST by direct ubiquitination. In agreement, stable expression of βTRCP-shRNA (targeting βTRCP1 and βTRCP2) in both human mammary epithelial cells (HMECs) and NIH3T3 cells resulted in a moderate but reproducible increase in REST protein abundance and half-life (Fig. 1g, lanes 2 and 3, and Supp. Fig. 4c), indicating that endogenous REST is regulated by βTRCP. These data indicate that SCFβTRCP controls REST by ubiquitin-mediated destabilization.
SCFβTRCP binds substrates in a phosphorylation-dependent manner6,10−14. Consistent with this, λ-phosphatase treatment abolished the interaction between REST and βTRCP and this was prevented by λ-phosphatase inhibitors (Fig. 2a). Notably, a dominant negative frame-shift mutant of REST found in human colon cancer cells4 failed to interact with βTRCP and exhibited substantially increased stability in cells (Supp. Fig. 6a), indicating the c-terminal half of REST is required for βTRCP recognition. Analysis of this region revealed a sequence highly similar to the phosphodegron found in Cdc25A, a well documented βTRCP-substrate11,12 (Fig. 2b). This putative degron includes a conserved DpSG motif that constitutes a critical interaction element within phosphodegrons for βTRCP15. Mass spectrometry was used to examine phosphorylation of REST within this region. To enable tryptic digestion of the peptide of interest, a N1022R substitution was introduced into REST that does not alter interaction with βTRCP or protein stability in cells (Supp. Figs. 5a-b). His-tagged RESTN1022R was co-expressed with dominant-negative Cul1 in 293T cells and purified under denaturing conditions (Supp. Fig. 5c). Analysis of phosphopeptides in RESTN1022R demonstrated that S1027 and S1030 within the MSEGSDDSGLHGARPVPQESSR peptide are phosphorylated both singly and in combination (Supp. Figs. 5c-g).
Figure 2. A conserved phosphodegron in REST is required for regulation by βTRCP.
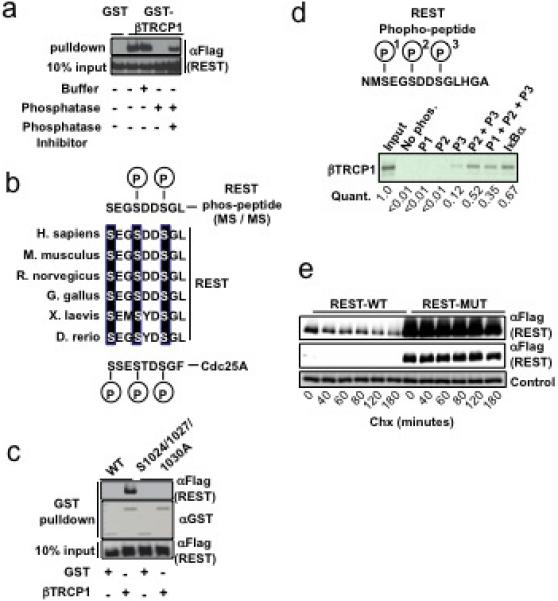
a, 293T cells were transfected with GST, GST-βTRCP -or Flag-REST expression plasmids. Flag-REST lysates were treated with buffer, λ-phosphatase, or λ-phosphatase + phosphatase-inhibitor as indicated. Flag-REST lysates were then mixed with GST- or GST-βTRCP lysates, precipitated with glutathione beads and immunoblotted with αFlag antibodies. b, Phosphorylation of the conserved REST degron in vivo. Sequence alignments of REST proteins (Hs REST residues 1024−1032) from several species and the phosphodegron from Hs CDC25A. Phospho-serines within the REST degron identified by MS/MS are shown in upper sequence. c, 293T cells expressing the indicated combinations of GST, GST-βTRCP1 (denoted on bottom), and Flag-REST mutants (denoted at top). GST-bound complexes were immunoblotted with αFlag (upper and lower panels) or αGST (middle). d, 35S-βTRCP1 was transcribed/translated in vitro and incubated with biotin-conjugated peptides spanning the REST degron (unphosphorylated or phosphorylated) or the IκB degron (phosphorylated). Peptide-associated-proteins were precipitated with streptavidin-conjugated beads, analyzed by SDS-PAGE, and quantified using a phosphoimager. Peptide sequence spanning the REST degron is shown in the top panel. e, 293T cells expressing wild-type or degron-mutant Flag-REST cDNAs were examined for Flag-REST protein half-life in a cycloheximide (Chx) timecourse.
To test the ability of the candidate REST-degron to interact with βTRCP, peptides spanning the degron were synthesized with phosphates at serines 1024, 1027, and 1030 alone or in combination. Individual serine-phosphorylation facilitated weak (S1030) or no interaction (S1024 or S1027) with βTRCP (Supp. Fig. 7). In contrast, peptides phosphorylated in combination at S1027+S1030 or S1024+S1027+S1030 associated with βTRCP (but not Fbw4) with an efficiency comparable to that of the well-established IκB phosphodegron peptide (Fig. 2d and Supp. Fig. 7). Mutation of each serine to alanine in the context of full-length REST resulted in decreased binding to βTRCP, and combined mutation of these critical serines completely abrogated the interaction with βTRCP (Fig. 2c and Supp. Fig. 6b). Notably, degron-mutant REST was substantially more stable than wild-type REST in cells (Fig. 2e). These data support the hypothesis that phosphorylation of the REST degron primes ubiquitination by SCFβTRCP, thereby promoting REST degradation.
The role of βTRCP in degradation of the REST tumor suppressor predicts that βTRCP overproduction might transform human cells. To examine this prediction, HMECs stably expressing human telomerase catalytic subunit (hTERT) and the SV40 LT oncogene (“TLM-HMECs,”16) were transduced with a control or GFP-βTRCP1-expressing retrovirus. Stable ectopic expression of βTRCP1 resulted in reduced REST abundance (Fig. 3a) and robust anchorage-independent proliferation (Fig. 3b), thus phenocopying REST loss-of-function4. This is consistent with a transgenic mouse model in which ectopic βTRCP1 expression in the mammary gland produced advanced breast cancer17. To determine whether REST degradation is critical for βTRCP1-mediated transformation, TLM-HMECs stably expressing βTRCP1 were transduced with retroviruses expressing wild-type or degron-mutant REST. Exogenous REST expression did not alter proliferation on an adhesive cell culture surface (Fig. 3c). In contrast, βTRCP1-induced anchorage-independent proliferation was severely impaired by restoring REST expression (Fig. 3d). Consistent with its increased stability, degron-defective REST suppressed βTRCP1-transformation more efficiently (Fig. 3d and Supp. Fig. 8). These data implicate REST as an essential target in βTRCP-driven oncogenic transformation.
Figure 3. βTRCP targets REST during oncogenic transformation.
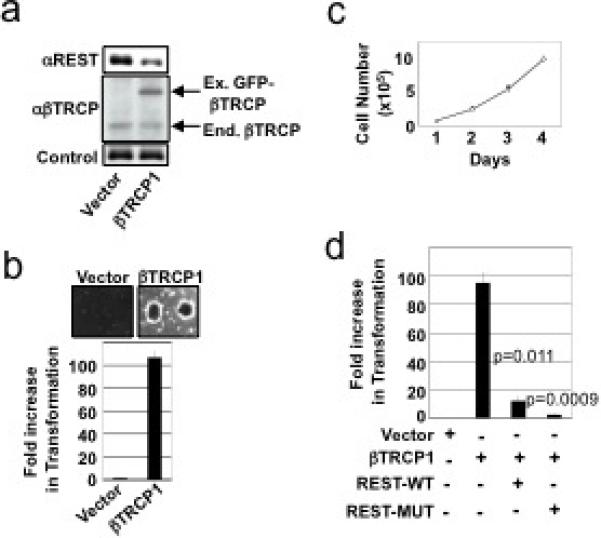
a, TLM-HMECs were transduced with control or GFP-βTRCP1-expressing retroviruses. Lysates were probed with antibodies against REST (upper panel), βTRCP (middle panel), or Vinculin (lower panel). b, Cells from a were analyzed for anchorage-independent colony formation. Assays were performed in quadruplicate (error bars +/− s.d.). Representative of 3 independent experiments is shown. c, HMECs were transduced with retroviruses expressing wild-type REST (REST-WT), degron-mutant REST (REST-MUT), and/or βTRCP1. Cell numbers were monitored for 4 days after plating on tissue-culture dishes. (open circles: vector-1+vector-2, closed circles: βTRCP1, open triangles: βTRCP1+vector-2, closed diamonds: βTRCP1+REST-WT, open squares: βTRCP1+REST-MUT) d, Cells from c were assessed for anchorage-independent colony formation. Assays were performed in triplicate (error bars +/− s.d.). Representative of 2 independent experiments is shown.
While REST is a well documented regulator of neuronal gene expression and has been proposed to restrain several steps in neurogenesis (reviewed in 3), its role in neurogenesis has not been tested genetically. Thus, we used embryonic stem (ES) cells to genetically examine the roles of REST and βTRCP in the differentiation program of neural stem and progenitor cells (reviewed in 18). For this we employed ES cells in which eGFP was recombined into the Sox1 locus19,20 (“46c cells”), a well characterized marker of early neural differentiation in vitro and in vivo.
We first confirmed that endogenous REST stability is regulated during neural differentiation of 46c cells. As shown in Figure 4a, REST half-life declined 2-fold in differentiated cells, consistent with the decreased REST stability observed in homogeneous neural stem cells (Fig. 1a). This decrease may be driven, in part, by a concomitant 13-fold increase in βTRCP1 expression (Fig. 4b). To test the role of REST and βTRCP in this differentiation program, 46c cells were transfected with control, REST, or βTRCP1 targeting siRNAs alone or in combination, and subsequently cultured in differentiation media and analyzed for neural differentiation by flow cytometric analysis of Sox1:eGFP fluorescence. Inactivation of REST promoted differentiation, correlating with the efficiency of REST knockdown (Fig. 3c and Supp. Figs. 10a-b), thus providing the first genetic evidence that REST negatively regulates early neural differentiation. Conversely, siRNAs that suppress βTRCP1 expression >90% (Supp. Fig. 11c) attenuate differentiation into the neural lineage (Fig. 3c). These results were confirmed in multiple time points (data not shown) and with multiple siRNAs (Supp. Figs. 10b,d). Importantly, simultaneous REST+βTRCP1 knockdown increased Sox1:eGFP-positive cells >5-fold relative to βTRCP1-siRNA alone (Fig. 4c), showing REST reduction restores neural differentiation in the absence of βTRCP. Similar results were observed by measuring the abundance of an independent neuronal marker, TUBB3 (Fig. 4d). Thus, down regulation of REST is a critical function of βTRCP during early neural differentiation.
Figure 4. The βTRCP-REST pathway controls neural differentiation.
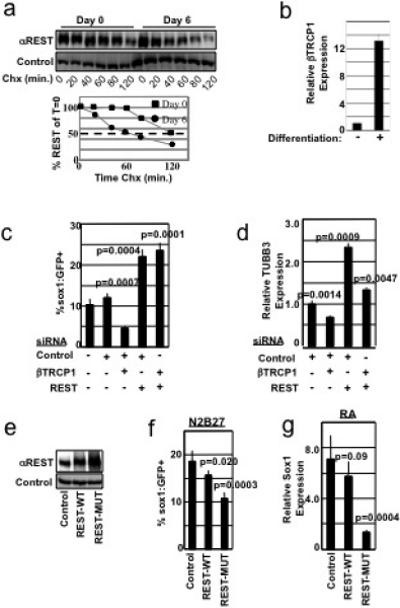
a, ES cells differentiated for 0 or 6 days were examined for REST protein half-life in a cycloheximide (Chx) timecourse. Differentiated lysates were analyzed at 3x the concentration of undifferentiated lysates. Quantitation of REST levels is shown in the lower panel. b, ES cells were differentiated for 0 or 6 days. βTRCP1 mRNA was analyzed by qRT-PCR, and normalized to GAPDH mRNA abundance. Experiment was performed in triplicate (error bars +/− s.d.). c, 46C cells transfected with the indicated combination of siRNAs were differentiated in N2B27 medium and analyzed for Sox1:GFP expression by flow cytometry. Experiments were performed in quadruplicate (error bars +/− s.d.) and is representative of 4 independent experiments. d, 46C cells from c were analyzed for expression of TUBB3 mRNA by qRT-PCR, and normalized to GAPDH mRNA abundance. Experiments were performed in triplicate (error bars +/− s.d.). e, 46C cells expressing control, Flag-REST-WT, or Flag-REST-MUT (triple point mutation in the REST-degron) cDNA were immunoblotted with αREST (upper panel) or αvinculin (lower panel) antibodies. Note: In this experiment, exogenous REST was expressed at levels higher than endogenous REST. f, 46C cells from e were cultured in N2B27 differentiation medium and analyzed for Sox1:GFP fluorescence. This experiment was performed in sextuplicate (error bars +/− s.d.) and is representative of 2 independent experiments. g, ES cells were infected as in e, differentiated into the neural lineage using an embryoid body-retinoic acid protocol, and analyzed for Sox1 mRNA by qRT-PCR (normalized to GAPDH mRNA abundance). Experiment was performed in triplicate (error bars +/− s.d.).
These data support the model that βTRCP regulates neural differentiation by facilitating REST degradation and predicts that a non-degradable REST would impede neural differentiation. To test this, we first examined the stability of wild-type or degron-mutant REST expressed in the context of neural differentiation. In this experiment, REST transgenes were expressed at levels much lower than endogenous REST to prevent alterations in differentiation kinetics (see below). Notably, the stability of endogenous REST and wild-type exogenous REST decreased similarly during neural differentiation (Supp. Fig. 10e and Fig. 4a). In contrast, degron-mutant REST was stable regardless of the cellular differentiation status (Supp. Fig. 10e). To test whether REST stabilization alters neural differentiation, 46c cells were transduced with high-titer retroviruses expressing wild-type or degron-mutant REST, resulting in a 1.5- and 2.6-fold increase in total REST (Fig. 4e). Notably, both transgenes attenuated neural differentiation, with the degron-mutant REST eliciting a more dramatic phenotype (Fig. 4f).
To further demonstrate REST's role in neural differentiation, we employed an independent neural differentiation assay. ES cells stably expressing wild-type or degron-mutant REST were differentiated by formation of embryoid bodies followed by stimulation with retinoic acid, a protocol routinely used to differentiate ES cells into the neuronal lineage21. In this context, non-degradable REST suppressed differentiation >5-fold as measured by mRNA expression of Sox1 (Fig. 4g). Collectively, these observations strongly link βTRCP function and REST degradation in controlling neural differentiation.
Here we demonstrate that REST is a labile protein targeted for ubiquitin-dependent proteasomal degradation by SCFβTRCP through a phospho-degron on REST. We show SCFβTRCP is a critical regulator of both physiologic and pathologic REST activities, constituting a new pathway controlling neural differentiation and cellular transformation (see Supp. Fig. 11). We provide the first genetic evidence that REST and SCFβTRCP regulate an early stage in neural specification. Our data are consistent with a model in which developmental cues induce degradation of REST, resulting in the derepression of proneural REST targets. The ability of REST to inhibit terminal differentiation of neurons also predicts that REST may promote proliferative properties in the neuronal lineage when overproduced or inappropriately stabilized. Consistent with this notion, REST is overexpressed in human medulloblastoma and ectopic REST expression in v-myc-immortalized neural stem cells promotes medulloblastoma formation in mice 22,23. Thus, the contrasting roles of REST as an oncogene and tumor suppressor are highly dependent on the developmental lineages.
βTRCP is overexpressed and oncogenic in epithelial cancers17,24,25 and we identified REST as a key target in this context. This suggests that pharmacologic inhibition of βTRCP may provide a means to restore REST tumor suppressor function in human cancer. The presence of a phosphodegron motif within REST suggests a role for upstream kinase(s) and/or phosphatase(s) that control REST degradation. We propose a model in which differentiation into the neural state is induced by this yet to be discovered signal transduction cascade that targets REST for degradation by SCFβTRCP, acting cooperatively with induction of βTRCP expression during neural differentiation. Conversely, hyperactivation of such pathway(s) priming REST degradation may be oncogenic in epithelial tissues and thus serve as new therapeutic targets in cancers with compromised REST function. Thus, exploration of these pathways will likely provide new opportunities for modulating neural stem cell and cancer cell behavior.
Supplementary Material
Supplemental Methods
Vectors
cDNAs encoding Flag-tagged human REST wild-type and frame-shift mutant have been previously described4. Point mutations in REST (pCMV-Flag-RESTS1024A, pCMV-Flag-RESTS1027A, pCMV-Flag-RESTS1030A, and pCMV-Flag-RESTS1024/1027/1030A) were generated with Stratagene mutagenesis kit by standard methods. REST cDNAs were cloned into lentiviral vectors pQCXIN (Clontech) and pHAGE-EF1α-puro (parental kindly provided by Richard Mulligan). Vectors encoding the GST-F-box proteins, dnCul1 (residues 1−452), βTRCPΔF, and shRNA targeting human βTRCP1 and βTRCP2 were previously described10,12. dnCul2 (residues 1−427), dnCul3 (residues 1−418), dnCul4 (residues 1−440), dnCul5 (residues 1−441) were prepared by cloning the respective N-terminal Cullin fragments into pcDNA3 (Invitrogen). shRNA sequence targeting both murine βTRCP1 and βTRCP2 (ATCAAGATCTGGGATAAAA) was cloned into pSuper-Retro-Puro (Oligoengine). pCMV-HA-Ub and pCMV-HA-UbK48 are gifts from Vishva Dixit (Genetech). Baculoviral expression vectors were generated in pBlueBac. Baculoviral HA-REST expression vector was generated by using the Gateway System (Invitrogen).
siRNA and DNA transfections
For the initial F-box siRNA screen, 272 siRNAs targeting 68 unique F-box proteins26 (4 siRNAs / F-box gene) were arrayed individually in 96-well format. 293T cells were transfected with 100 nM siRNAs using oligofectamine under standard conditions. On the subsequent day, cells were transfected with CMV-mRFP-REST plasmid DNA using mirus Trans-IT 293 under manufacturer recommended conditions. Cells were analyzed for mRFP fluorescence after 48 hours by flow cytometry. Subsequent testing of all candidate siRNAs was performed in triplicate and normalized to effects on mRFP (2 independent experiments). For interaction experiments, 293T cells were transfected with indictated plasmids with Lipofectamine 2000 (Invitrogen) per manufacturer's recommendations. For neural differentiation experiments, ES cells were plated in gelatin-coated 6-well plates at 0.5 × 106 cells/well and transfected with 100 nM siRNA using Lipofectamine 2000. 24 hours after transfection, cells were replated on gelatin-coated plates, and then cultured in N2B27 medium to induce neural differentiation. Experiments were performed in sextuplicate (error bars +/− s.d.) and are representative of at least 2 independent experiments.
Cell Culture
Human mammary epithelial cells (HMECs) expressing hTERT and SV40 LT (TLMHMECs)4,16 were cultured in mammary epithelial growth medium (MEGM, Cambrex). 293T, U2OS (human osteosarcoma cell line), and NIH3T3 cell lines were maintained in DMEM or McCoy's medium supplemented with 10% FBS and 50 μg/mL gentamycin. Neural stem cells and murine 46C ES cells with Sox1:GFP reporter were kindly provided by Dr. Austin Smith19,20. 46C cells were maintained in Knockout DMEM (Invitrogen) supplemented with 15% FBS, nonessential amino acids, 2 mM glutamine, 10 μM 2-mecaptoethanol, and 1000 units/ml ESGRO (Chemicon) on gelatin-coated tissue culture plates. 46C ES cells were induced to differentiate by culture in serum-free N2B27 medium (1:1 ratio of DMEM/F12 and Neurobasal medium, supplemented with N2 and B27 (Invitrogen))27. Neural stem cells were cultured in DMEM/F12 basal medium supplemented with insulin 25 μg/ml, Transferrin 100 μg/ml, progesterone 6 ng/ml, putrescine 16 μg/ml, sodium selenite 30 nM, BSA 50 μg/ml, FGF-2 10 ng/ml, EGF 10 ng/ml. For embryoid body formation, cells were plated in Corning ultra-low attachment 6-well plates at 5×105 cells per well in ES medium without LIF. Medium was changed every 2 days, and retinoic acid was added from day 4 to day 8 to 1 μM final concentration. Cells were harvested on day 8.
Retroviral infections of HMECs were performed with indicated viral supernatants in the presence of 8 μg/mL polybrene, and transduced cells were selected for resistance to the appropriate drug: puromycin (2.0 μg/mL), neomycin (200 μg/mL). ES cell infections were performed in the presence of 5 μg/mL polybrene and centrifuged for 1 hour (2000 rpm). Anchorage-independent proliferation assays were performed as previously described4. For each assay, the average of at least 3 replicates ± SD is shown. For growth curves, cells were seeded at a density of 5.0 × 104 per well in 6-well plates and cultured in MEGM. Cells were trypsinized and counted at the indicated time points (in triplicate with average ± SD shown). For protein half-life experiments, cells were treated with cycloheximide (100 μg/mL) for the indicated times and lysed in HLB buffer (see below).
Interaction Studies and Immunoblotting
HMECs, U20S, and ES cells were lysed in HLB buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% Triton-X 100, 1 mM DTT, supplemented with phosphatase- and protease-inhibitor cocktails (Calbiochem)) for 20 minutes with sonication. GST-pulldowns and immunoprecipitations were performed in lysis buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, and 1% NP-40. GSH-sepharose beads (15 μL per condition) were pre-washed in lysis buffer and incubated with 400 μg of lysate for 2 hours at 4°C while rocking. Beads were then washed 3−4 times in lysis buffer and bound protein eluted in 2X Laemmli Protein loading buffer containing SDS. Both GSH-bound samples and crude extract were resolved on 4−12% gradient SDS-PAGE gels. IP of endogenous REST and βTRCP was performed with 10 mg of U2OS whole cell lysate and precipitated with βTRCP antibody (Invitrogen 373400). Western blotting was performed with the following antibodies: γREST (Gail Mandel), αVinculin (Sigma,V9131), αRan (Sigma, R4777), αFlag (Sigma-A8592), αHA (Santa Cruz, F7), αGFP (Santa Cruz, FL), αβTRCP (Cell Signaling Technologies), αβTRCP (Invitrogen 373400), αCyclin E (Santa Cruz, HE12), αGST (Cell Signaling, 26H1).
In vitro binding assays utilized 35S-radiolabled recombinant F-box proteins that were in vitro transcribed and translated with T7 polymerase using TNT reticulocyte lysate (Promega) in the presence of 35S-methionine. Phosphorylated and unphosphorylated peptides spanning 14 residues of the REST phosphodegron were synthesized with N-terminal Biotin tags by Tufts Medical School Protein Core Facility (Biotin-N-M-S-E-GS-D-D-S-G-L-H-G-A, Biotin-N-M-pS-E-G-pS-D-D-pS-G-L-H-G-A, as well as singly and doubly phosphorylated peptides as indicated) and dissolved in PBS solution. Biotinylated and phosphorylated IκB-degron peptide is previously described (Winston et al., 1999). Peptide (1 μg) was preincubated with 10 μL of streptavidin agarose beads (Pierce) for 1 hr at 4°C in 400 μL NETN buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, 10 mM NaF, and 1 mM EDTA. Unbound peptide was washed from the streptavidin beads with NETN buffer and 35S-labeled F-box protein was incubated with the bead slurry for 2 hours at 4°C. Beads were washed 5 times with NETN buffer, and bound proteins were eluted in 2X Laemmli Protein loading buffer containing SDS, resolved on a 4−12% gradient SDS-PAGE gel, and detected by autoradiography. Quantitation of radiolabeled bands was performed using a phosphoscreen with a Typhoon Imager (Amersham) and ImageQuant TL software (Amersham).
In vitro ubiquitination
A mixture of bacculovirus expressing subunits of the SCFβTRCP complex (Flag-βTRCP, Cul1, Skp1, Rbx1) and HA-REST was transduced into exponentially growing SF9 insect cells in Xpress Insect cell medium (BioWhitaker) supplemented with 10% heat-inactivated serum. Forty hours post-transduction the holocomplex was purified at 4°C for 2 hrs through the Flag-βTRCP1 subunit from cleared lysate with 20 μL Flag-M2 beads (Sigma) in NETN buffer containing 50 mM Tris pH7.5, 150 mM NaCl, 0.5% NP-40, 10 mM NaF, 1 mM EDTA, 10 mM β-Glucophosphate, 10 mM NPP, and 0.1 μM okadaic acid. The bound holocomplex was washed three times with lysis buffer, twice with TBS (50 mM Tris pH7.5, 200 mM NaCl), and eluted in two batches with 0.5 μg/mL Flag-M2 peptide (Sigma) for 30 minutes at 4°C in TBS buffer. Elutions were combined and dialysed at 4°C first for 2 hours, then overnight against 500 mL of 50 mM Tris pH 7.5, 200 mM NaCl, 50% glycerol, 1 mM PMSF, and 1 mM DTT.
Eluted SCFβTRCP∼HA-REST holocomplex was incubated with 50 ng UBA1, 200 ng UbcH5, 2 mM ATP, 1 μM Ubiquitin Aldehyde, 50 ng Nedd8-activating enzyme NAE1 (Boston Biochem), 100 ng UbcH12, and 100 ng Nedd8 in ubiquitin reaction buffer containing 50 mM Tris pH7.5, 5 mM KCl, 5 mM NaF, 5 mM MgCl2, 0.5 mM DTT, 25 μM MG132, and energy regeneration mix (20 mM creatine phosphate and 2 μg/mL creatine kinase). The total 10 μL reaction was incubated at 30°C for 90 minutes, and the reaction was terminated with 2X Laemmli Protein loading buffer containing SDS. Reactions were resolved on 6% SDS-PAGE gels, transferred onto nitrocellulose, and immunoblotted with anti-HA.
26. Jin, J. et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 18, 2573−80 (2004).
27. Ying, Q. L. & Smith, A. G. Defined conditions for neural commitment and differentiation. Methods Enzymol 365, 327−41 (2003).
Acknowledgements
We thank G. Mandel and R. Truant for providing REST-C antibodies and mRFP-REST cDNA, respectively. We thank J. Luo, D. Nguyen, D. Lee, for suggestions and critical reading of the manuscript. We are grateful to R. Mulligan, A. Balazs, J. Jin, M. Sheng, and V. Dixit for providing reagents, and to D. Guardavaccaro and M. Pagano for communicating results prior to publication. T.F.W. is a fellow of the Susan G. Komen Foundation and is supported by grant PDF0403175. G.H. is a Helen Hay Whitney postdoctoral fellow. X.L.A. is supported by an N.I.H. Predoctoral fellowship (F31 NS054507). This work was supported by NIH grants to Y.S. (GM071004), J.W.H. (GM54137, AG11085), and a U.S. Army Innovator Award (W81XWH0410197) to S.J.E. S.J.E. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Chong JA, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–57. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- 2.Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–3. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- 3.Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15:500–6. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Westbrook TF, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–48. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 5.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and Its Corepressors Mediate Plasticity of Neuronal Gene Chromatin throughout Neurogenesis. Cell. 2005;121:645–57. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 7.Bai C, et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–74. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 8.Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–19. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 9.Guardavaccaro D, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4:799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 10.Shirogane T, Jin J, Ang XL, Harper JW. SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J Biol Chem. 2005;280:26863–72. doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- 11.Busino L, et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 12.Jin J, et al. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–74. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winston JT, et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13:270–83. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–36. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 15.Wu G, et al. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003;11:1445–56. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- 16.Zhao JJ, et al. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–95. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 17.Kudo Y, et al. Role of F-box protein betaTrcp1 in mammary gland development and tumorigenesis. Mol Cell Biol. 2004;24:8184–94. doi: 10.1128/MCB.24.18.8184-8194.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishikawa S, Jakt LM, Era T. Embryonic stem-cell culture as a tool for developmental cell biology. Nat Rev Mol Cell Biol. 2007;8:502–7. doi: 10.1038/nrm2189. [DOI] [PubMed] [Google Scholar]
- 19.Ying QL, Stavridis M, Griffiths D, Li M, Smith A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol. 2003;21:183–6. doi: 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- 20.Aubert J, et al. Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11836–41. doi: 10.1073/pnas.1734197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bain G, Kitchens D, Yao M, Huettner JE, Gottlieb DI. Embryonic stem cells express neuronal properties in vitro. Dev Biol. 1995;168:342–57. doi: 10.1006/dbio.1995.1085. [DOI] [PubMed] [Google Scholar]
- 22.Fuller GN, et al. Many human medulloblastoma tumors overexpress repressor element-1 silencing transcription (REST)/neuron-restrictive silencer factor, which can be functionally countered by REST-VP16. Mol Cancer Ther. 2005;4:343–9. doi: 10.1158/1535-7163.MCT-04-0228. [DOI] [PubMed] [Google Scholar]
- 23.Su X, et al. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol. 2006;26:1666–78. doi: 10.1128/MCB.26.5.1666-1678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saitoh T, Katoh M. Expression profiles of betaTRCP1 and betaTRCP2, and mutation analysis of betaTRCP2 in gastric cancer. Int J Oncol. 2001;18:959–64. [PubMed] [Google Scholar]
- 25.Noubissi FK, et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature. 2006;441:898–901. doi: 10.1038/nature04839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods
Vectors
cDNAs encoding Flag-tagged human REST wild-type and frame-shift mutant have been previously described4. Point mutations in REST (pCMV-Flag-RESTS1024A, pCMV-Flag-RESTS1027A, pCMV-Flag-RESTS1030A, and pCMV-Flag-RESTS1024/1027/1030A) were generated with Stratagene mutagenesis kit by standard methods. REST cDNAs were cloned into lentiviral vectors pQCXIN (Clontech) and pHAGE-EF1α-puro (parental kindly provided by Richard Mulligan). Vectors encoding the GST-F-box proteins, dnCul1 (residues 1−452), βTRCPΔF, and shRNA targeting human βTRCP1 and βTRCP2 were previously described10,12. dnCul2 (residues 1−427), dnCul3 (residues 1−418), dnCul4 (residues 1−440), dnCul5 (residues 1−441) were prepared by cloning the respective N-terminal Cullin fragments into pcDNA3 (Invitrogen). shRNA sequence targeting both murine βTRCP1 and βTRCP2 (ATCAAGATCTGGGATAAAA) was cloned into pSuper-Retro-Puro (Oligoengine). pCMV-HA-Ub and pCMV-HA-UbK48 are gifts from Vishva Dixit (Genetech). Baculoviral expression vectors were generated in pBlueBac. Baculoviral HA-REST expression vector was generated by using the Gateway System (Invitrogen).
siRNA and DNA transfections
For the initial F-box siRNA screen, 272 siRNAs targeting 68 unique F-box proteins26 (4 siRNAs / F-box gene) were arrayed individually in 96-well format. 293T cells were transfected with 100 nM siRNAs using oligofectamine under standard conditions. On the subsequent day, cells were transfected with CMV-mRFP-REST plasmid DNA using mirus Trans-IT 293 under manufacturer recommended conditions. Cells were analyzed for mRFP fluorescence after 48 hours by flow cytometry. Subsequent testing of all candidate siRNAs was performed in triplicate and normalized to effects on mRFP (2 independent experiments). For interaction experiments, 293T cells were transfected with indictated plasmids with Lipofectamine 2000 (Invitrogen) per manufacturer's recommendations. For neural differentiation experiments, ES cells were plated in gelatin-coated 6-well plates at 0.5 × 106 cells/well and transfected with 100 nM siRNA using Lipofectamine 2000. 24 hours after transfection, cells were replated on gelatin-coated plates, and then cultured in N2B27 medium to induce neural differentiation. Experiments were performed in sextuplicate (error bars +/− s.d.) and are representative of at least 2 independent experiments.
Cell Culture
Human mammary epithelial cells (HMECs) expressing hTERT and SV40 LT (TLMHMECs)4,16 were cultured in mammary epithelial growth medium (MEGM, Cambrex). 293T, U2OS (human osteosarcoma cell line), and NIH3T3 cell lines were maintained in DMEM or McCoy's medium supplemented with 10% FBS and 50 μg/mL gentamycin. Neural stem cells and murine 46C ES cells with Sox1:GFP reporter were kindly provided by Dr. Austin Smith19,20. 46C cells were maintained in Knockout DMEM (Invitrogen) supplemented with 15% FBS, nonessential amino acids, 2 mM glutamine, 10 μM 2-mecaptoethanol, and 1000 units/ml ESGRO (Chemicon) on gelatin-coated tissue culture plates. 46C ES cells were induced to differentiate by culture in serum-free N2B27 medium (1:1 ratio of DMEM/F12 and Neurobasal medium, supplemented with N2 and B27 (Invitrogen))27. Neural stem cells were cultured in DMEM/F12 basal medium supplemented with insulin 25 μg/ml, Transferrin 100 μg/ml, progesterone 6 ng/ml, putrescine 16 μg/ml, sodium selenite 30 nM, BSA 50 μg/ml, FGF-2 10 ng/ml, EGF 10 ng/ml. For embryoid body formation, cells were plated in Corning ultra-low attachment 6-well plates at 5×105 cells per well in ES medium without LIF. Medium was changed every 2 days, and retinoic acid was added from day 4 to day 8 to 1 μM final concentration. Cells were harvested on day 8.
Retroviral infections of HMECs were performed with indicated viral supernatants in the presence of 8 μg/mL polybrene, and transduced cells were selected for resistance to the appropriate drug: puromycin (2.0 μg/mL), neomycin (200 μg/mL). ES cell infections were performed in the presence of 5 μg/mL polybrene and centrifuged for 1 hour (2000 rpm). Anchorage-independent proliferation assays were performed as previously described4. For each assay, the average of at least 3 replicates ± SD is shown. For growth curves, cells were seeded at a density of 5.0 × 104 per well in 6-well plates and cultured in MEGM. Cells were trypsinized and counted at the indicated time points (in triplicate with average ± SD shown). For protein half-life experiments, cells were treated with cycloheximide (100 μg/mL) for the indicated times and lysed in HLB buffer (see below).
Interaction Studies and Immunoblotting
HMECs, U20S, and ES cells were lysed in HLB buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% Triton-X 100, 1 mM DTT, supplemented with phosphatase- and protease-inhibitor cocktails (Calbiochem)) for 20 minutes with sonication. GST-pulldowns and immunoprecipitations were performed in lysis buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, and 1% NP-40. GSH-sepharose beads (15 μL per condition) were pre-washed in lysis buffer and incubated with 400 μg of lysate for 2 hours at 4°C while rocking. Beads were then washed 3−4 times in lysis buffer and bound protein eluted in 2X Laemmli Protein loading buffer containing SDS. Both GSH-bound samples and crude extract were resolved on 4−12% gradient SDS-PAGE gels. IP of endogenous REST and βTRCP was performed with 10 mg of U2OS whole cell lysate and precipitated with βTRCP antibody (Invitrogen 373400). Western blotting was performed with the following antibodies: γREST (Gail Mandel), αVinculin (Sigma,V9131), αRan (Sigma, R4777), αFlag (Sigma-A8592), αHA (Santa Cruz, F7), αGFP (Santa Cruz, FL), αβTRCP (Cell Signaling Technologies), αβTRCP (Invitrogen 373400), αCyclin E (Santa Cruz, HE12), αGST (Cell Signaling, 26H1).
In vitro binding assays utilized 35S-radiolabled recombinant F-box proteins that were in vitro transcribed and translated with T7 polymerase using TNT reticulocyte lysate (Promega) in the presence of 35S-methionine. Phosphorylated and unphosphorylated peptides spanning 14 residues of the REST phosphodegron were synthesized with N-terminal Biotin tags by Tufts Medical School Protein Core Facility (Biotin-N-M-S-E-GS-D-D-S-G-L-H-G-A, Biotin-N-M-pS-E-G-pS-D-D-pS-G-L-H-G-A, as well as singly and doubly phosphorylated peptides as indicated) and dissolved in PBS solution. Biotinylated and phosphorylated IκB-degron peptide is previously described (Winston et al., 1999). Peptide (1 μg) was preincubated with 10 μL of streptavidin agarose beads (Pierce) for 1 hr at 4°C in 400 μL NETN buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40, 10 mM NaF, and 1 mM EDTA. Unbound peptide was washed from the streptavidin beads with NETN buffer and 35S-labeled F-box protein was incubated with the bead slurry for 2 hours at 4°C. Beads were washed 5 times with NETN buffer, and bound proteins were eluted in 2X Laemmli Protein loading buffer containing SDS, resolved on a 4−12% gradient SDS-PAGE gel, and detected by autoradiography. Quantitation of radiolabeled bands was performed using a phosphoscreen with a Typhoon Imager (Amersham) and ImageQuant TL software (Amersham).
In vitro ubiquitination
A mixture of bacculovirus expressing subunits of the SCFβTRCP complex (Flag-βTRCP, Cul1, Skp1, Rbx1) and HA-REST was transduced into exponentially growing SF9 insect cells in Xpress Insect cell medium (BioWhitaker) supplemented with 10% heat-inactivated serum. Forty hours post-transduction the holocomplex was purified at 4°C for 2 hrs through the Flag-βTRCP1 subunit from cleared lysate with 20 μL Flag-M2 beads (Sigma) in NETN buffer containing 50 mM Tris pH7.5, 150 mM NaCl, 0.5% NP-40, 10 mM NaF, 1 mM EDTA, 10 mM β-Glucophosphate, 10 mM NPP, and 0.1 μM okadaic acid. The bound holocomplex was washed three times with lysis buffer, twice with TBS (50 mM Tris pH7.5, 200 mM NaCl), and eluted in two batches with 0.5 μg/mL Flag-M2 peptide (Sigma) for 30 minutes at 4°C in TBS buffer. Elutions were combined and dialysed at 4°C first for 2 hours, then overnight against 500 mL of 50 mM Tris pH 7.5, 200 mM NaCl, 50% glycerol, 1 mM PMSF, and 1 mM DTT.
Eluted SCFβTRCP∼HA-REST holocomplex was incubated with 50 ng UBA1, 200 ng UbcH5, 2 mM ATP, 1 μM Ubiquitin Aldehyde, 50 ng Nedd8-activating enzyme NAE1 (Boston Biochem), 100 ng UbcH12, and 100 ng Nedd8 in ubiquitin reaction buffer containing 50 mM Tris pH7.5, 5 mM KCl, 5 mM NaF, 5 mM MgCl2, 0.5 mM DTT, 25 μM MG132, and energy regeneration mix (20 mM creatine phosphate and 2 μg/mL creatine kinase). The total 10 μL reaction was incubated at 30°C for 90 minutes, and the reaction was terminated with 2X Laemmli Protein loading buffer containing SDS. Reactions were resolved on 6% SDS-PAGE gels, transferred onto nitrocellulose, and immunoblotted with anti-HA.
26. Jin, J. et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 18, 2573−80 (2004).
27. Ying, Q. L. & Smith, A. G. Defined conditions for neural commitment and differentiation. Methods Enzymol 365, 327−41 (2003).