

Abstract
Mice lacking neurotrophin-3 (NT-3) have been shown previously to be born with severe sensory deficits. This study characterizes the developmental course of this deficit in the trigeminal sensory ganglion, which in NT-3 homozygous mutants contains only 35% of the normal number of neurons at birth. At embryonic day 10.5 (E10.5), normal numbers of neurons, as assessed by expression of neurofilament protein and of total cells, are present in the ganglia of mutant homozygotes. During the next 3 d (E10.5–E13.5), virtually all of the deficit develops, after which mutant animals retain only ∼30% the normal number of neurons. Quantification of neuronal and neuronal precursor numbers in normal and mutant animals reveals that neurons are specifically depleted in the absence of NT-3. A deficiency in precursor proliferation is only seen after most of the neuronal deficit has developed. Numbers of apoptotic cells in the ganglia of mutant animals are elevated during this same interval, indicating that the neuronal deficit is caused, in large part, by increased cell death of embryonic neurons.
To determine sources of NT-3 in the trigeminal system, we examined the expression pattern of β-galactosidase in mice, in which lacZ has replaced the NT-3 coding exon. E10.5–E11.5 embryos exhibit intense reporter expression throughout the mesenchyme and epithelia of the first branchial arch. β-galactosidase expression in E13.5 embryos is largely confined to the oral epithelium and the mesenchyme underlying the skin. Throughout the E10.5–E13.5 interval, the trigeminal ganglion and its targets in the CNS do not express reporter activity.
We conclude that NT-3 acts principally as a peripherally derived survival factor for early trigeminal neurons.
Keywords: neuronal development, neurotrophins, sensory neurons, apoptosis, mutant mouse
The neurotrophins are a family of related proteins, including NGF, BDNF, neurotrophin-3 (NT-3), and NT-4/5, required for the survival of many classes of neurons (for review, seeKorsching, 1993). Their major actions are mediated by a family of receptor tyrosine kinases called trkA, B, and C; each neurotrophin also interacts with another receptor, p75NTR (for review, see Bothwell, 1995). Examinations of mice with targeted mutations in the genes encoding neurotrophins or their receptors have revealed specific deficiencies at birth associated with each mutation (for review, see Fariñas & Reichardt, 1996). Characterization of neuronal losses at birth in the dorsal root ganglion has shown that animals lacking NT-3 exhibit complete elimination of proprioceptive neurons (Ernfors et al., 1994; Fariñas et al., 1994; Tessarollo et al., 1994; Tojo et al., 1995), but they additionally suffer severe losses in sensory neuron populations including neurons that are dependent on other neurotrophins postnatally (Fariñas et al., 1994). This suggests that NT-3 is required during embryogenesis for multiple populations of sensory neurons, which is consistent with the widespread expression of NT-3 and its major receptor trkC during embryogenesis (Ernfors et al., 1992; Tessarollo et al., 1993; Lamballe et al., 1994).
Of particular interest is the trigeminal ganglion, where a lack of NT-3 causes a reduction in the number of neurons at birth of >60% relative to wild-type animals. The trigeminal ganglion primarily supplies sensory innervation to derivatives of the first branchial arch including facial skin and the oral cavity. Interestingly, this ganglion does not contain proprioceptive neurons in wild-type animals. Thus, the deficits observed in animals lacking NT-3 must involve other neuronal classes. It is not known, however, how the trigeminal ganglion deficit is generated during the development of these animals. The development of this ganglion and its innervation patterns have been intensely studied as a model system for characterizing the roles of neurotrophins (for review, see Davies, 1994). The temporal and spatial patterns of neurotrophin and neurotrophin receptor expression have been mapped in great detail and correlated with stages of development within the ganglion and in innervation of target fields (Arumae et al., 1993;Buchman and Davies, 1993) (for review, see Davies, 1994).
Previous observations have revealed several actions of NT-3 that might contribute to the development of the trigeminal ganglion in vivo and account for aspects of the deficit observed in NT-3 mutant homozygotes. NT-3 has been shown to accelerate the differentiation of spinal sensory neurons from progenitor cells (Wright et al., 1992). NT-3 has been shown to promote survival of embryonic trigeminal neurons in vitro, some of which later become dependent on other neurotrophins (Buchman and Davies, 1993). NT-3 can act as a survival factor for neuroblasts in vitro (Birren et al., 1993; diCicco-Bloom et al., 1993; Karavanov et al., 1995), and some evidence indicates that it may also do so in vivo(ElShamy et al., 1995; ElShamy and Ernfors, 1996a,b). Finally, studies have shown that NT-3 application in vitro increases the proliferation of sensory neuron precursors (Memberg and Hall, 1995).
In the present work, we evaluate the possible roles of NT-3 by examining the details of development of the trigeminal ganglion in normal and NT-3-deficient mice. We find that the neuronal deficiency in animals lacking NT-3 appears during a comparatively short period of development that coincides with the peak of neurogenesis and axonal innervation of targets. We show that the neuronal deficit is associated with an abnormally high frequency of apoptosis. Examination of NT-3 expression indicates that NT-3 is derived from sources in the surrounding mesenchyme and target fields. The results indicate that the deficit reflects the loss of neurons dependent on obtaining this factor from peripheral sources.
MATERIALS AND METHODS
Mice with a targeted mutation in the NT-3 gene, in which the coding region of the lacZ gene replaces the coding exon for NT-3 (Fariñas et al., 1994), were obtained from our colony and bred out over the C57/Bl6 background. Animals were genotyped by DNA blot analysis as described (Fariñas et al., 1994).
Females in estrus were paired with males overnight and examined for vaginal plugs the following morning. For the purposes of staging embryos, pregnant females were regarded as having conceived at midnight. Some litters were additionally staged using the criteria ofTheiler (1989). Dams were killed by cervical dislocation at noon and the embryos dissected out and placed immediately into Carnoy’s fixative (60% ethanol, 30% chloroform, 10% acetic acid). Embryos were dehydrated, embedded in paraffin, sectioned at 7 μm on a rotary microtome, mounted in series, and stained with cresyl violet.
For immunohistochemistry, sections were rehydrated through a graded series of alcohols. Endogenous peroxidases were quenched using 10 mm Tris, pH 7.5, 150 mm NaCl (TBS) containing 10% methanol and 3% hydrogen peroxide. Sections were rinsed in TBS then blocked in TBS containing 10% normal goat serum, 0.1% Triton X-100 (Sigma, St. Louis, MO), 1% glycine, and 2–3% BSA (Sigma). Primary antibodies were added in blocking solution as follows: rabbit anti-neurofilament (NF)-150 kDa subunit (Chemicon, Temecula, CA, 1:2000); anti-BrdU (Novocastra, 1:100) (see below). Immunoreactivity was detected using the appropriate biotinylated secondary antibody and biotin–avidin–biotin peroxidase reagents from the Vectastain detection kit (Vector Labs, Burlingame, CA), following the manufacturer’s instructions.
Counts of neurons and cells. The trigeminal ganglion was mapped in paraffin series from three mutant and three wild-type animals for each stage analyzed. Every fifth section through the ganglion was photographed at high magnification, and positive profiles containing nucleoli were counted in the resulting montage. No correction was made in the counts for split nucleoli. The numbers of trigeminal precursors in embryos up to stage E13.5 were calculated as the average number of Nissl profiles minus the average number of neurofilament-positive profiles. (Satellite cells are not born until after these stages) (seeAltman and Bayer, 1982.)
Counts of pyknotic profiles. The density of pyknotic profiles was measured in Nissl-stained paraffin sections through the trigeminal of five wild-type and five mutant embryos at E11.5 and E13.5. Widely spaced (by at least 30 μm) sections representing 8–10% of the total cell number were photographed as above, and the number of pyknotic profiles was divided by the total number of cells within the sections. Care was taken to exclude red blood cells (Coggeshall et al., 1994).
Analysis of 5-bromo-2-deoxy-uridine (BrdU) incorporation.Pregnant dams were injected intraperitoneally with BrdU (Sigma) (50 mg/kg body weight) 2 hr before killing. The embryos were dissected, embedded, sectioned, and mounted as above.
Before staining with anti-BrdU antibody, sections were treated with 2N HCl in 0.05 m PBS, pH 7.2, at 37°C for 20 min; neutralized in 0.1 m borate buffer, pH 8.5, for 5 min; washed once in TBS; and then treated with peroxidase/methanol and stained following the protocol for immunohistochemistry described above.
LacZ staining of whole mounts and sections. Embryos up to age E13.5 were fixed for 1–2 hr in ice-cold 2% paraformaldehyde in PBS, pH 7.3. They were then either stained immediately for lacZ activity as whole mounts or frozen and cryosectioned at 10–30 μm for staining of sections.
Sections or whole mounts were placed into X-Gal staining solution (PBS, pH 7.3, containing 2 mm MgCl2, 0.02% NP-40 (Sigma), 0.01% sodium deoxycholate, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, and 1 mg/ml X-Gal (Boehringer, Indianapolis, IN). Specimens were developed overnight with shaking at 37°C, washed extensively with PBS, pH 7.3, and post-fixed in 4% paraformaldehyde in PBS, pH 7.3. Control material never showed color development under these conditions. Sections were then either immunostained for neurofilament (as above) or coverslipped with glycerol and stored.
In pilot experiments, the expression pattern of lacZ was compared in sections through the heads of heterozygous and homozygous mutant animals. No difference was found in the tissue distribution of lacZ product among animals of the different genotypes. Whole mounts were therefore performed on heterozygotes, whereas sections of homozygotes and heterozygotes were studied.
Comparison of maxillary and ophthalmic branches of the trigeminal ganglion. Seven-micron paraffin sections cut in the coronal plane through the heads of E13.5 embryos were immunostained for NF-150 as above. The areas of the ophthalmic and maxillary branches of the trigeminal nerve were measured at the level of the posterior margin of the optic chiasm using the Neurolucida computerized tracing system (Microbrightfield, Colchester, VT).
RESULTS
The NT-3 deficit emerges early in trigeminal development
To analyze the time course of the development of the trigeminal defect in animals lacking NT-3, we counted the numbers of neurons and the trigeminal ganglion of mutant and wild-type embryos at several stages of development. Neurons in E10.5, E11.5, and E13.5 embryos were counted as immunoreactive profiles in series stained for NF-150 kDa protein (NF-150), a ubiquitous early neuronal marker (Cochard and Paulin, 1984) (Fig. 1A,B). Neurons in E15.5 and postnatal day 0 (P0) animals were counted according to morphological criteria in Nissl-stained material. Total cells were counted in Nissl-stained material at E10.5, E11.5, and E13.5.
Fig. 1.
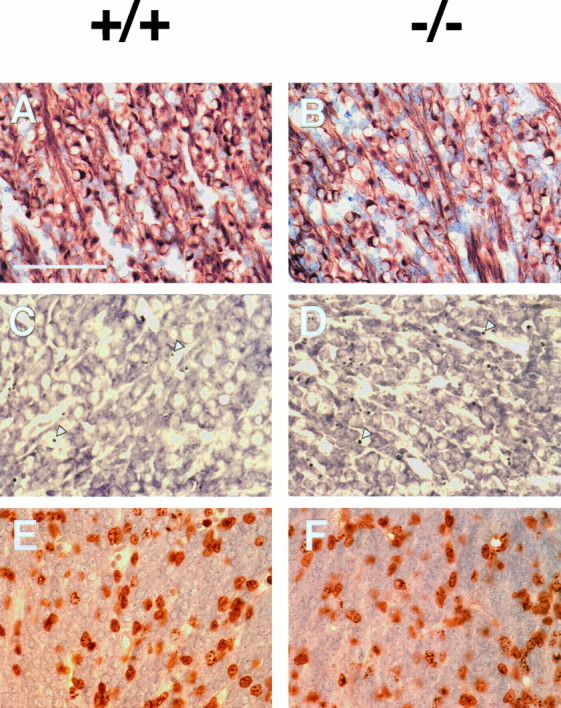
Summary of the trigeminal phenotype in embryos lacking NT-3. For each pair of photographs, representative sections from wild-type (A, C, E) and mutant (B, D,F) animals are compared. These observations are quantitated in Tables 1 and 2. A, B, E13.5 material stained for NF-150 and counterstained for cresyl violet. Neurons are depleted relative to overall trigeminal populations.C, D, E13.5 material stained for cresyl violet. The density of pyknotic profiles is greatly elevated in mutants. Arrowheads indicate red blood cells.E, F, E11.5 material stained for BrdU incorporation by proliferating cells. Proliferation is unchanged at this stage. Scale bar, 100 μm.
Mutant embryos at E10.5 showed no detectable difference in neuronal number when compared with wild-type embryos (Fig. 2, Table 1). Over the next days of development, during which most trigeminal neurons are born and extend axons to their peripheral targets (Davies and Lumsden, 1984), significant and increasing deficits in neuronal number were seen in mutant ganglia. At E11.5, mutants have only 60% as many neurofilament positive profiles as their wild-type counterparts. By E13.5, mutant animals have only 30% of the normal number of neurons. The number of neurons in mutant ganglia, and the deficit, remains roughly constant in size from E13.5 until birth. The trigeminal ganglia in heterozygous animals contained 52,210 ± 6821 neurons at E13.5, which is not significantly different from wild-type animals at this stage (wild-type embryos contain 48,755 ± 3943 neurons at E13.5). This suggests that NT-3 is not present in limiting amounts with respect to the survival of these neurons at this stage.
Fig. 2.

Numbers of trigeminal neurons (squares) and precursor cells (diamonds) in wild-type (filled symbols) and NT-3 (open symbols) animals during development. Neurons in E10.5, E11.5, and E13.5 animals were counted as profiles immunopositive for NF-150 kDa protein. Precursors for those stages were calculated as total (Nissl) cells minus neurons. E15.5 and P0 neurons were identified on the basis of morphology in Nissl material. The mean ± SD of counts from three separate animals are shown for each point plotted.
Table 1.
Subpopulations in the trigeminal ganglion in wild-type and NT-3 deficient mice
| E10.5 | E11.5 | E13.5 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Wild type | Mutant | Wild type | Mutant | Wild type | Mutant | ||||
| Neurons | 6093 ± 2065 | 5375 ± 1738 | 88% | 25545 ± 3562 | 14743 ± 5513* | 58% | 48755 ± 3943 | 15217 ± 1023‡ | 31% |
| Total cells | 24733 ± 1389 | 25542 ± 8151 | 100% | 61228 ± 10914 | 46270 ± 13104 | 77% | 82027 ± 12216 | 39192 ± 6604† | 48% |
| Precursors | 18640 ± 2488 | 20167 ± 8334 | 100% | 36833 ± 11480 | 31527 ± 14216 | 88% | 33272 ± 12836 | 23975 ± 6682 | 72% |
| % Neurons | 25 | 21 | 42 | 32 | 59 | 39 | |||
Percentages following entries for mutant animals show the mutant population as a percentage of wild type. Neurons is reported as the mean ± SD of counts of cells immunopositive for NF-150 in three animals for each group. Total cells is reported as the mean ± SD of counts of Nissl profiles in three animals for each group. Precursors is calculated as neurons minus total cells for each group. Uncertainty is calculated via propagation of errors. % Neurons is calculated as the percentage of neurons relative to total cells.
p < 0.05 (two-tailed Student’s t test);
p < 0.01;
p < 0.001.
To quantitate the numbers of precursor cells between E11.5 and E13.5 in normal and homozygous mutant embryos, we estimated the numbers of this population by subtracting neuronal numbers from the total numbers of cells present in the ganglion. (The major class of non-neuronal cells in the adult ganglion, the satellite cells, are not born until after these stages) (see Altman and Bayer, 1982) (see Discussion.) At E10.5 and E11.5 (Fig. 2; Table 1), the number of trigeminal precursor cells is similar in mutants and wild-type embryos. An ∼30% reduction in precursor numbers is seen in E13.5 mutant animals, although this difference is not statistically significant. This deficit occurs later in development compared with the defect in neurons, which is already substantial at E11.5 and is essentially complete at E13.5. Therefore, the absence of NT-3 affects neurons earlier and more severely than precursor cells. Neurons consequently represent a smaller fraction of all cells compared with wild type at both stages (Compare Fig. 1,A and B; Table 1).
Cell death is elevated in NT-3 mutants
The trigeminal ganglia of E10.5 mutant embryos contain normal numbers of cells and neurons. The subsequent deficit therefore could be attributable to a failure of precursors to proliferate or an increase in cell death. To investigate the latter possibility, we measured the density of pyknotic profiles at E11.5 and E13.5 (Table2). We found substantial numbers of pyknotic profiles in all stages and genotypes during the interval over which the deficit is occurring (data not shown). We found a significant, approximately twofold increase in density of pyknotic profiles in E11.5 mutants relative to wild type and a highly significant 2.5-fold increase at E13.5 (see Fig. 1C,D). Thus, the emergence of the neuronal deficit correlates with an abnormally high rate of cell death in mutant animals.
Table 2.
Proliferation and pyknosis in the trigeminal ganglion of wild-type and NT-3 deficient mice
| E11.5 | E13.5 | |||
|---|---|---|---|---|
| Wild type | Mutant | Wild type | Mutant | |
| BrdU+ | 10878 ± 2548 | 9047 ± 368 | 4528 ± 381 | 1820 ± 281† |
| % BrdU+ | 30.4 ± 0.7 | 28.7 ± 1.1 | 13.6 ± 1.1 | 7.6 ± 1.1† |
| % Pyknotic | 3.2 ± 1.5 | 6.1 ± 2.5* | 3.0 ± 0.5 | 8.2 ± 2.5† |
BrdU+ is reported as the mean ± SD of counts of cells immunopositive for BrdU incorporation in three animals in each group. % BrdU+ is calculated as the mean percentage ± SD of BrdU-positive cells relative to numbers of precursors. % Pyknotic is reported as the mean ± SD of the percentage of pyknotic profiles relative to total cells for five animals in each group.
p < 0.05, one-tailed Student’s t test;
p < 0.01.
To examine possible effects of the NT-3 deficiency on precursor proliferation, we also determined the number of cells that incorporate BrdU at different stages in normal and homozygous mutant animals (Table2, Fig. 2E,F). At E11.5, there is no significant difference in numbers of BrdU-positive cells between wild-type and mutant embryos. At E13.5, we observed a reduction in total numbers of BrdU-positive profiles in homozygous mutants; when this was normalized to the independently measured numbers of precursor cells, we found a highly significant, approximately twofold reduction in the intrinsic rate of proliferation. This indicates a change of precursor populations subsequent to elimination of neurons. Because neurogenesis is almost complete in mice by this stage (see Altman and Bayer, 1982), these changes seem unlikely to substantially reduce the final neuronal number but may reduce glial numbers.
NT-3 is expressed throughout the periphery of the trigeminal system
We took advantage of the β-galactosidase reporter construct inserted into the NT-3 locus to analyze the expression of NT-3 in the early trigeminal system by staining whole mounts of embryos using lacZ histochemistry (Fig. 3). Although our results provide greater resolution than published in situ hybridization studies (Arumae et al., 1993) (for review, see Davies, 1994), they are consistent with the patterns of expression found in those studies. We additionally stained some embryos using TuJ1 (Easter et al., 1993), a monoclonal antibody that recognizes the neuron-specific β3 isoform of tubulin to reveal axonal projections (Fig. 3B). At E9.5, when the first trigeminal neurons are born and begin to extend neurites (Davies and Lumsden, 1984; Easter et al., 1993), lacZ expression is already detectable in the first branchial arch (Fig.3A, arrow), the presumptive target of most trigeminal neurons. The midline of the roof of the mesencephalon is also intensely stained. By E10.5 (Fig. 3B), the reporter expression has greatly intensified and spread throughout the developing mandibular and maxillary processes. The olfactory pit (data not shown) and eye cup are stained to intermediate intensity. TuJ1 counterstaining reveals that the anterior margin of the trigeminal ganglion is surrounded by NT-3 expressing cells. At E11.5 (Fig. 3C), the expression pattern changes, with more intense expression toward the distal half of the maxillary (Fig. 3C, Mx) and mandibular processes. This trend is continued in E13.5 embryos, in which expression in the maxillary territory is most intense in the mystacial pad (Fig. 3D, MP) and distinctly less in the surrounding regions. The nostrils, ears, and mesenchyme surrounding the eyeball remain intensely stained. This pattern of staining remains similar for several days after E13.5 (data not shown) (see also Tojo et al., 1995).
Fig. 3.

Top. Changes in expression of a lacZ reporter construct inserted into the NT-3 locus. Whole mounts of heterozygous animals (see text). A, E9.5: staining is observed in the first branchial arch (arrow) and the anterior (a) and posterior (p) neuropores of the mesencephalon. B, E10.5 counterstained for TuJ1 to show axonal projections. The lacZ reaction was underdeveloped to allow visualization of axons. C, E11.5 embryo. Expression of NT-3 is strongest at the distal half of the maxillary process (Mx). D, E13.5 embryo. Reporter expression in the maxillary territory is strongest in the mystacial pad (MP) and distinctly less elsewhere.
To examine the trigeminal system in greater detail, we also stained cryostat sections from embryos with one or two copies of the gene replacement for lacZ activity, alone or in conjunction with NF-150 immunohistochemistry (Fig. 4). In E11.5 animals (Fig.4A,B), nearly every cell in the distal ends of the maxillary and mandibular processes, including the presumptive epidermis (Fig. 4A, ep), was stained. Lineage tracing studies (Trainor and Tam, 1995, and references therein) have shown that these structures are populated by cells of three different developmental origins that occupy segregated final locations within the arch. The epithelial lining is derived from ectoderm, whereas the subadjacent mesenchyme is derived from cells of neural crest origin. The deep mesenchymal (Fig. 4A,mch) interior is derived from paraxial mesoderm. The staining seen throughout the depth of these structures at this stage indicates that cells of all three lineages express NT-3 at this time. Thus, axons approaching their cutaneous targets are bathed in NT-3 along their entire trajectory (Fig. 4B). We found staining in other trigeminal targets, including the corneal ectoderm, the nostrils, and the oral epithelia (data not shown). In E12.5 animals (Fig. 4C), expression appears to be reduced in the presumptive epidermis, while remaining intense throughout the mesenchyme. By E13.5 (Fig.4E,F), reporter expression is no longer detectable in ectodermally derived targets such as the epidermis, instead being confined to mesenchyme adjacent to the epidermis (Fig. 4E), hair follicles (Fig.4F), and the eyeball (data not shown). Thus, NT-3 is transiently and intensely expressed in trigeminal target tissues from E11.5 to E13.5, which correlates with the developmental interval over which trigeminal neurons are lost in mutants.
Fig. 4.
Bottom. Changes in lacZ reporter expression during development (E,F). A, Maxillary process of an E11.5 embryo homozygous mutant. Intense reporter expression is seen in the presumptive epidermis (ep) and in the superficial (derived from neural crest) and deep (derived from mesoderm) mesenchyme (mch). B, E11.5, lower magnification of the maxillary process, immunostained for NF-150 to reveal axons. Axons approaching their cutaneous targets encounter NT-3-producing cells along their entire trajectory. Homozygous mutant. C, Maxillary process of an E12.5 homozygous mutant embryo. Intense expression persists in the mesenchyme, but the skin staining begins to weaken. D, Vicinity of the trigeminal in an E12.5 homozygous mutant embryo. NT-3 is expressed by mesenchyme surrounding the anterior edge of the trigeminal and by a few non-neuronal cells (arrows) within the ganglion. E,F, Mystacial pad of an E13.5 heterozygote sectioned at 20 μm. Expression is confined to the mesenchyme underlying the skin (E) and surrounding whisker follicles (F). Scale bar for each image, 50 μm.
In sections from embryos of stages E11.5–E13.5, the vast majority of trigeminal cells are negative for reporter expression (Fig.4D). Although others have shown expression of NT-3 in a small number of sensory neurons at later stages as assessed by a similar lacZ reporter (Tojo et al., 1996), we find no evidence for neuronal expression at the earlier stages during which the deficits in neuronal number are emerging in the trigeminal ganglia of animals lacking NT-3. We do find a very small number of positive cells, none of them neurons, within the ganglion at these stages (Fig.4D, arrows). In agreement with other reports (Arumae et al., 1993; Buchman and Davies, 1993), we find no evidence of NT-3 expression in the CNS targets of the trigeminal ganglion neurons (data not shown).
The mystacial pad of the snout, which is the most densely innervated cutaneous target in the adult mouse, is derived during development from the first branchial arch (Fig.3A,C, D,arrows). This structure expresses very high levels of NT-3 during the developmental interval over which trigeminal neurons are abnormally lost in mice lacking NT-3 (Buchman and Davies, 1993) (present results). We wondered whether NT-3 might be especially important within the first branchial arch for the maintenance of immature trigeminal neurons. If so, then the neurons innervating this tissue would be more affected by the absence of NT-3 than neurons supplying regions that express less NT-3. To test this idea, we compared the effect of the NT-3 mutation on the cross-sectional area of the ophthalmic nerve, which supplies areas outside the first branchial arch, which express much lower levels of NT-3 (Buchman and Davies, 1993) (see Fig. 3), and the maxillary nerve, which supplies the maxillary process of the first branchial arch including the presumptive mystacial pad.
We measured the cross-sectional area of the ophthalmic and maxillary nerves in paraffin sections cut in the coronal plane through E13.5 embryos in wild-type, heterozygous, and mutant embryos. (Fig.5, see Table 3) We found that neither nerve trunk was significantly reduced in area in heterozygous animals. However, the ophthalmic nerve in homozygous mutants was reduced by 60% in cross-sectional area, whereas the maxillary nerve was reduced by 50% (Table 3). This result suggests that neurons supplying targets outside the first branchial arch are depleted to a similar extent to neurons supplying targets within the arch. Thus, the intense expression of NT-3 in the first branchial arch likely does not correspond to a differential requirement by trigeminal neurons innervating those territories for NT-3.
Fig. 5.

Comparison of the peripheral branches of the trigeminal nerve in wild-type and mutant mice. A, Schematic drawing of an E13.5 embryo (adapted from Theiler, 1989) indicating the plane of section used for the camera lucida analysis. The thin ophthalmic branch (dorsal) and the thick maxillary branch (middle) were compared in wild-type, heterozygous, and mutant mice. The mandibular branch (ventral) travels obliquely to this plane and was not analyzed. ON, Optic nerve. B, Camera lucida drawing showing the ophthalmic and maxillary branches of the trigeminal nerve in a wild-type embryo. The ophthalmic nerve ishatched; the maxillary fascicles arecross-hatched. OX, Optic chiasm.C, Camera lucida drawing of the same complex in a mutant animal. The ophthalmic (hatched) and maxillary (cross-hatched) branches of the ganglion are both depleted (see Table 3). Scale bars, 100 μm.
Table 3.
Cross-sectional area (in μm2) of the maxillary and ophthalmic branches of the trigeminal in wild-type, heterozygous, or homozygous mutant mice
| +/+ | +/− | −/− | |
|---|---|---|---|
| Maxillary | 8486 ± 227 | 8564 ± 1630 | 4650 ± 16043-151 |
| Ophthalmic | 1958 ± 223 | 1761 ± 190 | 766 ± 2203-151 |
Each entry represents the mean ± SD of the measurements on three animals in E13.5 animals. Plane of section is indicated in Figure5A.
F3-151: p < 0.01, one-tailed Student’s t test.
DISCUSSION
Results presented in this paper show that the deficiency in neuronal numbers seen in the trigeminal ganglion in animals lacking NT-3 emerges rapidly over a short interval in the development of the animal. This period, between stages E10.5 and E13.5, is characterized in homozygous mutants by a progressive depletion of neurons from the pool of all trigeminal cells relative to wild-type embryos. During these stages, apoptotic cell death is elevated in the trigeminal ganglion of mutants relative to wild type; whereas neither the incorporation of BrdU into proliferating trigeminal cells nor the numbers of trigeminal precursor cells is reduced in mutants until after the birth of most neurons. Our lacZ-based assay for NT-3 expression shows that NT-3 is produced throughout the periphery of the trigeminal system but not within the ganglion or the CNS targets of ganglion neurons. We conclude that the neuronal defect seen in the trigeminal ganglion of mutant mice is attributable to the abnormal cell death of neurons. Thus, NT-3 acts in wild-type mice as a peripherally derived survival factor for early trigeminal ganglion neurons.
Neurons and precursors in the developing trigeminal ganglion
NT-3 has been proposed to perform a variety of actions in the developing nervous system (for review, see Korsching, 1993). To evaluate these potential roles, it was necessary for us to track the dynamics of both the neuronal pools and the precursor pools that give rise to them. As discussed below, we believe our measurements, centering around the expression of NF-150 by trigeminal neurons, can yield an accurate assessment of the relative dynamics of these two populations in the early ganglion.
We chose expression of NF-150, a member of the family of neuron-specific, middle molecular mass, intermediate-filament proteins, as our benchmark for identifying trigeminal neurons. During embryogenesis, these proteins are expressed by all sensory neurons, with the onset of expression at the cellular level concomitant with initial axonogenesis by recently born sensory neurons (Cochard and Paulin, 1984). Thus, although our counts based on NF-150 may omit some cells in the earliest stages of commitment to neuronal fate, it unambiguously identifies all neurons from an early stage.
Our observations indicate that the temporal expression of NF-150 by early trigeminal ganglion neurons is not disrupted in mice lacking NT-3. We find normal numbers of NF-150 expressing cells in E10.5 mutants, indicating the appropriate temporal expression of this antigen in the absence of NT-3 by the earliest trigeminal neurons. Additionally, our counts of total cell numbers at E11.5 and E13.5 (Table 1) suggest that the deficiencies seen in neurofilament counts of mutant embryos at these stages reflect the absence of cells rather than delay or failure of neurons to express NF-150 in mutant animals. If the absence of NT-3 were to cause a delay in the expression of NF-150, one might expect to see increased numbers of neurofilament-negative cells in mutant animals relative to wild-type animals. Instead, we find that the difference between wild-type and mutant embryos in total cell numbers is equal to or greater than the difference in neuronal counts at both E11.5 and E13.5. Thus, we conclude that the neurons are actually absent in mutants rather than present and failing to express NF-150. Finally, consistent with this conclusion, the development of the peripheral projections of trigeminal neurons in material stained for NF-150 appears to be temporally appropriate in mutant embryos (data not shown). Thus, NF-150 appears to mark an equivalent population of neurons in both wild-type and mutant embryos.
We selected the term “precursor” to designate members of the population (or populations) of proliferating cells in the trigeminal ganglion of midgestation embryos that give rise to the mature cell types found in the adult ganglion (i.e., principally neurons and satellite cells). Neurogenesis studies indicate that neurons are born from this pool over stages E9.5–E13.5, with a peak over stages E11–E12 (see Altman and Bayer, 1982). We believe that the quantification of the number of trigeminal ganglion cells that do not express neurofilament indeed provides a good estimate of the numbers of precursors at the stages examined for the purposes of this study. Cell cycle studies in early sensory ganglia show that essentially all neurofilament-negative cells can be labeled in E11.5 animals by repeated pulses of BrdU over a 9 hr period (I. Fariñas and L. Reichardt, unpublished observations), indicating that all of the neurofilament-negative cells counted in this study are indeed proliferating. By considering the numbers and proliferative properties of this pool at stage E11.5, we can appraise whether neurons are being born in normal numbers in mutant animals (see below).
NT-3 directly affects the neuronal population
In interpreting our results, we find no evidence for an effect of this mutation on the earliest events of gangliogenesis, because the E10.5 ganglion in mutant animals contains normal numbers of trigeminal cells and neurons. However, after this initial stage, we find a rapid depletion of the number of neurons in mutants relative to wild type. This decrease, seen in the absence of a detectable effect on the precursor population during neurogenesis, suggests that NT-3 acts directly on the neuronal population. Moreover, the increase in cell death seen at these stages strongly suggests that the loss of neurons is attributable to neuronal apoptosis.
Where is the NT-3 required by trigeminal neurons produced? The expression pattern of our lacZ reporter gene indicates that at E11.5, NT-3 is available along the entire trajectory of maxillary axons, including final target regions. We do not observe staining within the ganglion, but it is possible that NT-3 is available within the ganglion via diffusion from nearby mesenchymal cells (Figs. 3B,4D). The onset of the deficit in the trigeminal ganglion occurs between ages E10.5 and E11.5, a time span during which the first trigeminal axons are beginning to reach their targets (Figs.4B, 5B) (see also Davies and Lumsden, 1984). This suggests that the earliest trigeminal neurons may not require NT-3 for survival until their axons have reached the vicinity of their targets. Alternatively, the onset of the deficit in mutants might reflect a requirement for NT-3 by later born neurons. One prediction of this alternative idea is that the earliest neurons would be relatively unaffected by the loss of NT-3. This could be determined via a detailed neurogenesis study comparing wild type with mutant animals.
Developmental studies of trigeminal neurons in vitro have demonstrated a switchover in the neurotrophin dependence of early trigeminal neurons (Buchman and Davies, 1993). Cultures of early trigeminal neurons can survive in the presence of NT-3, BDNF, or NT-4, whereas most neurons from cultures of later trigeminal ganglia survive only in the presence of NGF. The switchover is observed in cultures from E11–E13 mice, which corresponds well to the stages at which we see neuronal losses in vivo in mice lacking NT-3. This switchover further corresponds to a period of rapid increase in NGF expression in the presumptive epidermis at the times (stages E12–E13) that we see a relative decrease in the expression of NT-3 in that tissue (Davies et al., 1987) (for discussion, see Davies, 1994). This raises the intriguing possibility that the survival of trigeminal neurons depends on obtaining NT-3 and NGF in sequence from the same tissue. Because the size of the trigeminal ganglion deficit is very large in animals lacking NT-3 and in animals lacking NGF, at least some trigeminal neurons must require both factors for survival (for discussion, see Fariñas and Reichardt, 1996). Whereas NGF and NT-3 use trkA and trkC, respectively, as their primary receptors, trigeminal neurons in this situation might, in fact, respond to both factors via the identical receptor, namely, trkA. trkA is expressed very early by most trigeminal neurons (Arumae et al., 1993) (unpublished observations), and both NT-3 and NGF are capable of signaling through this receptor (Clary and Reichardt, 1994; Davies et al., 1995). However, trigeminal neurons in cultures from stage E11–E13 animals do not survive in identical numbers in response to NT-3 and NGF (Buchman and Davies, 1993), suggesting complexities in the ligand-receptor system in the trigeminal ganglion. Both differential splicing of trkA and its coexpression with p75NTR have been shown to modulate the efficacy of trkA activation by NT-3 (Clary and Reichardt, 1994). Either mechanism might explain the changes in relative sensitivity of trigeminal neurons to NT-3 compared with NGF seen over this interval. Evaluation of this hypothesis would require additional study of the relationship between the extension of peripheral trigeminal axons in vivo and the expression of functional NT-3 receptors. In any event, the fact that the phenotypein vivo of mice lacking NT-3 results in incomplete elimination of trigeminal neurons suggests that other survival factors could be supporting these neurons at this stage.
Late disruption in precursor populations
It seems likely that lack of NT-3 does not primarily affect the generation of neurons by precursor cells during the interval over which the neuronal deficit is emerging. Our quantification of precursor numbers (Table 1, Fig. 2) indicates that these cells are present in normal numbers in mutant mice at E11.5 and that most of these cells survive throughout the interval of the deficit in the absence of NT-3. Consequently, NT-3 does not appear to be an essential survival factor for the majority of precursors in vivo. In addition, the numbers of trigeminal cells incorporating BrdU is not diminished in mutant animals at this stage (Table 2). Thus, precursor cells are not significantly diminished in numbers or slowed in proliferation by the absence of NT-3 at E11.5 when neurogenesis is at its maximum (seeAltman and Bayer, 1982). This suggests that neurons are being generated in normal numbers at this stage. Therefore, the simplest inference from these findings is that the neuronal defect seen in the trigeminal ganglion of E11.5 mutants does not arise primarily from a diminished generation of neurons by precursors. Instead, the progressive decrease of neurons as percentage of all cells in mutants, relative to wild-type embryos, indicates that the lack of NT-3 directly affects neuronal numbers via the neurons themselves.
Although not seen in earlier embryos, we do observe a change in the intrinsic rate of proliferation of precursors at E13.5. (Table 2). Because this change occurs after the onset of the neuronal deficiencies, which are already seen in E11.5 animals, we cannot determine whether they represent direct or indirect effects of the absence of NT-3. By E13.5, almost all neurons have been born, and the changes seen in E13.5 animals in themselves could reflect decreased generation of satellite cells (see Altman and Bayer, 1982). Consistent with this hypothesis, NT-3 has been shown to act as a mitogen for oligodendrocyte precursors (Barres et al., 1994). Therefore, the proliferative changes seen at E13.5 probably do not contribute substantially to the deficit in neuronal numbers.
Our results provide strong evidence that neurons are the population most affected by the absence of NT-3 in the developing trigeminal ganglion. Our quantification of neuronal and non-neuronal pools clearly shows that the former pool is specifically depleted in animals lacking NT-3 in the absence of effects on precursor populations. After the initial review of this paper, another study (ElShamy and Ernfors, 1996b) was published that argues that precursors, not neurons, are the cells primarily affected in the absence of NT-3. They observe colabeling of BrdU label with some TUNEL-positive cells and observe an increased proportion, versus wild type, of TUNEL-positive cells colabeled with anti-nestin, a marker for neural progenitors.
The two sets of observations might be reconciled if the neurons that die in the trigeminal ganglion of NT-3 knock-out mice at E11.5 have committed to do so shortly after their final mitosis. A 5–6 hr BrdU labeling protocol was used in their experiments, and other markers for progenitor cells have been shown to persist in newborn neurons (Cochard and Paulin, 1984; Memberg and Hall, 1995). However, in our analysis of development of the deficiency in the dorsal root ganglia of NT-3 mutant mice, we also have evidence indicating that there may be methodological problems associated with the nonstandard BrdU labeling protocol used by these authors (I. Fariñas, C. Yoshida, C. Backus, and L. Reichardt, unpublished observations).
In conclusion, our results indicate that lack of NT-3 results in abnormal elimination of trigeminal neurons early in development, with a subsequent disruption of precursor populations, possibly as an indirect effect of the loss of neurons. Thus, NT-3 acts as a peripherally derived survival factor for these neurons in the wild-type mouse. Further understanding of the cellular events associated with the neuronal requirement for NT-3 will require additional investigation of the relationship between the defects seen in knock-out mice and the expression of functional NT-3 receptors by trigeminal neurons.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant MH 48200 and The Howard Hughes Medical Institute. G.W. received support from NIH Training Grant GM 07449. I.F. is the recipient of a Human Frontier Science Program Fellowship. L.F.R. is an Investigator of The Howard Hughes Medical Institute. We thank Dr. Peter O’Hara for use of his Neurolucida setup and Larry Ackerman for help with photography. G.W. wishes to dedicate this work to the memory of his mother.
Correspondence should be addressed to Dr. Reichardt, P.O. Box 0724/HHMI, University of California, San Francisco, 513 Parnassus, San Francisco, CA 94143-0724.
REFERENCES
- 1.Altman J, Bayer S. Development of the cranial nerve ganglia and related nuclei in the rat. Adv Anat Embryol Cell Biol. 1982;74:1–90. doi: 10.1007/978-3-642-68479-1. [DOI] [PubMed] [Google Scholar]
- 2.Arumae U, Pirvola U, Palgi J, Kiema T-R, Palm K, Moshnyakov M, Ylikoski J, Saarma M. Neurotrophins and their receptors in rat trigeminal system during maxillary nerve growth. J Cell Biol. 1993;122:1053–1065. doi: 10.1083/jcb.122.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barres BA, Raff MC, Gaese F, Bartke I, Dechant G, Barde Y-A. A crucial role for neurotrophin-3 in ologodendrocyte development. Nature. 1994;367:371–375. doi: 10.1038/367371a0. [DOI] [PubMed] [Google Scholar]
- 4.Birren SJ, Lo LC, Anderson DJ. Sympathetic neurons undergo a developmental switch in trophic dependence. Development. 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- 5.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 6.Buchman VL, Davies AM. Different neurotrophins are expressed and act in developmental sequence to promote the survival of embryonic sensory neurons. Development. 1993;118:989–1001. doi: 10.1242/dev.118.3.989. [DOI] [PubMed] [Google Scholar]
- 7.Clary DO, Reichardt LF. An alternatively spliced form of the nerve growth factor receptor trkA confers an enhanced response to neurotrophin-3. Proc Natl Acad Sci USA. 1994;91:11133–11137. doi: 10.1073/pnas.91.23.11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cochard P, Paulin D. Initial expression of neurofilaments and vimentin in the central and peripheral nervous system of the mouse embryo in vivo. J Neurosci. 1984;4:2080–2094. doi: 10.1523/JNEUROSCI.04-08-02080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coggeshall RE, Pover CM, Fitzgerald M. Dorsal root ganglion cell death and surviving cell numbers in relation to the development of sensory innervation in the rat hindlimb. Dev Brain Res. 1994;82:193–212. doi: 10.1016/0165-3806(94)90163-5. [DOI] [PubMed] [Google Scholar]
- 10.Davies AM. The role of neurotrophins in the developing nervous system. J Neurobiol. 1994;25:1334–1348. doi: 10.1002/neu.480251103. [DOI] [PubMed] [Google Scholar]
- 11.Davies AM, Lumsden A. Relation of target encounter and neuronal death to nerve growth factor responsiveness in the developing mouse trigeminal system. J Comp Neurol. 1984;223:124–127. doi: 10.1002/cne.902230110. [DOI] [PubMed] [Google Scholar]
- 12.Davies AM, Bandtlow C, Heumann R, Korsching S, Rohrer H, Thoenen H. Timing and site of nerve growth factor synthesis in developing skin in relation to innervation and expression of the receptor. Nature. 1987;326:353–358. doi: 10.1038/326353a0. [DOI] [PubMed] [Google Scholar]
- 13.Davies AM, Minichiello L, Klien R. Developmental changes in NT3 signalling via trkA and trkB in embryonic neurons. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiCicco-Bloom E, Friedman WJ, Black IB. NT-3 stimulates sympathetic neuroblast proliferation by promoting precursor survival. Neuron. 1993;11:1101–1111. doi: 10.1016/0896-6273(93)90223-e. [DOI] [PubMed] [Google Scholar]
- 15.Easter SS, Ross LS, Frankfurter A. Initial tract formation in the mouse Brain. J Neurosci. 1993;13:285–299. doi: 10.1523/JNEUROSCI.13-01-00285.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ElShamy WM, Ernfors P. A local action of neurotrophin-3 prevents the death of proliferating sensory neuron precursor cells. Neuron. 1996a;16:963–972. doi: 10.1016/s0896-6273(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 17.ElShamy WM, Ernfors P. Requirement of neurotrophin-3 for the survival of proliferating trigeminal cells. Development. 1996b;122:2405–2414. doi: 10.1242/dev.122.8.2405. [DOI] [PubMed] [Google Scholar]
- 18.ElShamy WM, Linnarsson S, Lee K-F, Jaenisch R, Ernfors P. Prenatal and postnatal requirements of NT-3 for sympathetic neuroblast survival and innervation of specific targets. Development. 1995;122:491–500. doi: 10.1242/dev.122.2.491. [DOI] [PubMed] [Google Scholar]
- 19.Ernfors P, Merlio J-P, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- 20.Ernfors P, Lee K-F, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 21.Fariñas I, Reichardt LF. Neurotrophic factors and their receptors: implications of genetic studies. Semin Neurosci. 1996;8:133–143. [Google Scholar]
- 22.Fariñas I, Jones KR, Backus C, Wang X-W, Reichardt LF. Targeted mutation of the neurotrophin-3 gene results in severe sensory and sympathetic deficits. Nature. 1994;369:658–661. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- 23.Karavanov A, Sainio K, Palgi J, Saarma M, Saxen L, Sariola H. Neurotrophin-3 rescues neuronal precursors from apoptosis and promotes neuronal differentiation in the embryonic metanephric kidney. Proc Natl Acad Sci USA. 1995;92:11279–11283. doi: 10.1073/pnas.92.24.11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamballe F, Smeyne RJ, Barbacid M. Developmental expression of trkC, the neurotrophin-3 receptor, in the mammalian nervous system. J Neurosci. 1994;14:14–28. doi: 10.1523/JNEUROSCI.14-01-00014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Memberg SP, Hall AK. Proliferation, differentiation, and survival of rat sensory neuron precursors in vitro require specific trophic factors. Mol Cell Neurosci. 1995;6:323–335. doi: 10.1006/mcne.1995.1025. [DOI] [PubMed] [Google Scholar]
- 27.Tessarollo L, Tsoulfas P, Martin-Zanca D, Gibert D, Jenkins NA, Copeland NG, Parada LF. TrkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- 28.Tessarollo L, Vogel KS, Palko ME, Reid SW, Parada LF. Targeted mutation in the neurotrophin-3 gene results in loss of muscle sensory neurons. Proc Natl Acad Sci USA. 1994;91:11844–11848. doi: 10.1073/pnas.91.25.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Theiler K. The house mouse: atlas of embryonic development. Springer; New York: 1989. [Google Scholar]
- 30.Tojo H, Kaisho Y, Nakata M, Matsuoka K, Kitagawa M, Abe T, Takami K, Yamamoto M, Shino A, Igarishi K, Aizawa S, Shiho O. Targeted disruption of the neurotrophin-3 gene with lacZ induces loss of trkC-positive neurons in sensory ganglia but not in spinal cords. Brain Res. 1995;669:163–175. doi: 10.1016/0006-8993(94)01219-8. [DOI] [PubMed] [Google Scholar]
- 31.Tojo H, Takami K, Kaisho Y, Nakata M, Abe T, Shiho O, Igarashi K. Analysis of neurotrophin-3 expression using the lacZ reporter gene suggests its local mode of neurotrophic activity. Neuroscience. 1996;71:221–230. doi: 10.1016/0306-4522(95)00445-9. [DOI] [PubMed] [Google Scholar]
- 32.Trainor PA, Tam PPL. Cranial paraxial mesoderm and neural crest cells of the mouse embryo: co-distribution in the craniofacial mesenchyme but distinct segregation in branchial arches. Development. 1995;121:2569–2582. doi: 10.1242/dev.121.8.2569. [DOI] [PubMed] [Google Scholar]
- 33.Wright EM, Vogel KS, Davies AM. Neurotrophic factors promote the maturation of developing sensory neurons before they become dependent on these factors for survival. Neuron. 1992;9:139–150. doi: 10.1016/0896-6273(92)90229-7. [DOI] [PubMed] [Google Scholar]