

Abstract
To investigate the role of p185her2/neu / ErbB3 signaling in pituitary tumor function we examined these receptors in human prolactinomas. Immunofluorescent p185her2/neu was detected in almost all (7/8), and ErbB3 expression in a subset (4/8) of tumors (7 adenomas and one carcinoma). Quantitative PCR also showed abundant ErbB3 mRNA in tumor specimens derived from a rarely encountered prolactin-cell carcinoma. Activation of p185c-neu / ErbB3 signaling with heregulin, the ErbB3 ligand, in rat lactosomatotroph (GH4C1) tumor cells specifically induced prolactin (PRL) mRNA expression ~ 5-fold and PRL secretion ~ 4-fold, while growth hormone (GH) expression was unchanged. Heregulin (6 nM) induced tyrosine phosphorylation and ErbB3 and p185c-neu heterodimerization, with subsequent activation of intracellular ERK and Akt. The Akt signal was specific to ErbB3 activation by heregulin, and was not observed in response to EGF activation of EGFR. Gefitinib, the tyrosine kinase inhibitor, suppressed heregulin-mediated p185c-neu / ErbB3 signaling to PRL. Heregulin induction of PRL was also abrogated by transfecting cells with siRNA directed against ErbB3. Pharmacological inhibition of heregulin-induced PI3K / Akt (with LY294002) and ERK (with U0126) signaling, as well as siRNA-mediated MAPK1 downregulation showed ERK signaling as the primary transducer of heregulin signaling to PRL. These results demonstrate ErbB3 expression in human prolactinomas and a novel ErbB3-mediated mechanism for PRL regulation in experimental lactotroph tumors. Targeted inhibition of upregulated p185c-neu / ErbB3 activity could be useful for the treatment of aggressive prolactinomas resistant to conventional therapy.
Keywords: Pituitary tumor, Prolactinoma, GH4C1 cells, PRL, heregulin, p185c-neu, ErbB3
INTRODUCTION
Prolactinomas arising from pituitary lactotrophs account for ~40% of all pituitary tumors (1, 2). Excess prolactin (PRL) secretion may lead to infertility, sexual dysfunction and osteoporosis (1). Prolactinoma mass effects may lead to local compressive symptoms including visual field defects and hypopituitarism. Tumorous lactotrophs usually express functional dopamine (D2) receptors, enabling medical therapy with dopamine agonists to suppress PRL synthesis and secretion and tumor growth (1). However, dopamine agonist resistance encountered in a subset of patients with PRL-secreting pituitary adenomas limits therapeutic efficacy (3). Effective treatments are also needed for invasive refractory macroprolactinomas and for those aggressive tumors rarely undergoing malignant transformation resulting in a poor prognosis (4-6).
Mechanisms underlying pituitary tumor aggressive behavior and malignant transformation are largely unclear. Dopamine agonist resistance may be due to decreased D2 receptor or differential isoform expression, and or disrupted autocrine growth factor signaling (1, 3). ErbB receptors comprise 4 subtypes (EGFR, p185her2/neu, ErbB3 and ErbB4) (7), and the kinase-deficient ErbB3 requires dimerization with p185her2/neu to effect signaling (8). Reports of increased ErbB receptor member expression particularly in aggressive pituitary tumors and pituitary carcinomas (9-13) could reflect tumor progression to a more dedifferentiated state and associated loss of growth control. For such patients, targeted ErbB receptor inhibition could provide an alternative medical control of tumor growth and or hormone secretion, as recently demonstrated for experimental rat prolactinomas (14).
Discovery of aberrant ErbB receptor activation in human cancers has led to selective therapeutic targeting particularly of EGFR and p185her2/neu (ErbB2) with monoclonal antibodies and small compound tyrosine kinase inhibitors (TKIs) (7). However, recent studies examining mechanisms for TKI resistance have highlighted the importance of kinase-inactive ErbB3 expression, and associated activation of the phosphatidyl-3-OH kinase (PI(3)K) / Akt survival pathway (15, 16).
Here we show ErbB3 receptor expression in a subset of human prolactinomas and report the functional role of p185c-neu / ErbB3 signaling in experimental rat lacto-somatotroph hormone regulation.
MATERIALS AND METHODS
Materials
Ham's F10 media, fetal bovine serum (FBS), penicillin, streptomycin and amphotericin B were purchased from Invitrogen (Carlsbad, CA). EGF was from Sigma (St. Louis, MO), and heregulin (NRG1-β1/HRG1-β1) from R&D Systems (Minneapolis, MN). Gefitinib (iressa) was purchased from Biaffin GmbH & Co (Kassel, Germany), U0126 from Promega (Madison, WI) and LY294002 from Calbiochem (San Diego, CA).
Cell cultures
GH4C1 rat lacto-somatotroph cells were purchased from the American Type Culture Collection (ATCC). After synchronization by serum starvation (medium containing 0.2% BSA for ~ 24 hrs), cells were plated in 100 mm dishes (~ 1.5 x 106 cell density), 6-well plates (~ 0.5 x 106 cell density) or 24-well plates (~ 50,000 cell density) and treatment agents added with fresh serum-depleted medium (with 0.2% BSA) and samples collected as indicated.
Pituitary glands obtained from female Wistar-Furth rats (140-160 g body weight) were dissociated to single-cell suspensions by enzymatic degradation of the extracellular adhesion proteins with the Neural Tissue Dissociation Kit (Papain) according to the manufacturer's instructions (Miltenyi Biotech GmbH; Bergisch Gladbach, Germany). Animal procedures were performed according to the guidelines of the Institutional Animal Care and Use Committee. Cells were incubated in complete medium (DMEM containing 10% FBS and antibiotics) for 24 hrs and then in serum-free medium (0.3% BSA) for 8 hrs, followed by treatment with heregulin for indicated times. At the end of each experiment, supernatants were collected for GH and PRL assays and cells were disolved in TRIZOL® reagent (Invitrogen) and stored at -80°C.
siRNA transfections
GH4C1 cells grown in 6-well plates to ~ 50% confluency were serum-starved overnight and transfected with 100 pmol Silencer® Select Negative Control #1 (Scr) siRNA (Cat. Nr. 4390843), Silencer Select® Pre-designed rat MAPK1 siRNA (Cat. Nr. 4390771; ID: s138100), ErbB2 (Neu) siRNA (Cat. Nr. 4390771, ID: s127710), ErbB3 siRNA (Cat. Nr. 4390771, ID: s131557; Applied Biosystems / Ambion, Austin, TX) or both ErbB2 (Neu) and ErbB3 siRNA (50 pmol each) for 24 hrs using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's instructions. After an additional 8 hr incubation (serum-free medium), treatments were added and samples collected at indicated times.
Templates for probes and Northern Blot Analysis
RNA extraction was performed using TRIZOL® reagent (Invitrogen) according to the manufacturer's instructions. The β-actin probe was a 1.076 kb fragment of the mouse β-actin gene (Ambion, Austin, TX). Probes for rat GH and PRL were generated and Northern blot analysis performed as previously described (14).
Immunoprecipitation and Western blotting
After completion of treatments, cells were placed on ice and washed with cold PBS. For whole cell protein extraction, cells were lysed in 150 μl RIPA buffer (Sigma) and for immunoprecipitation (IP) experiments, cells were lyzed in 850 μl modified RIPA buffer (1% Triton X-100, 1% deoxycholate, 0.1% NaDodSO4, 0.15M NaCl, 0.01M sodium phosphate pH 7.4, 2mM EDTA, 10mM sodium pyrophosphate, 400μM sodium orthovanadate), containing complete protease inhibitor cocktail tablets (Roche Molecular Biochemicals) and phosphatase inhibitor cocktail 2 (Sigma). Lysates were centrifuged at 13,000 x g for 20 min at 4 °C and protein concentrations determined by Bradford's method (Bio-Rad, Richmond, CA). ~ 1 mg protein was immunoprecipitated with rabbit polyclonal anti-EGFR (1005), anti-ErbB3 (C-17; 2 μg; Santa Cruz Biotechnology, CA) and with monoclonal antibody 7.16.4 (17) (3 μg; a kind gift from Dr. Greene, University of Pennsylvania) which reacts specifically with rat p185 molecules. Pre-clearing was performed with A/G PLUS-Agarose beads (20 μl; Sigma) overnight at 4°C. IP with appropriate antibody titers was performed for 1 hr prior to addition of A/G PLUS-Agarose beads (20 μl) overnight at 4°C. Immunoprecipitates were washed 1x in lysis buffer and five times in washing buffer and resuspended in SDS sample buffer pH 6.8 as described (18).
Western blot analysis was performed according to the guidelines of NuPAGE® electrophoresis system protocol (Invitrogen). In brief, whole cell lysates (~ 50 μg protein per lane) or IP samples were heated for 5 min at 100°C, respectively. Proteins were separated on NuPAGE® 4-12% Bis-Tris gels and electro-transferred for 1 hr to PVDF (Invitrogen). Membranes were blocked for 1 hr in 2% nonfat dry milk (or 5% BSA) in TBS-T buffer, and incubated overnight with primary antibody. The following primary antibodies were used: mouse anti-pERK1/2, rabbit anti-ERK1/2 (1:400; Santa Cruz Biotechnology), mouse monoclonal anti-pTyr (PY99), rabbit polyclonal anti-EGFR (1005), anti-Neu (C-18), anti-ErbB3 (C-17; 1:200; Santa Cruz Biotechnology), rabbit monoclonal anti-pAkt (phospho S473; 1:1000; Abcam, Cambridge, MA), rabbit polyclonal anti-Akt and anti-GAPDH (1:1000; Cell Signaling, Danvers, MA). After washing with TBS-T, membranes were incubated with peroxidase conjugated secondary antibody for 1.5 hrs (2% nonfat dry milk or 5% BSA in TBS-T buffer). Blots were washed and hybridization signals measured by ECL detection system (Amersham).
Immunofluorecence
Tumor specimens were fixed in 10% formalin and embedded in paraffin. After deparaffinization of the sections, antigen retrieval was performed using citrate and permeabilization by 0.1% Triton X. Slides were blocked in 10% goat serum in 1% BSA-PBS and then incubated with primary antibody overnight at 4°C. The following antibodies were used: Rabbit polyclonal anti-Neu (C-18) and anti-ErbB3 (C-17) (1:100; Santa Cruz Biotechnology). Following washes, slides were incubated with Alexa Fluor goat anti-rabbit 488 (H+L) secondary antibody (1:500; Invitrogen) for 2 hrs at RT. Nuclei were stained using 1:500 Topro-3 iodide 1mM solution (1:250 in PBS, Molecular Probes, Inc., Eugene, OR) for 2 hrs at RT, and following such, slides were mounted with Prolong Gold anti-fade reagent (Invitrogen). Confocal microscope images were obtained using a TCS-SP confocal scanner (Leica Microsystems, Mannheim, Germany). To detect contributions of autofluorescence in these paraffin embedded tissues, a spectral imaging approach was used. The confocal spectrophotometer was set to detect specific FITC fluorescence ranging from 505 to 540 nm. A second channel detecting autofluorescence with wavelength from 560 to 600 nm was used, and both channels color coded and merged. Green represents specific fluorescence from FITC and red, autofluorescence. The staining was strong and autofluorescence was very low in comparison to the specific signal. Only erythrocytes showed appreciable autofluorescence and appear dark orange in the images. A Leica PlanApo 20x 0.7 N.A. lens was used for overview images and a PlanApo 40x 1.2 N.A. for high magnification images.
Quantitative real time PCR
Total RNA was extracted with Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer instructions. Total RNA was reverse transcribed into first-strand cDNA using MMLV Reverse Transcriptase (Invitrogen) according to the manufacturer's protocol. Quantitative (Q)-PCR reactions were carried out in the iQ5 Multicolor Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA) as follows: 95°C for 3 min, 1 cycle; 95°C for 15s, 58°C for 1 min, 40 cycles. Melting curve analysis was performed to ensure product specificity. Certified RT2 qPCR Primer Assays for human ErbB2 and ErbB3 were purchased from SuperArray (Cat. Nr. PPH00209B and PPH00463B; Frederick, MD). Human Housekeeping Gene Primer Set (10 genes) was from Real Time Primers (Elkins Park, PA). This set included primers for β-glucuronidase (GUSB), β-actin (ACTB), glyceraldehydes-3-phosphate dehydrogenase (GAPDH), transferring receptor (TFRC), phosphor-glycerate kinase 1 (PGK1), hypoxanthin phosphoribosyl-transferase (HPRT1), peptidyl-propyl-isomerase A (PP1A), ribosomal protein L13A (RPL13A), TATA box binding protein (TBP) and β2-microglobulin (B2M).
Hormone assays
RIA for rat GH and PRL were performed using reagents provided by the National Hormone and Peptide Program, National Institute of Diabetes and Digestive and Kidney Diseases (Harbor-UCLA Medical Center, Torrance, California, USA) as described (14).
Statistical analysis
Cell cycle phases were analyzed by ModFit LT software (Version 2.0, Becton Dickinson). NIH Image 1.59 software was used for densitometric analysis of specific bands in blots and comparisons evaluated using 2-tailed Student's t test. Results are expressed as mean ± SE of independently performed experiments. Statistical significance was set at p <0.05.
RESULTS
ErbB family receptor expression and activation in rat pituitary GH4C1 cells
The functional role of pituitary prolactinoma ErbB3 expression was examined in rat lactosomatotroph GH4C1 cells. Immunoprecipitation and immunoblotting with receptor-specific antibodies showed expression of EGFR (175 kd), p185c-neu (185 kd) and ErbB3 (185 kd) in GH4C1 cells. The EGFR ligand, EGF (5 nM), induced EGFR and p185c-neu tyrosine phosphorylation, but did not result in ErbB3 phosphorylation. In contrast, heregulin (6 nM), the ErbB3 and ErbB4 ligand, induced tyrosine phosphorylation of ErbB3 and p185c-neu but not that of EGFR (Fig. 1A). Thus ErbB1-3 receptors appear functional in GH4C1 cells, and p185c-neu likely heterodimerizes with both activated EGFR and ErbB3.
Fig. 1. Effects of heregulin on PRL and GH.

A) ErbB receptor expression in GH4C1 cells: GH4C1 cells were serum-starved overnight, treated with EGF (5 nM) or HRG (6 nM) for 10 min and total protein extracted. Immunoprecipitations (IP) were performed with (Aa) EGFR (1005), (Ab) p185 (Ab 7.16.4) and (Ac) ErbB3 (C-17) antibodies as indicated. Immunoblotting (IB) was performed with pTyr (PY99) antibody (upper panels). Subsequently, membranes were stripped and re-blotted with EGFR (1005), Neu (C-18) and ErbB3 (C-17) antibodies (lower panels).
B-C) Time and dose-dependent effects of heregulin on PRL and GH expression: GH4C1 cells were serum-starved for 24 hrs and subsequently treated with indicated HRG concentrations for up to 48 hrs. B) Total RNA was extracted at the times indicated and GH mRNA expression determined by Northern blot. Subsequently, membranes were stripped and reblotted with specific probes for PRL and β-actin, respectively. The ratio of GH or PRL mRNA vs. β-actin mRNA was calculated by densitometric analysis of each treatment group. The GH / β-actin ratio and PRL / β-actin ratio of the control group at 24 hrs was set as 1.0, and relative mRNA expression levels normalized (mean ± SE; upper panel). Results shown are representative of three independently performed experiments (lower panel). C) GH and PRL secretion in the culture medium were determined by RIA. GH and PRL secretion levels of the control group at 24 hrs were set as 1, and relative secretion levels normalized (mean ± SE of three independently performed experiments). *, p<0.05; **, p<0.01; ***, p<0.001.
Increasing heregulin concentrations selectively induced PRL mRNA expression up to ~ 5-fold at 48 hrs (p<0.05), with no observed effect on GH mRNA expression (Fig. 1B). Dose-dependent heregulin-mediated induction of PRL secretion was also confirmed in the cell culture medium (Fig. 1C). Heregulin increased PRL release ~ 4-fold at 48 hrs (p<0.05), with no effect on GH secretion. Heregulin did not alter GH4C1 cell proliferation, as assessed by cell cycle analysis and cell proliferation assays (not shown). Heregulin did not induce PRL in nontumorous mixed primary rat pituitary cell cultures derived from female Wistar-Furth rats (not shown).
Heregulin-mediated p185c-neu / ErbB3 signaling
In time course experiments (Fig. 2A), treatment of GH4C1 cells with heregulin (6 nM) induced rapid (within 5 min) ErbB3 tyrosine phosphorylation. High levels of receptor phosphorylation were observed for up to 60 min, and gradually declined thereafter. Heregulin also induced marked p185c-neu tyrosine phosphorylation for up to 30 min. Similar to the patterns of immunoprecipitated receptor activation, heregulin induced ERK and Akt phosphorylation in whole cell extracts as detected by Western blot. While ERK activation occurred both in response to EGFR (with EGF; Fig. 2B) and ErbB3 (with heregulin; Fig. 2A), induction of Akt signaling was specific for p185c-neu / ErbB3 signaling. Formation of heregulin-mediated p185c-neu/ ErbB3 heterodimers was confirmed by immunoprecipitation (Fig. 2C).
Fig. 2. Heregulin-mediated signaling.
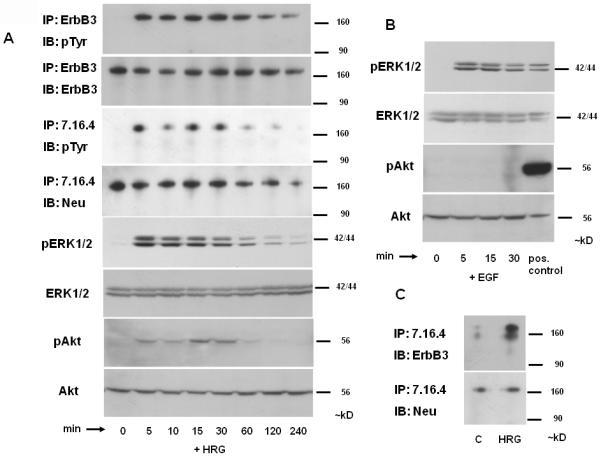
GH4C1 cells were serum-starved overnight, and treated with HRG (6 nM; A, C) or EGF (5 nM; B) for indicated times (in C for 10 min). Immunoprecipitations (IP) were performed with ErbB3 (C-17) and p185 (Ab 7.16.4) antibodies and immunoblotting (IB) with pTyr (PY99) (A) or ErbB3 (C-17) (C) antibodies. Subsequently, membranes were stripped and re-blotted with ErbB3 (A) or Neu (C-18) antibodies (A, C). ERK1/2 phosphorylation, total ERK1/2, Akt phosphorylation and total Akt as detected by Western blot of total protein extracts are shown (A - B). bFGF-treated (10 min; 30 ng/ml) pituitary folliculostellate TtT/GF cells were used as a positive control for phosphorylated Akt (B). A representative of 3 independently performed experiments is depicted.
As gefitinib blocks heregulin signaling by inducing formation of inactive heterodimers between ErbB3 and other ErbB receptor members (19, 20), we examined effects of the tyrosine kinase inhibitor on heregulin-induced ErbB receptor activation and downstream signaling. As shown in Fig. 3A, pretreatment with increasing gefitinib concentrations suppressed both receptor activation and signaling. Interestingly, inhibition of heregulin-induced ERK and Akt phosphorylation was observed at the lowest concentration tested (0.1 μM), while suppression of p185c-neu and ErbB3 tyrosine phosphorylation was observed with higher gefitinib doses (0.5 and 5 μM). Gefitinib also dose-dependently prevented heregulin-induced p185c-neu / ErbB3 heterodimerization (Fig. 3B) and PRL secretion (Fig. 3C).
Fig. 3. Gefitinib suppresses heregulin-induced signaling.
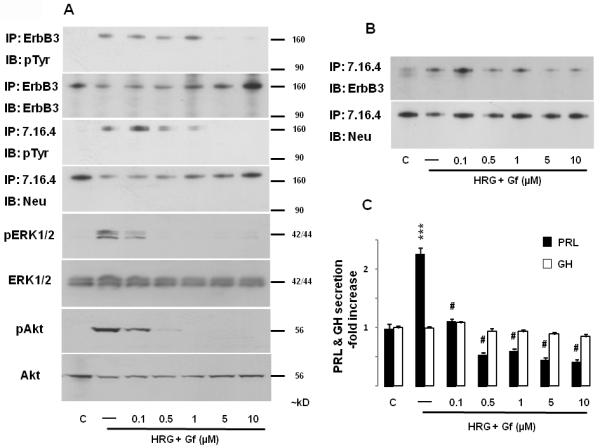
GH4C1 cells were serum-starved overnight, pre-treated with gefitinib (Gf) (indicated concentrations) for 45 min prior to induction with HRG (6 nM; 10 min). Immunoprecipitations and immunoblotting were performed as for Fig. 2. GH and PRL secretion in the culture medium were determined as for Fig. 1. ***, p<0.001 vs. control group; #, p<0.001 vs. HRG only group. A representative of 3 independently performed experiments is shown.
Functional role of pituitary p185c-neu / ErbB3 heterodimer signaling
To further explore mechanisms for the observed heregulinmediated effects through p185c-neu and ErbB3 receptor activation, we employed siRNA to selectively down-regulate these respective ErbB receptor members. Under these experimental conditions, we observed up to 60% reduction of p185c-neu and ErbB3 expression, which was associated with decreased total tyrosine phosphorylation in response to heregulin (Fig. 4A). With down-regulated p185c-neu and or ErbB3 levels, heregulin-induced PRL mRNA (Fig. 4B) and PRL hormone (Fig. 4C) expression were attenuated to almost basal levels, while no effects on GH were observed, demonstrating both the specificity of, and requirement for, both p185c-neu and ErbB3 for heregulin-mediated PRL induction.
Fig. 4. Pituitary p185c-neu / ErbB3 heterodimer signaling.
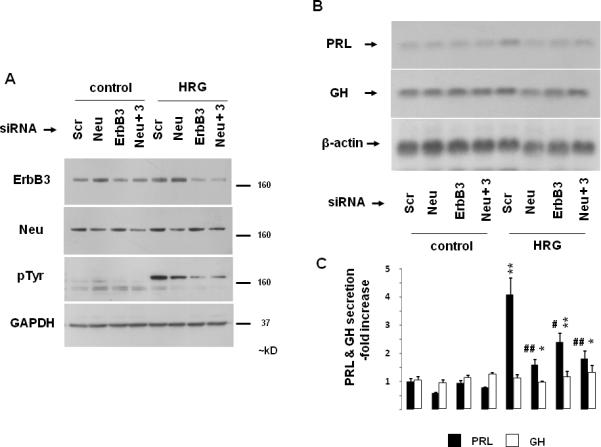
GH4C1 cells were serum-starved overnight, transfected with negative control (Scr), p185c-neu (Neu), ErbB3 (100 pmol) or both p185c-neu and ErbB3 (50 pmol each) siRNA (24 hrs). After an additional 8 hr (serum-free medium), cells were treated with HRG (6 nM) for 10 min (A) or 48 hrs (B - C). Expression levels of ErbB3, p185c-neu, pTyr and GAPDH loading control as determined by Western blot of whole cell extracts are depicted (A). GH and PRL mRNA expression and protein secretion were determined as for Fig. 1. Representative of 3 independently performed experiments is shown (mean ± SE). *, p<0.05; **, p<0.01 vs. Scr control group; #, p<0.05; ##, p<0.01 vs. Scr HRG group.
As heregulin induces ErbB receptor-mediated ERK and Akt signaling (Fig. 2A and 3A) we used a pharmacologic approach to block ERK or Akt activation, to identify pathways, specifically mediating heregulin action on the lactotroph. The PI3K inhibitor LY294002 dose-dependently suppressed heregulin-induced Akt phosphorylation (Fig. 5Aa), confirming activity of the inhibitor used. However, at the same concentrations, LY294002 had no effect on heregulin-induced PRL mRNA (Fig. 5Ab) and PRL hormone (Fig. 5Ac) expression. Dose-dependent effects of the MEK inhibitor U0126 on heregulin-induced ERK phosphorylation are shown in Fig. 5Ad. In contrast to PI3K/Akt signaling blockade, the MEK inhibitor dose-dependently suppressed heregulin-induced PRL mRNA expression (Fig. 5Ae) and PRL secretion (Fig. 5Af). The specific involvement of ERK in mediating heregulin-induced PRL expression was confirmed by siRNA experiments (Fig. 5B). Suppression of MAPK1 by siRNA attenuated heregulin-induced (10 min) ERK phosphorylation, PRL mRNA and secreted hormone levels. These results thus indicate that heregulin-induced PRL appears to be mediated by ERK signaling.
Fig. 5. ERK signaling mediates heregulin-induced PRL.

A) GH4C1 cells were serum-starved overnight and pre-treated for 45 min with the PI3K inhibitor LY294002 (Aa-c) or the MEK inhibitor U0126 (Ad-f). Cells were treated with HRG (6 nM) for 10 min (Aa and Ad) or 48 hrs (Ab-c, Ae-f). Phosphorylated Akt and total Akt (Aa), as well as phosphorylated ERK and total ERK (Ad) were detected by Western blot in whole protein extracts. GH and PRL mRNA expression and protein secretion were determined by Northern blot (Ab, Ae) and RIA (Ac, Af) as for Fig. 1. A representative of 3 independently performed experiments is shown (mean ± SE). *, p<0.05 vs. control group; #, p<0.05 vs. HRG only (-) group.
B) GH4C1 cells were serum-starved overnight, transfected with negative control (Scr) or rat MAPK1 (100 pmol) siRNA (24 hrs). After an additional 8 hr (serum-free medium), cells were treated with HRG (6 nM) for 10 min (Ba) or 48 hrs (Bb-c). Phosphorylated ERK and GAPDH loading control were detected by Western blot in whole protein extracts. GH and PRL mRNA expression and protein secretion were determined by Northern blot and RIA as for Fig. 1. A representative of 3 independently performed experiments is shown (mean ± SE). ***, p<0.001 vs. Scr siRNA control group; #, p<0.001 vs. Scr HRG group.
ErbB2 and ErbB3 expression in human PRL-secreting pituitary adenomas
Immunofluorescence staining for ErbB2 (p185her2/neu) and ErbB3 was performed in 8 human PRL-secreting tumors (Suppl. Table). Human breast carcinoma tissue samples were used as immunoreactive controls, while antibody-specific blocking peptides (sc-284-P and sc-285-P) suppressed the fluorescent signal, confirming the specificity of the antibody reactions (Suppl. Fig. 1 and 2).
Two patients (cases 7-8) underwent multiple surgeries due to dopamine agonist resistance while case 8 eventually evolved into a carcinoma with loss of PRL immunoreactivity. Positive ErbB2 staining with varying intensities was observed in 7/8 tumor specimens, while tumor tissue obtained from 4 cases was immunoreactive for ErbB3 (Fig. 6A), and also exhibited a Ki-67 index of ≥ 4% (Suppl. Table).
Fig. 6. ErbB2 and ErbB3 expression in human prolactinomas.
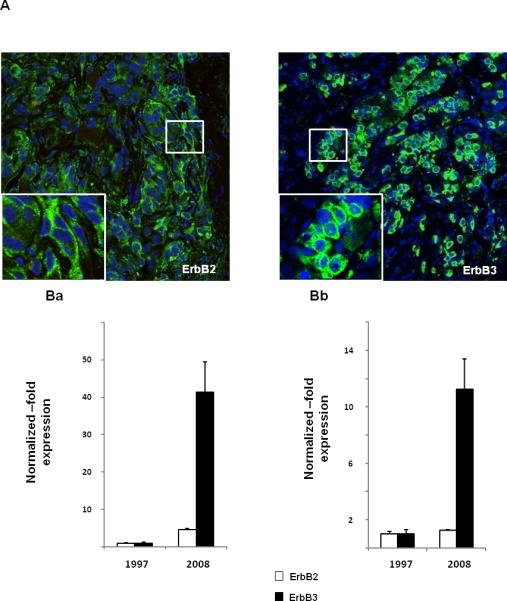
A) ErbB2 and ErbB3 immunostaining in a benign human prolactinoma: Fluorescent confocal microscopy images of ErbB2 (left panel) and ErbB3 (right panel) in a human PRL-secreting adenoma (tumor 2 from suppl. table). ErbB receptor expression in green (Alexa 488) and nucleic acid staining (TO-PRO-3) in blue. The field size is 375 μm and 75 μm for the insert.
B) ErbB2 and ErbB3 mRNA expression in malignantly transformed prolactinoma: Quantitative PCR analysis for ErbB2 and ErbB3 mRNA expression in tumor specimens derived from the same patient who initially presented with a benign prolactinoma (1997) which underwent malignant transformation (2008). Ba) Internal normalization was performed with 2 housekeeping genes (GAPDH and TFRC) which were unchanged between the two specimens. Bb) Internal normalization was performed with 10 housekeeping genes (GUSB, ACTB, GAPDH, TFRC, PGK1, HPRT, PP1A, RPL13A, TBP and B2M).
ErbB2 and ErbB3 mRNA expression were compared by quantitative PCR analysis of 2 tumor specimens derived from a patient who initially underwent surgery for an invasive but histologically benign prolactinoma (specimen obtained in 1997). Tissue was not available for immunostaining. Progressive resistance to high dose dopamine agonists necessitated 4 consecutive transsphenoidal pituitary surgeries due to tumor remnant re-growth. Gamma-knife radiotherapy temporarily stabilized the tumor mass and PRL levels, however, subsequent tumor enlargement caused loss of unilateral vision. The surgical tumor specimen obtained in 2008 showed a Ki-67 index of 20%. When we employed 2 housekeeping genes (GAPDH and TFRC) for internal normalization, ErbB2 and ErbB3 mRNA expression levels were observed to be elevated ~ 5 and 41-fold respectively in the 2008 specimen (Fig. 6Ba). When 10 housekeeping genes (most of which also exhibited upregulation) were evaluated in the assay, ErbB2 and ErbB3 expression levels were shown to be increased ~ 1 and 11-fold, respectively (Fig. 6Bb).
DISCUSSION
Pituitary adenomas account for ~15% of primary intracranial neoplasms and are discovered in up to 25% of unselected autopsy specimens (2). PRL-secreting adenomas account for ~40% of all pituitary tumors (1). Indications for prolactinoma therapy include hyperprolactinemia-associated hypogonadism, infertility and osteoporosis, as well as central compressive effects (1, 21). Drug intolerance or development of dopamine agonist resistance may constrain medical therapy of benign prolactinomas (3). Effective drug therapies are also required for recurrent invasive macroprolactinomas or those that undergo rare malignant transformation and are resistant to attempts at surgery and radiation (4-6).
We show here that human prolactinomas express ErbB3 receptors, and that heregulin, an ErbB3 ligand, induces PRL gene expression. Expression of other ErbB receptor family members EGFR (22-27) and p185her2/neu (11, 13, 25) have been reported in the normal anterior pituitary. EGFR (9, 10, 13, 25-27) and p185her2/neu (11, 25, 28, 29) expression in pituitary adenomas have been associated with more invasive tumor phenotypes (9-13), suggesting a role for these receptors in aggressive tumor behavior. Estrogen-mediated rat lactotroph hyperplasia is also associated with upregulation of pituitary TGFα, an EGFR ligand (30), and lactotroph TGFα overexpression in transgenic mice results in hyperplasia and pituitary adenoma formation (31). EGFR tyrosine kinase inhibition was shown to inhibit EGF-induced rat lactotroph differentiation and tumor growth (14), indicating a role for EGFR signaling in prolactinoma regulation.
ErbB3 activation and subsequent PI3K / Akt signaling may contribute to TKI resistance in breast and lung cancer (15, 16). p185her2/neu and ErbB3 are functionally incomplete receptors. The extracellular domain of p185her2/neu appears to be devoid of ligand-binding activity, while ErbB3 contains a non-functional kinase domain devoid of catalytic kinase activity. However, the p185her2/neu / ErbB3 heterodimer is the most active signaling dimer in the ErbB family (8). Ligand induction leads to ErbB3 trans-phosphorylation (32), the only ErbB receptor which couples directly to PI3K / Akt (33). Although pituitary heregulin (glial growth factor) expression has been reported (34), ErbB3 expression and the role of p185her2/neu / ErbB3 signaling in pituitary tumorigenesis were not heretofore known.
Rat lactosomatotroph GH4C1 cells are shown here to express functional ErbB receptor members (Fig. 1A), and p185c-neu tyrosine phosphorylation was observed in response to both EGFR and ErbB3 activation, suggesting p185c-neu as the preferred dimerization partner in these cells. p185c-neu tyrosine phosphorylation was detected only in response to ligand induction, suggesting expression of the non-mutated proto-oncogenic p185c-neu, similar to the observed absence of p185her2/neu activating mutations in human pituitary adenomas (28). Heregulin induction of PRL (Fig. 1B-C), suggests a functional role for p185c-neu / ErbB3 signaling in PRL but not GH control, consistent with known GH regulation (35). In contrast to EGFR and p185her2/neu (11, 13, 22-27), non-tumorous pituitary lactotrophs likely do not express ErbB3, as we did not observe PRL regulation by heregulin in primary rat pituitary cell cultures. ErbB receptor-mediated PRL regulation may be selective for transformed lactotrophs with altered intracellular signaling and loss of dopaminergic control. Indeed, GH3 / GH4C1 cells do not express functional D2 dopamine receptors (36) and are resistant to dopamine agonists, similar to drug-resistant aggressive prolactinomas.
Aberrant Akt (37) and MAPK (38) activity in human pituitary tumors, and heregulinmediated ErbB3 and p185c-neu tyrosine phosphorylation and receptor heterodimerization we observed, was associated with ERK and Akt activation. EGF, the EGFR ligand, also activated ERK but not PI3K / Akt signaling, as previously shown for GH3 cells (14), demonstrating the requirement of pituitary ErbB3 activation for PI3K / Akt signaling induction. As targeted ErbB3-specific therapies are not currently available, we employed pharmacological inhibition of the receptor with gefitinib, an EGFR tyrosine kinase inhibitor, which induces formation of inactive heterodimers between ErbB3 and other ErbB receptor members, thereby blocking heregulin signaling (19, 20). Indeed, gefitinib treatment suppressed ErbB3 and p185c-neu tyrosine phosphorylation and downstream signaling. Dose-dependent differences of gefitinib inhibition on receptor (higher doses) vs. ERK and Akt activation (lower doses) are likely due to differences in assay sensitivities, as the blots for immunoprecipitated ErbB3 and p185c-neu show total tyrosine phosphorylation load, and are not reflective of specific tyrosine residues which signal for ERK and Akt. Although low doses of gefitinib could inhibit trans-activated EGFR on ERK signaling by heregulin, the ligand did not appear to induce EGFR trans-activation (Fig. 1A).
Individual p185c-neu and ErbB3 downregulation suppressed heregulin effects, indicating the requirement of functional pituitary p185c-neu / ErbB3 signaling for heregulin action on PRL. In the presence of heregulin, specific pharmacologic and siRNA-mediated inhibition of ERK but not Akt signaling mediated PRL regulation, consistent with known Ras/Raf/MAPK/Ets-1-mediated PRL regulation in GH3/GH4 cells (39, 40). However, downstream involvement of additional Ras/MAPK-independent mechanisms (as has been shown for EGF-mediated PRL regulation (41, 42) cannot be excluded. In contrast to PRL-releasing peptide (PrRP) and insulin, however, which signal to the proximal rat PRL promoter in a pathway that involves PI3K/Akt (43), p185c-neu / ErbB3-mediated PI3K/Akt activation does not appear to induce lactotroph differentiation but could reflect changes leading to a more aggressive cell phenotype.
The unavailability of functional human pituitary cell lines and scarcity of surgically obtained human prolactinoma specimens renders mechanistic analysis of heregulin signaling in human prolactinomas an experimental challenge. We examined p185her2/neu and ErbB3 expression in 7 benign prolactinomas and one carcinoma (Hardy class 4) by immunofluorescence and demonstrate p185her2/neu expression in the majority of tumors (7/8). In addition, we now also show ErbB3 expression in a subset of these tumors (4/8), a novel finding which may indicate altered patterns of tumor differentiation. Tumors which were immunoreactive for ErbB3 expressed relatively high levels of Ki67, a marker of increased pituitary cell proliferation (44). ErbB3 immunoreactivity was also detected in an aggressive carcinoma specimen. Quantitative PCR analysis of available RNA samples derived from a single individual who initially presented with a benign prolactinoma which recurred as a malignant prolactinoma with dopamine resistance and failed surgery and radiation, revealed dramatic upregulation of ErbB3 mRNA expression in the second tumor sample.
In addition to potential efficacy of temozolomide therapy (45, 46) and the development of new generation PRL receptor antagonists (47), these results highlight the importance of examining ErbB receptor expression in surgical prolactinoma specimens. Benign prolactinomas usually respond effectively to medical therapy with dopamine agonists, and drug resistance may be an early sign of aggressive tumor behavior. In very rare cases of malignant tumor transformation during the observed course of the disease, and resistance to conventional therapy, information on progressive ErbB receptor status would provide an additional tool for determining therapy options for controlling tumor cell growth and excess PRL secretion.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Kolya Wawrowsky from the Cedars-Sinai Imaging Core for the confocal images and Patricia Lin from the Flow Cytometry Core. This study was supported by a scholarship from the Deutsche Forschungsgemeinschaft (VL 55 / 1-1), by NIH grant CA 075979 (SM) and the Doris Factor Molecular Endocrinology Laboratory.
Financial support: This study was supported by a scholarship from the Deutsche Forschungsgemeinschaft (VL 55 / 1-1), by NIH grant CA 075979 (SM) and the Doris Factor Molecular Endocrinology Laboratory.
Footnotes
Disclosure statement: The authors have nothing to disclose.
References
- 1.Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27:485–534. doi: 10.1210/er.2005-9998. [DOI] [PubMed] [Google Scholar]
- 2.Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112:1603–18. doi: 10.1172/JCI20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olafsdottir A, Schlechte J. Management of resistant prolactinomas. Nat Clin Pract Endocrinol Metab. 2006;2:552–61. doi: 10.1038/ncpendmet0290. [DOI] [PubMed] [Google Scholar]
- 4.Kaltsas GA, Nomikos P, Kontogeorgos G, Buchfelder M, Grossman AB. Clinical review: Diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab. 2005;90:3089–99. doi: 10.1210/jc.2004-2231. [DOI] [PubMed] [Google Scholar]
- 5.Scheithauer BW, Kurtkaya-Yapicier O, Kovacs KT, Young WF, Jr., Lloyd RV. Pituitary carcinoma: a clinicopathological review. Neurosurgery. 2005;56:1066–74. discussion -74. [PubMed] [Google Scholar]
- 6.Kars M, Roelfsema F, Romijn JA, Pereira AM. Malignant prolactinoma: case report and review of the literature. Eur J Endocrinol. 2006;155:523–34. doi: 10.1530/eje.1.02268. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Berezov A, Wang Q, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–8. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzahar E, Waterman H, Chen X, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996;16:5276–87. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeRiche VK, Asa SL, Ezzat S. Epidermal growth factor and its receptor (EGF-R) in human pituitary adenomas: EGF-R correlates with tumor aggressiveness. J Clin Endocrinol Metab. 1996;81:656–62. doi: 10.1210/jcem.81.2.8636285. [DOI] [PubMed] [Google Scholar]
- 10.Jaffrain-Rea ML, Petrangeli E, Lubrano C, et al. Epidermal growth factor binding sites in human pituitary macroadenomas. J Endocrinol. 1998;158:425–33. doi: 10.1677/joe.0.1580425. [DOI] [PubMed] [Google Scholar]
- 11.Nose-Alberti V, Mesquita MI, Martin LC, Kayath MJ. Adrenocorticotropin-Producing Pituitary Carcinoma with Expression of c-erbB-2 and High PCNA Index: A Comparative Study with Pituitary Adenomas and Normal Pituitary Tissues. Endocr Pathol. 1998;9:53–62. doi: 10.1007/BF02739952. [DOI] [PubMed] [Google Scholar]
- 12.Roncaroli F, Nose V, Scheithauer BW, et al. Gonadotropic pituitary carcinoma: HER-2/neu expression and gene amplification. Report of two cases. J Neurosurg. 2003;99:402–8. doi: 10.3171/jns.2003.99.2.0402. [DOI] [PubMed] [Google Scholar]
- 13.Onguru O, Scheithauer BW, Kovacs K, et al. Analysis of epidermal growth factor receptor and activated epidermal growth factor receptor expression in pituitary adenomas and carcinomas. Mod Pathol. 2004;17:772–80. doi: 10.1038/modpathol.3800118. [DOI] [PubMed] [Google Scholar]
- 14.Vlotides G, Siegel E, Donangelo I, Gutman S, Ren S, Melmed S. Rat Prolactinoma cell growth regulation by Epidermal Growth Factor receptor ligands. Cancer Res. 2008;68:6377–86. doi: 10.1158/0008-5472.CAN-08-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 16.Sergina NV, Rausch M, Wang D, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–41. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drebin JA, Stern DF, Link VC, Weinberg RA, Greene MI. Monoclonal antibodies identify a cell-surface antigen associated with an activated cellular oncogene. Nature. 1984;312:545–8. doi: 10.1038/312545a0. [DOI] [PubMed] [Google Scholar]
- 18.Dobashi K, Weiner DB, Greene MI. Differential regulation of oncogenic and cellular p185 by serine/threonine kinases. DNA. 1989;8:723–32. doi: 10.1089/dna.1989.8.723. [DOI] [PubMed] [Google Scholar]
- 19.Anido J, Matar P, Albanell J, et al. ZD1839, a specific epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, induces the formation of inactive EGFR/HER2 and EGFR/HER3 heterodimers and prevents heregulin signaling in HER2-overexpressing breast cancer cells. Clin Cancer Res. 2003;9:1274–83. [PubMed] [Google Scholar]
- 20.Hirata A, Hosoi F, Miyagawa M, et al. HER2 overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in lung cancer cells. Cancer Res. 2005;65:4253–60. doi: 10.1158/0008-5472.CAN-04-2748. [DOI] [PubMed] [Google Scholar]
- 21.Casanueva FF, Molitch ME, Schlechte JA, et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol (Oxf) 2006;65:265–73. doi: 10.1111/j.1365-2265.2006.02562.x. [DOI] [PubMed] [Google Scholar]
- 22.Birman P, Michard M, Li JY, Peillon F, Bression D. Epidermal growth factor-binding sites, present in normal human and rat pituitaries, are absent in human pituitary adenomas. J Clin Endocrinol Metab. 1987;65:275–81. doi: 10.1210/jcem-65-2-275. [DOI] [PubMed] [Google Scholar]
- 23.Peillon F, Le Dafniet M, Garnier P, et al. Receptors and neurohormones in human pituitary adenomas. Horm Res. 1989;31:13–8. doi: 10.1159/000181080. [DOI] [PubMed] [Google Scholar]
- 24.Halper J, Parnell PG, Carter BJ, Ren P, Scheithauer BW. Presence of growth factors in human pituitary. Lab Invest. 1992;66:639–45. [PubMed] [Google Scholar]
- 25.Chaidarun SS, Eggo MC, Sheppard MC, Stewart PM. Expression of epidermal growth factor (EGF), its receptor, and related oncoprotein (erbB-2) in human pituitary tumors and response to EGF in vitro. Endocrinology. 1994;135:2012–21. doi: 10.1210/endo.135.5.7956924. [DOI] [PubMed] [Google Scholar]
- 26.Kontogeorgos G, Stefaneanu L, Kovacs K, Cheng Z. Localization of Epidermal Growth Factor (EGF) and Epidermal Growth Factor Receptor (EGFr) in Human Pituitary Adenomas and Nontumorous Pituitaries: An Immunocytochemical Study. Endocr Pathol. 1996;7:63–70. doi: 10.1007/BF02739916. [DOI] [PubMed] [Google Scholar]
- 27.Theodoropoulou M, Arzberger T, Gruebler Y, et al. Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J Endocrinol. 2004;183:385–94. doi: 10.1677/joe.1.05616. [DOI] [PubMed] [Google Scholar]
- 28.Ezzat S, Zheng L, Smyth HS, Asa SL. The c-erbB-2/neu proto-oncogene in human pituitary tumours. Clin Endocrinol (Oxf) 1997;46:599–606. doi: 10.1046/j.1365-2265.1997.1921003.x. [DOI] [PubMed] [Google Scholar]
- 29.Botelho CH, Magalhaes AV, Mello PA, Schmitt FC, Casulari LA. Expression of p53, Ki-67 and c-erb B2 in growth hormone-and/or prolactin-secreting pituitary adenomas. Arq Neuropsiquiatr. 2006;64:60–6. doi: 10.1590/s0004-282x2006000100013. [DOI] [PubMed] [Google Scholar]
- 30.Borgundvaag B, Kudlow JE, Mueller SG, George SR. Dopamine receptor activation inhibits estrogen-stimulated transforming growth factor-alpha gene expression and growth in anterior pituitary, but not in uterus. Endocrinology. 1992;130:3453–8. doi: 10.1210/endo.130.6.1534540. [DOI] [PubMed] [Google Scholar]
- 31.McAndrew J, Paterson AJ, Asa SL, McCarthy KJ, Kudlow JE. Targeting of transforming growth factor-alpha expression to pituitary lactotrophs in transgenic mice results in selective lactotroph proliferation and adenomas. Endocrinology. 1995;136:4479–88. doi: 10.1210/endo.136.10.7664668. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Mei Y, Liu X, Zhou M. Neuregulin-1 only induces trans-phosphorylation between ErbB receptor heterodimer partners. Cell Signal. 2007;19:466–71. doi: 10.1016/j.cellsig.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 33.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 34.Lemke GE, Brockes JP. Identification and purification of glial growth factor. J Neurosci. 1984;4:75–83. doi: 10.1523/JNEUROSCI.04-01-00075.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopchick JJ. Growth Hormone. In: Leslie J, DeGroot J, Larry Jameson, editors. Endocrinology. Fifth Edition Vol 1. Elsevier Saunders; Philadelphia: 2006. pp. 579–99. [Google Scholar]
- 36.Cronin MJ, Faure N, Martial JA, Weiner RI. Absence of high affinity dopamine receptor in GH3 cells: a prolactin-secreting clone resistant to the inhibitory action of dopamine. Endocrinology. 1980;106:718–23. doi: 10.1210/endo-106-3-718. [DOI] [PubMed] [Google Scholar]
- 37.Musat M, Korbonits M, Kola B, et al. Enhanced protein kinase B/Akt signalling in pituitary tumours. Endocr Relat Cancer. 2005;12:423–33. doi: 10.1677/erc.1.00949. [DOI] [PubMed] [Google Scholar]
- 38.Hubina E, Nanzer AM, Hanson MR, et al. Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur J Endocrinol. 2006;155:371–9. doi: 10.1530/eje.1.02213. [DOI] [PubMed] [Google Scholar]
- 39.Conrad KE, Oberwetter JM, Vaillancourt R, Johnson GL, Gutierrez-Hartmann A. Identification of the functional components of the Ras signaling pathway regulating pituitary cell-specific gene expression. Mol Cell Biol. 1994;14:1553–65. doi: 10.1128/mcb.14.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YH, Jue SF, Maurer RA. Thyrotropin-releasing hormone stimulates phosphorylation of the epidermal growth factor receptor in GH3 pituitary cells. Mol Endocrinol. 2000;14:1328–37. doi: 10.1210/mend.14.9.0512. [DOI] [PubMed] [Google Scholar]
- 41.Pickett CA, Gutierrez-Hartmann A. Epidermal growth factor and Ras regulate gene expression in GH4 pituitary cells by separate, antagonistic signal transduction pathways. Mol Cell Biol. 1995;15:6777–84. doi: 10.1128/mcb.15.12.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben-Jonathan N, Chen S, Dunckley JA, Lapensee C, Kansra S. Estrogen Receptor {alpha} mediates the Epidermal Growth Factor -stimulated prolactin expression and release in lactotrophs. Endocrinology. 2009;150:795–802. doi: 10.1210/en.2008-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duval DL, Gutierrez-Hartmann A. PRL-releasing peptide stimulation of PRL gene transcription--enter AKT. Endocrinology. 2002;143:11–2. doi: 10.1210/endo.143.1.8647. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber S, Saeger W, Ludecke DK. Proliferation markers in different types of clinically non-secreting pituitary adenomas. Pituitary. 1999;1:213–20. doi: 10.1023/a:1009933820856. [DOI] [PubMed] [Google Scholar]
- 45.Kovacs K, Horvath E, Syro LV, et al. Temozolomide therapy in a man with an aggressive prolactin-secreting pituitary neoplasm: Morphological findings. Hum Pathol. 2007;38:185–9. doi: 10.1016/j.humpath.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 46.Neff LM, Weil M, Cole A, et al. Temozolomide in the treatment of an invasive prolactinoma resistant to dopamine agonists. Pituitary. 2007;10:81–6. doi: 10.1007/s11102-007-0014-1. [DOI] [PubMed] [Google Scholar]
- 47.Goffin V, Touraine P, Culler MD, Kelly PA. Drug Insight: prolactin-receptor antagonists, a novel approach to treatment of unresolved systemic and local hyperprolactinemia? Nat Clin Pract Endocrinol Metab. 2006;2:571–81. doi: 10.1038/ncpendmet0270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.