

Abstract
Integrin-linked kinase (Ilk) is a scaffold and kinase that links integrin receptors to the actin cytoskeleton and to signaling pathways involved in cell adhesion, migration, and extracellular matrix deposition. Targeted deletion of Ilk from embryonic mouse dorsal forebrain neuroepithelium results in severe cortical lamination defects resembling cobblestone (type II) lissencephaly. Defects in adult mutants include neuronal invasion of the marginal zone, downward displacement of marginal zone components, fusion of the cerebral hemispheres, and scalloping of the dentate gyrus. These lesions are associated with abundant astrogliosis and widespread fragmentation of the basal lamina at the cortical surface. During cortical development, neuronal ectopias are associated with severe disorganization of radial glial processes and displacement of Cajal-Retzius cells. Lesions are not seen when Ilk is specifically deleted from embryonic neurons. Interestingly, targeted Ilk deletion has no effect on proliferation or survival of cortical cells or on phosphorylation of two Ilk substrates, Pkb/Akt and Gsk-3β, suggesting that Ilk does not regulate cortical lamination via these enzymes. Instead, Ilk acts in vivo as a major intracellular mediator of integrin-dependent basal lamina formation. This study demonstrates a critical role for Ilk in cortical lamination and suggests that Ilk-associated pathways are involved in the pathogenesis of cobblestone lissencephalies.
Keywords: integrin-linked kinase, extracellular matrix, cortical development, cobblestone lissencephaly, radial glia, reelin
Introduction
Mammalian cortical development proceeds in a highly orchestrated manner in which neurons migrate along radial glial fibers toward the surface of the brain to form a multilayered “inside-out” cortex. Integrin receptors are critical for several aspects of this process. The interaction between migrating neurons and radial glia, and the timely detachment of neurons at the end of migration involves α3β1 integrin (Anton et al., 1999; Sanada et al., 2004; Schmid et al., 2004). In contrast, integrin subunits α6 and β1 are involved in basal lamina assembly and radial glial end-feet anchorage at the cortical surface, and their absence results in abnormalities of cortical lamination in mutant mice (Georges-Labouesse et al., 1998; Graus-Porta et al., 2001). The mechanisms by which the various functions of integrins are coordinated inside cells during corticogenesis are not known.
Integrin-linked kinase (Ilk), a scaffolding protein and serine/threonine protein kinase, is a key effector of integrin function. Ilk exists as a multiprotein complex, binding the cytoplasmic portion of β1 and β3 integrins as well as adaptor proteins such as PINCH and α-parvins that regulate the actin cytoskeleton (Grashoff et al., 2004; Wu, 2004; Hannigan et al., 2005). Loss-of-function studies in Caenorhabditis elegans indicate that PAT-4/Ilk recruits integrins to adhesion complexes (Mackinnon et al., 2002). In Drosophila, loss of Ilk function leads to detachment of filamentous actin from the plasma membrane (Zervas et al., 2001), revealing an important role for Ilk in actin stabilization at integrin attachment sites. In vitro, localization of the multiprotein Ilk complex to focal adhesions regulates extracellular matrix deposition and provides an important physical connection between integrins and the actin cytoskeleton (Guo and Wu, 2002).
Ilk also has kinase activity in vitro and is able to induce phosphorylation of proteins such as Pkb/Akt, a downstream effector in a phosphatidylinositol 3 kinase-dependent Rac1 signaling pathway that regulates both integrin-associated rearrangements of actin filaments and cell migration (Qian et al., 2005). In addition, inhibition of Gsk-3β by Ilk was suggested as a major mechanism for neurite extension and axonal growth in vitro (Mills et al., 2003; Zhou et al., 2004). Although Ilk has been implicated in cell adhesion, migration, and extracellular matrix modification, its role in the development of the CNS has not been studied.
To determine the essential function of Ilk in brain development, we targeted Ilk for deletion in the mouse dorsal forebrain using cre/lox technology. The resulting mutant has severe cortical lamination defects with overmigration of neurons into the marginal zone, breakdown of the basal lamina, disorganization of radial glia, and displacement of Cajal-Retzius (CR) cells. The defects resemble those seen in mouse mutants of integrins, dystroglycan, and other genes involved in basal lamina assembly and suggest that Ilk is a major intracellular mediator of integrin and/or dystroglycan-mediated basal lamina assembly during brain development. Strikingly, absence of Ilk does not appear to perturb the proliferation, survival, or differentiation of neuronal precursors, although Ilk has been shown previously to regulate these functions in other cell types in vitro.
Materials and Methods
Mouse lines. The Ilk floxed mice (Ilkfl/fl) have been described previously (Terpstra et al., 2003). The Emx1IREScre mice were obtained from Jessica Gorski and Kevin Jones (University of Colorado, Boulder, CO), and the Nex-cre mice were obtained from Sandra Goebbels and Klaus-Armin Nave (Max-Planck-Institute, Göttingen, Germany). The pattern of recombination promoted by the Emx1IREScre allele has been described previously (Gorski et al., 2002). Emx1-cre;Ilkfl/fl mutants were compared in all experiments with sex-matched Emx1-cre;Ilkfl/+, Ilkfl/+, or Ilkfl/fl littermates. Similarly, Nex-cre;Ilkfl/fl mutants were compared with Nex-cre; Ilkfl/+, Ilkfl/+, or Ilkfl/fl sex-matched littermates. The animals examined were of a mixed background. All animals were handled in accordance with protocols approved by the University of California, San Francisco Committee on Animal Research.
Antibodies. Ilk monoclonal antibody (mAb) (BD Transduction Laboratories, Lexington, KY), neuronal-specific nuclear protein (NeuN) mAb (Chemicon, Temecula, CA), calbindin polyclonal antibody (pAb) (Swant, Bellinzona, Switzerland), Englebreth-Holm-Swarm laminin pAb (Sigma, St. Louis, MO), GFAP mAb (Chemicon), dystrophin MANDRA1 mAb (Sigma), chondroitin sulfate proteoglycan (CSPG) (CS-56) mAb (Sigma), RC2 mAb and Otx1 mAb (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), calretinin pAb (Chemicon), reelin mAb G10 (Andre Goffinet, University of Louvain, Brussels, Belgium), phospho-histone pAb (Upstate Biotechnology, Lake Placid, NY), Ki67 mAb (Novocastra, Newcastle upon Tyne, UK), Akt-[pS473] pAb (Cell Signaling Technology, Beverly, MA), Akt mAb (Cell Signaling Technology), Gsk-3β-[pS9] pAb (Cell Signaling Technology), and Gsk-3β mAb (BD Transduction Laboratories).
Histological analysis. Mice were deeply anesthetized with 2.5% avertin and perfused with 4% paraformaldehyde in PBS. Embryos and neonates were decapitated, and heads (embryos) or brains (neonates) were submerged in 4% paraformaldehyde in PBS. Tissues for frozen sliding microtome sections (40 μm) or cryostat sections (10 μm) were submerged in 30% sucrose overnight at 4°C and then either cut directly or first embedded in Optimal Cutting Temperature Compound (Tissue-Tek, Miles, Elkhart, IN). Sections for Nissl staining were dehydrated overnight in 70% EtOH and stained with 0.1% cresyl violet/0.5% acetic acid. Sections were then rinsed in dH2O, 70% ethanol, 95% ethanol, and chloroform and differentiated with 1.7% acetic acid in 95% EtOH.
Immunohistochemistry. Frozen sections were blocked in 3% goat serum and 0.1% Tween 20 and were subjected to heat-based antigen retrieval in 10 mm citrate buffer, pH 6.0, using the 34700 BioWave Microwave (Ted Pella, Redding, CA). For DAB immunohistochemistry, sections were quenched in 1% H2O2 before blocking. Sections were incubated in primary antibodies for 2 h at room temperature or overnight at 4°C and then incubated with biotinylated mouse or rabbit secondary antibodies (Vector Laboratories, Burlingame, CA), ABC solution (Vector Laboratories), and 0.05% diaminobenzidine/0.0003% H2O2. For fluorescent labeling of sections, either goat anti-mouse or anti-rabbit Alexa 488 and 594 (Molecular Probes, Eugene, OR) were used as secondary reagents. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays were performed using the In Situ Cell Death Detection Kit (Boehringer Manheim, Mannheim, Germany) according to the instructions of the manufacturer.
Golgi staining. Modified Golgi-Cox impregnation of neurons was performed using the FD Rapid GolgiStain kit (FD NeuroTechnologies, Ellicott City, MD) according to the instructions of the manufacturer. Briefly, 2-month-old nonperfused mouse brains were immersed in impregnation solution for 2 weeks, transferred to “solution C” for 2 d, and cut at 100 μm on the cryostat. Sections were mounted on 3% gelatin-coated slides and allowed to dry before staining with silver nitrate solution, “solution D and E.” Sections were dehydrated through descending alcohol series and mounted with DPX mounting medium (VWR Scientific, West Chester, PA).
Western blotting. Cortices from embryonic day 14.5 (E14.5) mice were dissected and lysed in modified radioimmunoprecipitation assay buffer [10 mm Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 mm Tris, pH 7.5, 100 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 1 mm NaVO4, 1 mm NaF, and 1× Complete Mini protease cocktail (Roche Products, Welwyn Garden City, UK)]. Precleared lysates (2.5 μg) were resolved on 4-15% gradient SDS-PAGE gels (Bio-Rad, Hercules, CA). Proteins were transferred onto Immobilon-P transfer membranes (Millipore, Bedford, MA), blocked with 5% BSA, and incubated with primary antibodies overnight at 4°C. Membranes were incubated with HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), followed by ECL reagent (Amersham Biosciences, Arlington Heights, IL). Membrane stripping was performed using the Restore Western Blot Stripping Buffer (Pierce, Rockford, IL).
Statistical analysis. Statistical significance was determined using the Student's t test in all studies.
Results
Generation of conditional knock-out mice with dorsal forebrain-restricted Ilk deletion
To gain insight into the role of Ilk in the CNS, we deleted Ilk from the vast majority of cells in the dorsal forebrain by crossing mice expressing cre recombinase driven by the Emx1 promoter to mice expressing a floxed Ilk allele (Gorski et al., 2002; Terpstra et al., 2003). Emx1 cre recombinase activity is first observed in precursors of neurons and glia at embryonic day 9 (Gorski et al., 2002), before initiation of cortical neurogenesis (Kubo and Nakajima, 2003). Deletion of Ilk from the dorsal forebrain was confirmed by immunofluorescence and Western blot analysis, which showed a significant decrease of Ilk in the targeted regions (Fig. 1). Residual Ilk staining was attributable to its presence in cells not derived from the dorsal forebrain neuroepithelium, such as inhibitory interneurons, vascular endothelial cells, and meningeal cells.
Figure 1.

Expression pattern of Ilk in control and forebrain-specific Emx1- cre; Ilkfl/fl mutant littermates. A, B, Coronal sections (10 μm) from an E14.5 littermate control (A) and Ilk mutant (B) immunostained with an Ilk antibody showed selective absence of Ilk from the cortex, but not the midbrain, of the Ilk mutant. Residual Ilk staining was attributable to the presence of Ilk in meninges, interneurons, and blood vessels, which are not targeted by the Emx1 promoter-driven cre. C, Western blot analysis of E14.5 forebrain extracts (2.5 μg) showed a significant decrease of Ilk expression in an Ilk mutant compared with a littermate control. β-Tubulin was used as a loading control in separately loaded lanes. The Ilk protein blot was also stripped and reprobed with Akt, showing comparable protein levels in the two samples (see the same extracts in Fig. 9). Mutant genotypes were Emx1- cre; Ilkfl/fl. Littermate control genotypes were Ilkfl/+. Con, Control; Mut, mutant. Scale bar, 100 μm.
Mutant mice were born at the expected Mendelian ratios and were indistinguishable from littermates at the time of weaning. However, at ∼4-5 months of age, homozygous mutants began showing progressive weight loss and premature death. In addition, mutant animals were sterile.
Targeted deletion of Ilk results in severe cortical lamination defects
Analysis of brains from adult Ilk mutants showed severe defects of cortical lamination. Coronal sections of mutant brains showed multiple regions of cell invasion into the marginal zone and downward expansion of hypocellular regions into the underlying cortex (Fig. 2). There was extensive midline fusion of the cerebral hemispheres (Fig. 2F), and the dentate gyrus of the hippocampus showed an abnormal, scalloped appearance with displacement of granule cells toward the pial surface (Fig. 2I). Staining with anti-NeuN antibody demonstrated that neurons were the major constituent of the ectopias, which were most prominent in medial and caudal regions of the cortex (Fig. 3A-D). Dense accumulations of neurons were found just below hypocellular downward expansions of marginal zone components (Fig. 3B,D). Calbindin-positive interneurons were found within these dense accumulations (Fig. 3F,H), suggesting misplacement of these cells from their usual location beneath the marginal zone (Fig. 3E,G). Otx1-expressing neurons that are localized normally to cortical layers V and VI were misplaced within ectopic outgrowths toward the surface of the cortex (Fig. 3J). Analysis of neuronal dendritic processes using microtubule-associated protein 2 (MAP2) immunostaining (Fig. 3K,L) showed that, although in control brains there was an orderly arrangement of dendritic bundles coursing through the cortex and terminating at the marginal zone (Fig. 3K), in mutant brains dendritic processes were severely disorganized, veering toward and terminating at misplaced hypocellular marginal zone-like regions within the cortex (Fig. 3L). A similar pattern was observed with Golgi impregnation in which apical dendrites in mutant brains lacked the orderly, parallel arrangement of dendrites seen in control brains (Fig. 3M,N). There were no obvious defects in spine formation between control and mutant brains (Fig. 3O,P).
Figure 2.
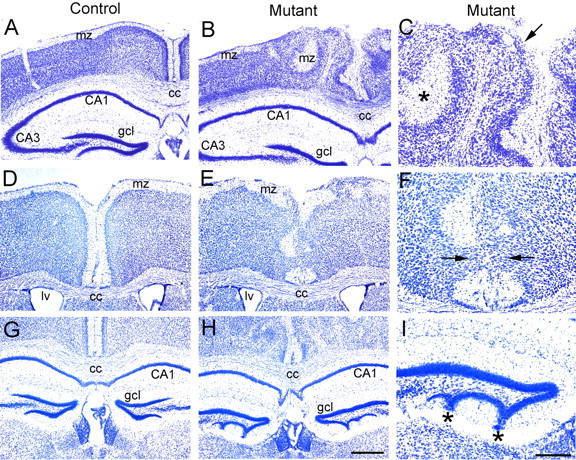
Deletion of Ilk from dorsal forebrain results in cortical lamination defects in Emx1-cre; Ilkfl/fl mutants. Nissl-stained coronal sections (40 μm) from adult littermate controls (A, D, G) and Ilk mutants (B, C, E, F, H, I). C, F, and I represent close-up views of regions shown in B, E, and H. A-F, Control brains showed a highly organized, multilayered cortex with a hypocellular marginal zone at the surface of the brain (A, D). In contrast, mutant brains showed severe disorganization of cortical lamination that was most apparent near the midline (B, E). Neurons invaded and obliterated the marginal zone (arrow, C), and hypocellular marginal zone regions were displaced deep into the cortex of mutant brains (asterisk, C). Midline fusion of the two hemispheres with complete obliteration of the marginal zone was observed in the mutant (E; arrows, F) but not in the control littermate (D). G-I, Whereas the granule cell layer (gcl) in the control hippocampus was well separated from the pial surface of the dentate gyrus (G), the mutant hippocampus showed scalloping of the granule cell layer in the lower portion of the dentate gyrus with displacement of granule cells extending to the pial surface (H; asterisks, I). Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Ilkfl/+ (A) or Emx1-cre; Ilkfl/+ (D, G). cc, Corpus callosum; gcl, granule cell layer; mz, marginal zone; lv, lateral ventricle. Scale bars: (in H) A, B, D, E, G, H, 500 μm; (in I) C, F, I, 250 μm.
Figure 3.
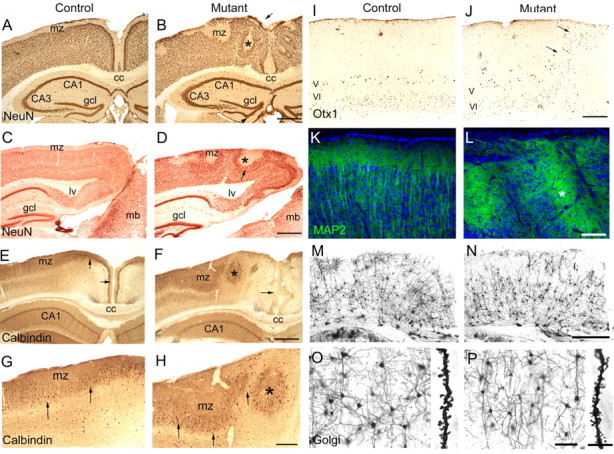
Analysis of the cellular composition of cortical ectopias in Emx1-cre; Ilkfl/fl adult mutants. Coronal (A, B, E-P) and sagittal (C, D) sections (40 μm, A-L; 100 μm, M-P) from adult littermate controls (A, C, E, G, I, K, M, O) and Ilk mutants (B, D, F, H, J, L, N, P). A-D, In control animals, NeuN immunostaining highlighted the laminar organization of the cortex in which neurons were excluded from hypocellular marginal zone regions at the surface of the brain (A, C). In Ilk mutants, ectopic neurons invaded and obliterated the marginal zone (arrow, B). Hypocellular regions were found deep within the mutant cortex (asterisk, B, D), and neurons piled up underneath these hypocellular regions (arrow, D). The granule cell layer of the dentate gyrus had an abnormal, undulating appearance in the mutant (arrowhead, B). E-H, In control animals, calbindin staining highlighted interneurons that were widely scattered but also formed a distinct layer beneath the marginal zone (arrows, E, G). In midline regions of mutant brains, this calbindin-positive layer was absent (arrow, F). Instead, interneurons were found at the periphery of misplaced hypocellular marginal zone regions (asterisk, F, H). In lateral regions of mutant brains, interneurons were distributed in a similar manner to controls, although the calbindin-positive layer below the marginal zone had an abnormal, wavy appearance in the mutant (arrows, H). I, J, Otx1 antibody identifies a subset of neurons localized to layers V and VI of controls (I) (Frantz et al., 1994). In mutant brains, many of the Otx1-expressing neurons were mislocalized to more superficial layers of the cortex within ectopic outgrowths (arrows, J). K, L, MAP2 staining in control brains highlighted linear bundles of dendrites coursing radially through the cortex and filling the marginal zone (K). In the mutant, the arrangement of dendrites was severely disorganized, with many dendrites seen veering away from the surface of the cortex to terminate and fill misplaced hypocellular marginal-zone like regions (asterisk, L). M, N, Golgi staining in control brains highlighted the orderly arrangement of cortical neurons and their processes (M). The orderly, radial arrangement of apical dendrites was disturbed in mutant sections, especially near the midline (N, right side of image). There were no apparent differences between mutant and control brains in the distribution and branching of basal dendrites (O, P) or in the density or morphology of dendritic spines (O, P, insets). Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Ilkfl/+ (A, E, G, I) or Emx1-cre; Ilkfl/+ (C, K, M, O). cc, Corpus callosum; gcl, granule cell layer; mb, midbrain; mz, marginal zone; lv, lateral ventricle. Scale bars: A-F, M, N, 500 μm; G-J, 200 μm; K, L, O, P, 100 μm; insets in O, P, 20 μm.
Absence of Ilk results in fragmentation of the basal lamina
Fragmentation of the basal lamina is frequently observed after deletion of extracellular matrix constituents or receptors in mice (Georges-Labouesse et al., 1998; Costell et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Moore et al., 2002; Beggs et al., 2003; Poschl et al., 2004). We therefore examined the basal lamina in Ilk mutant mice. Using anti-laminin, we observed extensive fragmentation of the basal lamina throughout the entire dorsomedial cortical surface of adult Ilk mutants (Fig. 4A,B). Fragmentation was also prominent around large blood vessels penetrating into the cortex from the pia-meninges but was not seen in smaller cortical blood vessels (Fig. 4A,B). Mutant brains also showed marked astrogliosis, as assessed using anti-GFAP, that extended far from the pial surface into deep layers of the cortex (Fig. 4C,D). Midline regions of mutant brains showed complete loss of basal lamina accompanied by fusion of the hemispheres, as highlighted by dystrophin immunostaining (Fig. 4E,F).
Figure 4.
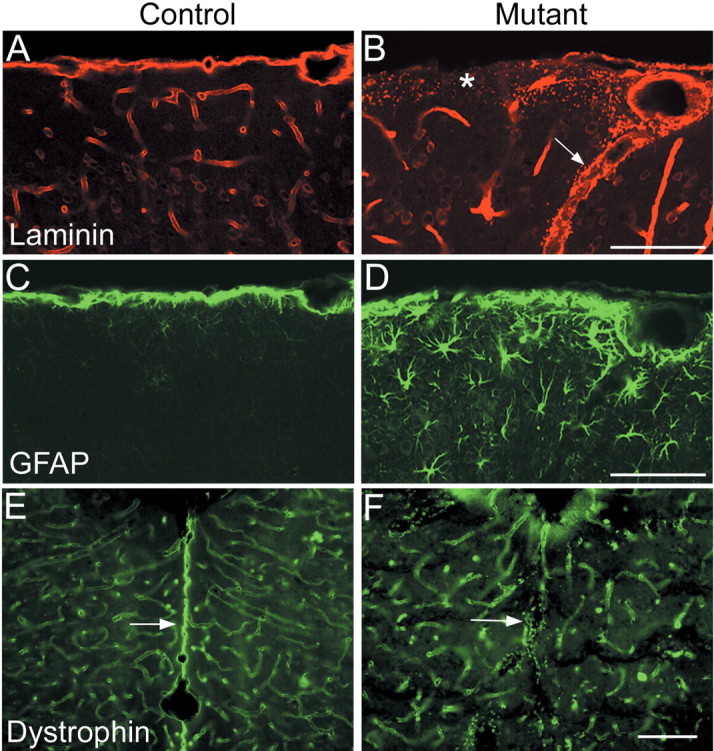
Deletion of Ilk results in basal lamina defects in adult Emx1-cre; Ilkfl/flmutant mice. Coronal sections (40 μm) from adult littermate controls (A, C, E) and Ilk mutants (B, D, F). A, B, In controls, immunostaining using anti-pan-laminin showed a smooth layer of basal lamina-associated laminin at the surface of the brain, as well as in basal lamina surrounding blood vessels (A). In the Ilk mutant, there was widespread fragmentation of laminin at the cortical surface with almost complete absence of laminin in some regions (asterisk, B). Laminin fragmentation was also seen associated with large blood vessels (arrow, B) but not small blood vessels. C, D, In control animals, there was a thin layer of GFAP staining that was restricted to the surface of the brain (C). In mutant brains, there was abundant astrogliosis, with GFAP reactivity extending deep into the cortex (D). E, F, Dystrophin staining was seen in small blood vessels as well as at the pial surface of control brains, in which it accentuated the separation of the two hemispheres (arrow, E). In Ilk mutants, midline fusion of the hemispheres was accompanied by a significant fragmentation and loss of dystrophin staining (arrow, F). Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Emx1-cre; Ilkfl/+ (A, C) or Ilkfl/+ (E). Scale bars, 100 μm.
Ectopias and basal lamina fragmentation originate during embryonic cortical development
To examine early events that lead to the observed defects in brain development, we examined Ilk mutants during embryonic cortical development. Mutant E14.5 embryos showed numerous small neuronal ectopias that invaded the marginal zone and were associated with irregularities of the cortical plate (Fig. 5B). However, as assessed by chondroitin sulfate proteoglycan expression, the preplate split normally into the marginal zone and subplate (Fig. 5C,D). Basal lamina fragmentation was also readily detectable at this stage of cortical development. Large gaps in the basal lamina that were associated with neuronal ectopias and contained small scattered fragments of laminin were evident in these embryos, especially in mediocaudal regions of the developing brain (Fig. 5E,F). Fusion of the hemispheres with splitting and fragmentation of the basal lamina were observed in mediorostral regions of the mutant cortex (data not shown). In more lateral regions, fragmentation of the basal lamina was less frequent, although small, scattered fragments of laminin could be found in these regions throughout the thickness of the cortical plate (Fig.5H). Ectopias were also rare in lateral regions. These studies showed that fragmentation of the basal lamina coincides temporally and spatially with formation of neuronal ectopias early in embryonic development. Additional studies at postnatal day 0 (P0) showed that the cortical lamination defects, including neuronal ectopias and basal lamina breakdown, were well developed by birth and resembled defects seen in the adult (data not shown).
Figure 5.
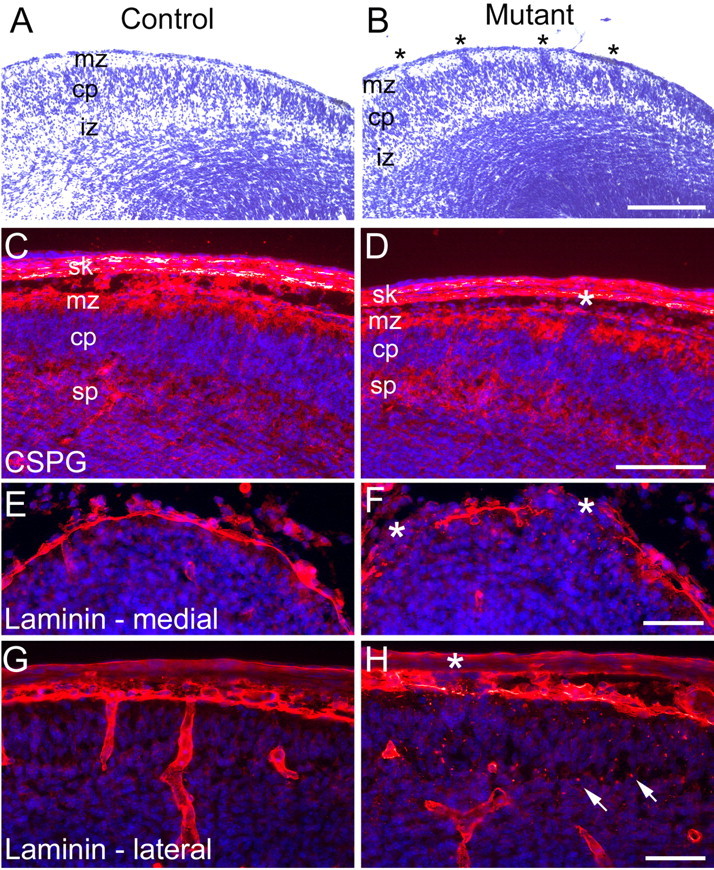
Ectopias and basal lamina defects are present during the embryonic development of Emx1-cre; Ilkfl/fl mutants. Coronal sections (10 μm) from littermate control E14.5 embryos (A, C, E, G) and Ilk mutants (B, D, F, H). Immunofluorescent sections (C-H) were costained with 4′,6′-diamidino-2-phenylindole (DAPI) (blue) to highlight nuclei. A, Nissl-stained control brains showed a distinct cortical plate with an overlying hypocellular marginal zone. B, In Ilk mutants, there were multiple small ectopias arising from the cortical plate that invaded the marginal zone (asterisks, B). C, D, Immunostaining for chondroitin sulfate proteoglycan showed normal splitting of the preplate into the marginal zone and subplate in both control (C) and mutant (D) brains. In the mutant, there were occasional ectopias invading the marginal zone (asterisk, D). E, G, Immunostaining of sections from control embryos showed an uninterrupted layer of surface laminin at the midline (E) and more laterally (G). In the Ilk mutant, midline sections showed multiple, large gaps in the continuity of surface laminin that were associated with neuronal ectopias (asterisks, F). In more lateral brain sections of the mutant (H), breakdown of surface laminin was less frequent (asterisk, H). However, multiple fragments of laminin were seen scattered throughout the cortical plate (arrows, H). Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Ilkfl/+. cp, Cortical plate; iz, intermediate zone; mz, marginal zone; sk, skull; sp, subplate. Scale bars: A, B, 200 μm; C, D, 100 μm; E-H, 50 μm.
Radial glial morphology is disrupted in the absence of Ilk
Previous studies have shown that proper anchorage of radial glial end feet is dependent on the presence of an intact basal lamina and a functioning adhesion system (Hartmann et al., 1998; Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003). To determine whether Ilk is important for radial glial development and function, we examined radial glia in Ilk mutant E14.5 embryos. Mutants showed severe morphologic abnormalities of radial glia that occurred beyond sites of surface basal lamina disruption (Fig. 6). Whereas in control animals radial glia were aligned in parallel arrays throughout the cortical plate, terminating in well defined end feet at the pial surface, in mutant embryos, radial glial fibers were severely disorganized, with excessive crisscrossing and branching seen throughout the targeted region (Fig. 6A,B). At the pial surface of mutants, radial glia often terminated in tangled heaps of fibers (Fig. 6B), and, occasionally, radial glial end feet were retracted from the pial surface (Fig. 6F). Radial glia were abnormal even in lateral regions in which the basal lamina was intact (Fig. 6C,D), although small laminin fragments were seen scattered throughout the cortical plate in these regions (best seen in Fig. 5H). In midline regions, in which basal lamina gaps were common, abnormal radial glial fibers were observed veering away from these gaps and terminating on remaining patches of laminin (Fig. 6G). These studies suggest that abnormalities of radial glial end feet may contribute to defects in basal lamina assembly in the Ilk mutant.
Figure 6.
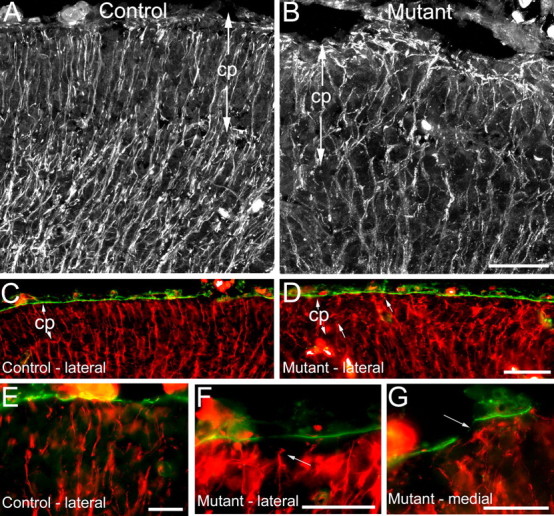
Absence of Ilk during embryonic cortical development leads to disorganization of radial glia in Emx1-cre; Ilkfl/fl mutants. Coronal sections (10 μm) of E14.5 littermate control (A, C, E) and Ilk mutants (B, D, F, G) were immunostained with RC2 antibody (white or red) to visualize radial glial fibers. Some sections were costained with pan-laminin antibody (green, C-G). A, In control brains, radial glia showed a parallel arrangement of fibers that terminated in well defined end feet at the pial surface. B, Radial glia of mutants appeared excessively branched and disorganized and formed tangles at the pial surface. C, D, Costaining of sections for both RC2 (red) and laminin (green) revealed that, despite severe disorganization of radial glial fibers in mutant brains, surface laminin was intact in many lateral regions (D) and was indistinguishable from control brains (C). E-G, Control sections showed a parallel arrangement of radial glial fibers reaching laminin at the pial surface (E). In mutant brains, radial glial end feet were occasionally seen detached from the pial surface (arrow, F). In medial regions of mutant brains, in which laminin breakdown was common, radial glia veered away from gaps in the basal lamina toward remaining segments of laminin (arrow, G). Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Ilkfl/+. cp, Cortical plate. Scale bars: A-D, 50 μm; E-G, 20 μm.
Positioning of Cajal-Retzius cells is defective in Ilk mutants
Cajal-Retzius cells secrete reelin, a signaling molecule that is critical for proper positioning of neurons (Tissir and Goffinet, 2003). A defect in localization of CR cells may lead to aberrant neuronal migration caused by mislocalized release of reelin. Previous studies have suggested that integrin adhesion is important for CR cell localization (Graus-Porta et al., 2001). Because CR cells are targeted by Emx promoter-driven cre (Gorski et al., 2002), we asked whether any defects of CR cell localization or reelin secretion are associated with the absence of Ilk in these cells. Using both reelin and calretinin to identify CR cells, we found that, in control E14.5 embryos and P0 neonates, there was a relatively continuous single layer of CR cells at the marginal zone (Fig. 7A,I). In contrast, mutant embryos showed multiple gaps in the CR cell layer, especially in mediocaudal regions (Fig. 7B,F). These gaps were associated with large neuronal ectopias (Fig. 7B,H) and corresponded to breaks in the basal lamina (Fig. 7D). The distribution of CR cells changed dramatically in mutant neonates, in which CR cells were found in ectopic clusters deep within the cortex, mostly in midline regions (Fig. 7K,L). Cortical areas containing the CR cell clusters were hypocellular but were underlain by dense bands of neurons (Fig. 7K,L) as also seen with anti-NeuN (Fig. 3B,D). These findings suggest that, although CR cells form and migrate normally in the absence of Ilk during embryonic cortical development, sporadic breakdown of the basal lamina results in loss of CR cells from these regions. Loss and displacement of CR cells during the course of cortical development may contribute to the abnormal positioning of neurons seen in the adult mutant cortex.
Figure 7.
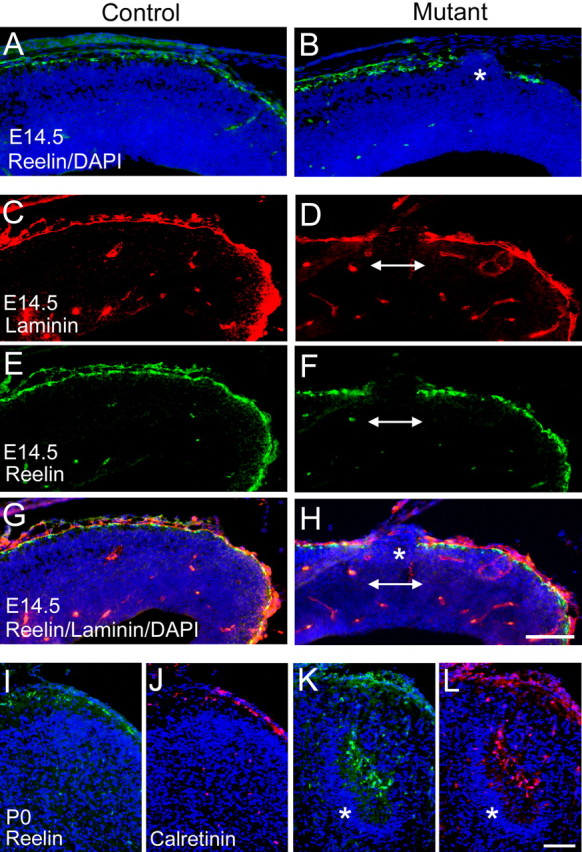
The CR cell layer is disrupted in the absence of Ilk in Emx1-cre; Ilkfl/fl mutants. Coronal sections (10 μm) from E14.5 littermate control (A, C, E, G) and E14.5 Ilk mutants (B, D, F, H) as well as P0 littermate controls (I, J) and P0 Ilk mutants (K, L) were immunostained with antibodies to the extracellular protein reelin (A, B, E, F, I, K) and cytoplasmic protein calretinin (J, L) to analyze the integrity of the CR cell layer. Selected sections were costained with DAPI (blue) to visualize nuclei (A, B, G-L) and laminin to evaluate the integrity of the basal lamina (C, D, G, H). C and E represent the same section viewed at different wavelengths and are shown combined in G. Similarly, D and F are combined in H. A-H, In control E14.5 embryos, immunostaining for reelin (A) demonstrated a mostly uninterrupted layer of CR cells at the surface of the cortex. In contrast, in mutant brains, prominent gaps in the CR cell layer and reelin expression were seen (B; double arrow, F, H). These gaps were associated with neuronal ectopias (asterisk, B, H) and in fragmentation of the basal lamina (double arrow, D, H). I-L, In control P0 neonates, CR cells were localized to a single layer at the surface of the brain (I, J). In mutant P0 neonates, clusters of CR cells were found within hypocellular marginal zone-like regions deep in the mutant cortex (K, L). These CR cell clusters, most frequently found in midline regions, were underlain by bands of piled neurons (asterisk, K, L). Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Emx1-cre; Ilkfl/+. Scale bars, 100 μm.
Deletion of Ilk from postmitotic neurons does not result in lamination defects
Our initial targeting strategy resulted in ablation of Ilk from both neuronal and glial precursors. To determine whether Ilk functions in neurons to control cortical lamination, we generated conditional mutant mice in which Ilk was specifically deleted from postmitotic neurons using a transgenic mouse line with cre recombinase driven by the Nex promoter. The Nex-cre transgene is expressed as early as E11, and, by E12.5, expression is robust throughout the forebrain (Shimizu et al., 1995) (S. Goebbels and K.-A. Nave, unpublished results). The resulting mutants showed no obvious behavioral difference from controls. Nissl staining did not reveal any obvious cortical lamination defects in mutants (Fig. 8A-D). No differences in GFAP or synaptophysin staining were seen between the mutant and control animals (data not shown). Golgi staining revealed a normal distribution and morphology of neurons in the mutants with no significant abnormalities of dendritic projection, branching, or spine formation (Fig. 8E-H). These results suggest that glia-derived Ilk, and not neuronal Ilk, is the major contributor to the lamination defects observed in Emx1-cre;Ilkfl/fl mutants.
Figure 8.
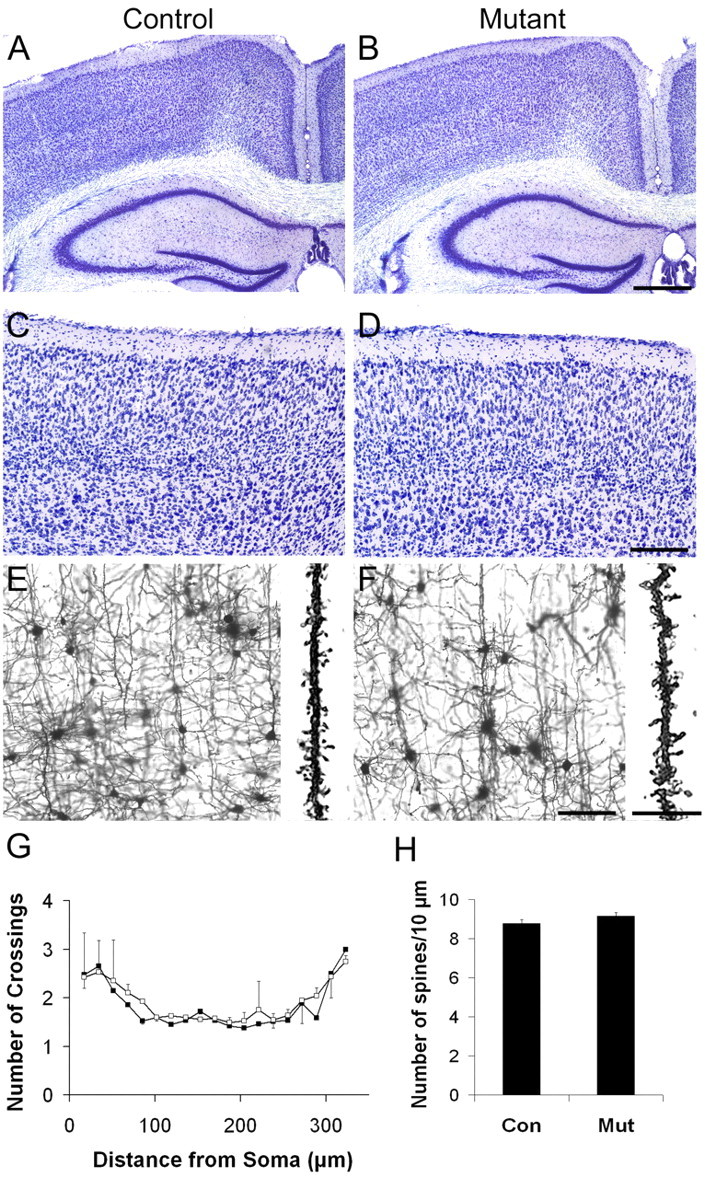
Deletion of Ilk from postmitotic neurons does not result in lamination defects in Nex-cre; Ilkfl/fl mutants. Targeted deletion of Ilk was achieved by crossing Ilk floxed mice with mice carrying cre recombinase under control of the Nex promoter. A-D, Coronal Nissl-stained sections (40 μm) from adult littermates (A, C) and Nex-cre; Ilkfl/fl mutants (B, D) showed no obvious defects of cortical or hippocampal lamination. E-H, Golgi-stained sections (100 μm) from cortices of adult littermates (E) or Nex-cre; Ilkfl/fl mutants (F) showed no obvious defects of dendritic projection, branching, or spine formation in the mutant (E, F). G, Quantification of branching patterns of projecting apical dendrites in Golgi-stained cortices using Scholl analysis showed no significant differences in branch complexity between mutant and control cortical neurons (average of p = 0.69, range of 0.22-1.00; n = 2 pairs). Twenty neurons per animal in two pairs of animals were analyzed. Data are expressed as mean ± variance. H, Quantification of spine density in projecting apical dendrites of Golgi-stained cortices showed no significant differences between mutant and control cortical neurons (8.78 ± 0.2 in the control vs 9.15 ± 0.18 in the mutant; p = 0.31; n = 2 pairs). Twenty neurons per animal were analyzed. Data are expressed as mean ± variance. Paramedian regions of cortex are shown in E and F. Mutant genotypes were Nex-cre; Ilkfl/fl. Control littermate genotypes were Ilkfl/+ (A, C) or Ilkfl/fl (E). The two control genotypes were combined for quantification of dendritic branching and spine density (G, H). Con, Control; Mut, mutant. Scale bars: A, B, 500 μm; C, D, 200 μm; E, F, 100 μm; insets in E, F, 20 μm.
Ablation of Ilk does not affect proliferation or survival of cortical cells or phosphorylation levels of two in vitro Ilk targets, Pkb/Akt and Gsk-3β
Ilk is a serine/threonine kinase known to phosphorylate target proteins such as Pkb/Akt and Gsk3-β in vitro (Delcommenne et al., 1998; Persad et al., 2001b). Through phosphorylation of these targets, Ilk has been implicated in control of proliferation and apoptosis (Chun et al., 2003; Wu, 2004) as well as in neurite extension, axon elongation, cell migration, and actin rearrangement (Mills et al., 2003; Zhou et al., 2004). To determine whether phosphorylation of the two most prominent substrates of Ilk is reduced in the absence of this kinase in vivo, we examined the phosphorylation levels of Pkb/Akt and Gsk-3β in control and Emx1- cre;Ilkfl/fl mutant E14.5 dorsal forebrain extracts (Fig. 9A). We used phospho-specific antibodies against Akt-[pS473] and Gsk-3β-[pS9], which represent the reported sites of Ilk phosphorylation (Persad et al., 2001a,b). There were no significant differences in protein levels or phosphorylation levels of either of these two targets between the mutant and control extracts (Fig. 9A). Similar studies at P0 also showed no differences in protein levels or phosphorylation of these two Ilk targets (data not shown). Consistent with the above findings, mutant embryos showed no significant changes in proliferation of cortical cells, as assessed by phospho-histone staining (Fig. 9B-D). There were also no differences in proliferation as assessed by Ki67 staining (data not shown). In addition, there was no significant increase in the number of apoptotic cells in mutant brains compared with controls, as assessed by TUNEL assays (Fig. 9E,F).
Figure 9.
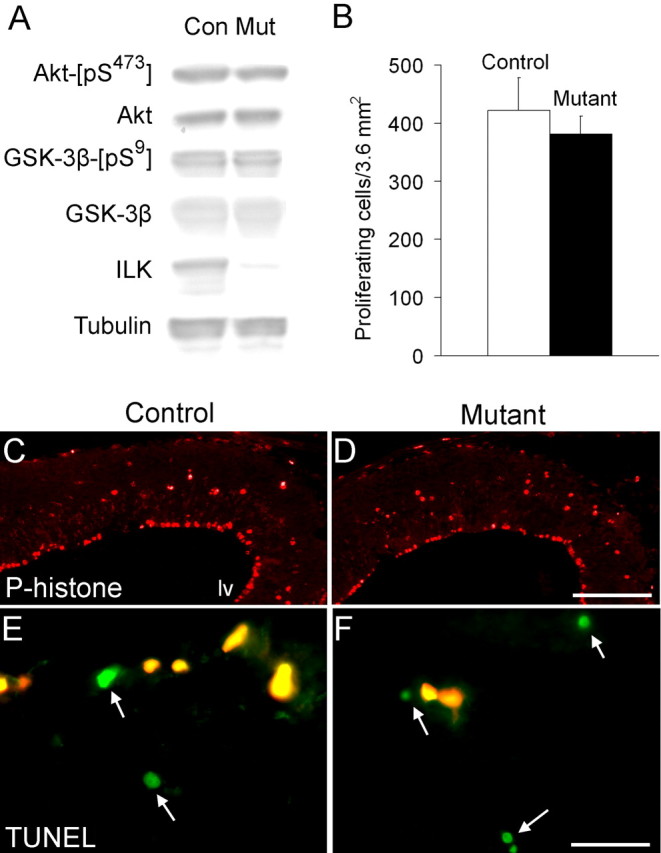
Ilk deletion has no effect on phosphorylation or function of Ilk targets, Pkb/Akt and Gsk-3β, in Emx1-cre; Ilkfl/fl mutants. A, Western blot analysis of cortical extracts (2.5 μg) from E14.5 Emx1-cre; Ilkfl/fl mutants and control littermates was performed using phospho-specific antibodies against Akt-[pS473] and Gsk-3β-[pS9], the reported in vitro phosphorylation target sites of Ilk (Persad et al., 2001a,b). Equal amounts of mutant and control extract (2.5 μg) were probed with Ilk in separate lanes. Membranes probed with Ilk, Akt-[pS473], and Gsk-3β-[pS9] were stripped and reprobed with Akt, tubulin, and Gsk-3β, respectively, to compare protein levels. Although a significantly decreased level of Ilk expression was seen in the mutant extract, there were no detectable differences in protein levels or phosphorylation levels of Pkb/Akt or Gsk-3β between mutant and control extracts. The Ilk and β-tubulin blots are the same as shown in Figure 1. B-D, Anti-phospho-histone, which labels a subset of proliferating cells from G2 to early telophase (Weissman et al., 2003), was used to assess proliferation levels in control and mutant embryonic brains. Staining of coronal sections (10 μm) from E14.5 littermate controls (C) and E14.5 Ilk mutants (D) with anti-phospho-histone did not show any obvious differences in proliferation levels between the two strains. B, Quantification of phosphohistone-labeled nuclei in E14.5 sections did not show any statistically significant differences in proliferation levels between control and mutant brains (422 ± 56 proliferating cells/3.6 mm2 in the control vs 382 ± 30 proliferating cells/3.6 mm2 in the mutant; p = 0.34; n = 3 pairs). Twelve cortical fields were analyzed per animal; data are expressed as mean ± SD. E, F, TUNEL assays did not show any differences in numbers of apoptotic cells between mutant and control brains (green cells, arrows, E, F). Yellow staining cells are nonspecifically fluorescing red blood cells that fluoresce in both the green and red (nonspecific) channels. Quantification of labeled nuclei showed 292 apoptotic cells/21.7 mm2 in the mutant versus 252 apoptotic cells/21.7 mm2 in the control in one pair of animals. Seventy-two high-power fields were analyzed per strain. Mutant genotypes were Emx1-cre; Ilkfl/fl. Control littermate genotypes were Ilkfl/+. Con, Control; Mut, mutant; lv, lateral ventricle. Scale bars: C, D, 500 μm; E, F, 20 μm.
Discussion
In this study, we demonstrated a critical role for Ilk in cortical development. Mice lacking Ilk showed cortical lamination defects with breakdown of the basal lamina and abnormalities of radial glia and CR cell positioning. In contrast to in vitro studies, we found that cell proliferation, survival, and differentiation were not affected by the absence of Ilk, and that phosphorylation levels of two major Ilk targets, Pkb/Akt and Gsk-3β, were not reduced in the embryonic cortex of Ilk mutants.
Role of Ilk in formation and maintenance of the basal lamina
Ilk most likely regulates cortical lamination by affecting formation or maintenance of the basal lamina. Basal lamina assembly takes place at the interface between the glia limitans and the meninges and depends on both of these neighboring structures for proper execution (Sievers et al., 1994; Shearer and Fawcett, 2001; Beggs et al., 2003). Meningeal fibroblasts secrete components of the basal lamina, which then bind to receptors on radial glial end feet of the glia limitans to be assembled into a functioning basal lamina (Sievers et al., 1994; Shearer and Fawcett, 2001; Beggs et al., 2003). Defects of several proteins involved in basal lamina assembly, including laminin, perlecan, collagen, dystroglycan, and integrins, cause basal lamina abnormalities associated with neuronal migration defects (Georges-Labouesse et al., 1998; Miner et al., 1998; Costell et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Moore et al., 2002; Beggs et al., 2003; Poschl et al., 2004). The Ilk mutant shares many of the cortical phenotypes with these mutants. Our studies showed extensive breakdown of the basal lamina and displacement of neurons toward the cortical surface. In regions in which the basal lamina was intact, cortical lamination was normal, although neurons and glia lacked Ilk. In addition, glial processes appeared disorganized throughout the cortical plate, and occasional radial glial end feet were seen retracted from the pial surface. These abnormal radial glia may provide inadequate support for matrix assembly at the pial surface. In fact, glia appear to be the primary cell type responsible for the defect in the Ilk mutant because ablation of Ilk specifically from postmitotic neurons did not result in lamination defects. These findings support a role for Ilk within radial glia to promote basal lamina assembly.
Mechanism of Ilk action during cortical development
Ilk is probably involved in matrix assembly through its interaction with β1 integrin cytoplasmic domain (Hannigan et al., 1996). Integrin β1 heterodimers bind several basal lamina constituents and play key roles in basal lamina assembly (Henry et al., 2001; Lohikangas et al., 2001; Li et al., 2002). As in other cell types, Ilk may act at radial glial end feet as a scaffold to link cell surface integrin receptors to the actin cytoskeleton. Rearrangement of the actin cytoskeleton is required for laminin assembly after engagement of integrin and dystroglycan receptors (Colognato et al., 1999), and Ilk is likely involved in this process. Interestingly, although phosphorylation of Pkb/Akt and Gsk-3β by Ilk was shown to be important in vitro for actin reorganization, cell migration, and neurite extension (Mills et al., 2003; Zhou et al., 2004; Qian et al., 2005), phosphorylation of these proteins was not reduced by the absence of Ilk in vivo during cortical development. It remains to be determined whether the kinase activity of Ilk is required for its role in basal lamina assembly in the forebrain in vivo.
Our studies did not reveal major defects of proliferation or cell survival in the absence of Ilk. In contrast, several studies have shown Ilk to be important for proliferation of cells such as chondrocytes and granule cell precursors (Grashoff et al., 2003; Terpstra et al., 2003) (J. Mills, A. Niewmierzycka, A. Oloumi, B. Rico, R. St-Arnaud, I. Mackenzie, N. Mawji, L. F. Reichardt, and S. Dedhar, unpublished observations) and in survival of cells, such as endothelial cells and hippocampal neurons (Gary et al., 2003; Friedrich et al., 2004). The conflicting results most likely reflect differences of in vivo versus in vitro conditions plus tissue-specific differences of Ilk function. For example, cerebellar granule cells, which migrate in an outside-in manner, may rely much more heavily than cortical cells on interactions with surface laminin for proliferation (Blaess et al., 2004). In contrast, proliferation of cortical neuronal precursors, which begin their migration at the subventricular zone, may depend on mechanisms that do not rely on integrin signaling, such as calcium wave propagation (Weissman et al., 2004).
Role of Cajal-Retzius cells in cortical lamination defects of Ilk mutants
A major feature observed in Ilk mutants was mislocalization of CR cells during embryonic cortical development. Our results indicate that retention of CR cells at the marginal zone may rely on Ilk and integrin-dependent adhesion to an intact basal lamina. Indeed, gaps in the CR cell layer of Ilk mutant embryos were consistently associated with gaps in the basal lamina. Although the Emx promoter-driven cre targets CR cells (Gorski et al., 2002), it is unlikely that deletion of Ilk from CR cells contributed directly to their mislocalization. CR cells migrated normally and were evenly distributed at the marginal zone in all regions except those with breakdown of the basal lamina.
Sporadic loss of CR cells and reelin from regions of basal lamina breakdown may contribute to the formation of neuronal ectopias. We found that ectopic outgrowths were consistently associated with gaps in the CR cell layer. Reelin has been shown to direct neuronal migration (Dulabon et al., 2000), and its loss in Ilk mutants may allow neurons to overmigrate. Later in development, displacement of the remaining CR cells to deep regions of the cortex may further contribute to the severe, scalloped appearance of the Ilk mutant brain. Indeed, in the Ilk mutant neonate, dense aggregates of neurons appear below displaced clusters of CR cells, suggesting that mislocalized reelin secretion results in retention of large subsets of neurons in deep regions of the cortex.
Finally, loss of CR cells may contribute to the abnormal radial glial scaffold observed in the Ilk mutants. However, radial glial abnormalities were widespread, whereas CR cell loss was limited to small, sporadic regions, suggesting that radial glial defects precede the loss of CR cells.
Comparison of the Ilk mutant to other basal lamina-related mutants
The lamination defects in the Ilk mutant were similar to those described in several other mouse mutants deficient in various gene products involved in basal lamina assembly. For example, the Ilk mutant shares many cortical features with the β1 integrin conditional mutant (Graus-Porta et al., 2001), indicating that Ilk may be a major cytoplasmic mediator of β1 integrin function during cortical development. In contrast, a conditional mutant of another integrin-associated protein kinase, focal adhesion kinase (Fak) (Beggs et al., 2003), had a milder phenotype than either the β1 integrin or Ilk mutants and did not show loss or displacement of CR cells. The apparently normal localization of CR cells in the Fak mutant indicates that the degree of laminin breakdown in the absence of Fak may be insufficient to eliminate or displace CR cells from their normal positions. Additional differences were also observed between the Ilk and Fak mutants. The Ilk mutant exhibited very early and severe morphologic radial glial abnormalities, whereas the Fak mutant showed partial agenesis of the corpus callosum that was not present in the Ilk mutant. The divergent phenotypes indicate that these cytoplasmic proteins are involved in distinct signaling pathways. Interestingly, the Ilk mutant also shared many features with the conditional dystroglycan mutant (Moore et al., 2002), indicating that Ilk may be involved in cytoplasmic pathways, including regulation of actin dynamics, common to both the integrins and the dystrophin-glycoprotein complex.
Role of Ilk in the pathogenesis of congenital muscular dystrophies
The Ilk mutant cortex resembles cobblestone (type II) lissencephaly seen in brains of congenital muscular dystrophy patients. These conditions include Muscle-Eye-Brain disease, Walker-Warburg syndrome, and Fukuyama congenital muscular dystrophy (Fukuyama et al., 1981; Haltia et al., 1997; Kobayashi et al., 1998; Cormand et al., 2001). Affected patients present with a myriad of abnormalities, including cobblestone cortex, severe mental retardation, seizures, muscular dystrophy, and cerebellar and ocular abnormalities (Muntoni and Voit, 2004). Several genes responsible for these diseases were found to code for glycosyltransferases, with α-dystroglycan being a major target of glycosylation (Kobayashi et al., 1998; Yoshida et al., 2001; Michele et al., 2002; Moore et al., 2002). However, the genes mutated in many patients remain unidentified. Integrins and dystroglycans are laminin receptors that play synergistic roles in laminin matrix assembly (Henry et al., 2001; Li et al., 2002). In addition, both surface receptor systems connect to the actin cytoskeleton through several adaptor molecules. The disorganized cortex and basal lamina fragmentation seen in forebrains of Ilk mutants suggest that Ilk signaling may be perturbed in congenital muscular dystrophies and, consequently, may play a role in their pathogenesis. Interestingly, additional mouse mutants with targeted deletion of Ilk recently generated in our laboratory show eye, cerebellar, and skeletal muscle abnormalities similar to those seen in congenital muscular dystrophy patients (A. Niewmierzycka and L. F. Reichardt, unpublished observations). Future studies will clarify the role played by Ilk and related proteins in this group of diseases.
Footnotes
This work was supported by National Research Service Award Grant AR051669-01 (A.N.) and National Institutes of Health Grant R01 NS19090 (L.F.R). We thank Andre Goffinet for anti-reelin and Peter Klein for anti-Gsk-3β and anti-Gsk-3β-[pS9], Jessica Gorski and Kevin Jones for the Emx1IREScre mice, Sandra Goebbels and Klaus Armin Nave for the Nex-cre mice, and members of the Reichardt laboratory for helpful discussions. The RC2 antibody, developed by Miyuki Yamamoto, and the Otx1 antibody, developed by Susan K. McConnell, were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa (Iowa City, IA).
Correspondence should be addressed to Louis F. Reichardt, 1550 4th Street, Room 284A, University of California, San Francisco, San Francisco, CA 94143-2611. E-mail: lfr@cgl.ucsf.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257022-10$15.00/0
References
- Anton ES, Kreidberg JA, Rakic P (1999) Distinct functions of alpha3 and alpha(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron 22: 277-289. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF (2003) FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 40: 501-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess S, Graus-Porta D, Belvindrah R, Radakovits R, Pons S, Littlewood-Evans A, Senften M, Guo H, Li Y, Miner JH, Reichardt LF, Muller U (2004) β1-Integrins are critical for cerebellar granule cell precursor proliferation. J Neurosci 24: 3402-3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T (2003) Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J Cell Biol 163: 397-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato H, Winkelmann DA, Yurchenco PD (1999) Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol 145: 619-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormand B, Pihko H, Bayes M, Valanne L, Santavuori P, Talim B, Gershoni-Baruch R, Ahmad A, van Bokhoven H, Brunner HG, Voit T, Topaloglu H, Dobyns WB, Lehesjoki AE (2001) Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology 56: 1059-1069. [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R (1999) Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol 147: 1109-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA 95: 11211-11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES (2000) Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27: 33-44. [DOI] [PubMed] [Google Scholar]
- Frantz GD, Weimann JM, Levin ME, McConnell SK (1994) Otx1 and Otx2 define layers and regions in developing cerebral cortex and cerebellum. J Neurosci 14: 5725-5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich EB, Liu E, Sinha S, Cook S, Milstone DS, MacRae CA, Mariotti M, Kuhlencordt PJ, Force T, Rosenzweig A, St-Arnaud R, Dedhar S, Gerszten RE (2004) Integrin-linked kinase regulates endothelial cell survival and vascular development. Mol Cell Biol 24: 8134-8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama Y, Osawa M, Suzuki H (1981) Congenital progressive muscular dystrophy of the Fukuyama type: clinical, genetic and pathological considerations. Brain Dev 3: 1-29. [DOI] [PubMed] [Google Scholar]
- Gary DS, Milhavet O, Camandola S, Mattson MP (2003) Essential role for integrin linked kinase in Akt-mediated integrin survival signaling in hippocampal neurons. J Neurochem 84: 878-890. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A (1998) Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol 8: 983-986. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22: 6309-6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Aszodi A, Sakai T, Hunziker EB, Fassler R (2003) Integrin-linked kinase regulates chondrocyte shape and proliferation. EMBO Rep 4: 432-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Thievessen I, Lorenz K, Ussar S, Fassler R (2004) Integrin-linked kinase: integrin's mysterious partner. Curr Opin Cell Biol 16: 565-571. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367-379. [DOI] [PubMed] [Google Scholar]
- Guo L, Wu C (2002) Regulation of fibronectin matrix deposition and cell proliferation by the PINCH-Ilk-CH-IlkBP complex. FASEB J 16: 1298-1300. [DOI] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U (2002) A critical function of the pial basement membrane in cortical histogenesis. J Neurosci 22: 6029-6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltia M, Leivo I, Somer H, Pihko H, Paetau A, Kivela T, Tarkkanen A, Tome F, Engvall E, Santavuori P (1997) Muscle-eye-brain disease: a neuropathological study. Ann Neurol 41: 173-180. [DOI] [PubMed] [Google Scholar]
- Hannigan G, Troussard AA, Dedhar S (2005) Integrin-linked kinase: a cancer therapeutic target unique among its Ilk. Nat Rev Cancer 5: 51-63. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S (1996) Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379: 91-96. [DOI] [PubMed] [Google Scholar]
- Hartmann D, Ziegenhagen MW, Sievers J (1998) Meningeal cells stimulate neuronal migration and the formation of radial glial fascicles from the cerebellar external granular layer. Neurosci Lett 244: 129-132. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP (2001) Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. J Cell Sci 114: 1137-1144. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394: 388-392. [DOI] [PubMed] [Google Scholar]
- Kubo K, Nakajima K (2003) Cell and molecular mechanisms that control cortical layer formation in the brain. Keio J Med 52: 8-20. [DOI] [PubMed] [Google Scholar]
- Li S, Harrison D, Carbonetto S, Fassler R, Smyth N, Edgar D, Yurchenco PD (2002) Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J Cell Biol 157: 1279-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohikangas L, Gullberg D, Johansson S (2001) Assembly of laminin polymers is dependent on beta1-integrins. Exp Cell Res 265: 135-144. [DOI] [PubMed] [Google Scholar]
- Mackinnon AC, Qadota H, Norman KR, Moerman DG, Williams BD (2002) C. elegans PAT-4/Ilk functions as an adaptor protein within integrin adhesion complexes. Curr Biol 12: 787-797. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP (2002) Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418: 417-422. [DOI] [PubMed] [Google Scholar]
- Mills J, Digicaylioglu M, Legg AT, Young CE, Young SS, Barr AM, Fletcher L, O'Connor TP, Dedhar S (2003) Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J Neurosci 23: 1638-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR (1998) Roles for laminin in embryogenesis:exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol 143: 1713-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418: 422-425. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Voit T (2004) The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 14: 635-649. [DOI] [PubMed] [Google Scholar]
- Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S (2001a) Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J Cell Biol 153: 1161-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S (2001b) Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem 276: 27462-27469. [DOI] [PubMed] [Google Scholar]
- Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U (2004) Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 131: 1619-1628. [DOI] [PubMed] [Google Scholar]
- Qian Y, Zhong X, Flynn DC, Zheng JZ, Qiao M, Wu C, Dedhar S, Shi X, Jiang BH (2005) Ilk mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene 24: 3154-3165. [DOI] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai L-H (2004) Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron 42: 197-211. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES (2004) α3β1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development 131: 6023-6031. [DOI] [PubMed] [Google Scholar]
- Shearer MC, Fawcett JW (2001) The astrocyte/meningeal cell interface—a barrier to successful nerve regeneration? Cell Tissue Res 305: 267-273. [DOI] [PubMed] [Google Scholar]
- Shimizu C, Akazawa C, Nakanishi S, Kageyama R (1995) MATH-2, a mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal, is specifically expressed in the nervous system. Eur J Biochem 229: 239-248. [DOI] [PubMed] [Google Scholar]
- Sievers J, Pehlemann FW, Gude S, Berry M (1994) Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J Neurocytol 23: 135-149. [DOI] [PubMed] [Google Scholar]
- Terpstra L, Prud'homme J, Arabian A, Takeda S, Karsenty G, Dedhar S, St-Arnaud R (2003) Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J Cell Biol 162: 139-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM (2003) Reelin and brain development. Nat Rev Neurosci 4: 496-505. [DOI] [PubMed] [Google Scholar]
- Weissman T, Noctor SC, Clinton BK, Honig LS, Kriegstein AR (2003) Neurogenic radial glial cells in reptile, rodent and human: from mitosis to migration. Cereb Cortex 13: 550-559. [DOI] [PubMed] [Google Scholar]
- Weissman TA, Riquelme PA, Ivic L, Flint AC, Kriegstein AR (2004) Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43: 647-661. [DOI] [PubMed] [Google Scholar]
- Wu C (2004) The PINCH-Ilk-parvin complexes: assembly, functions and regulation. Biochim Biophys Acta 1692: 55-62. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 1: 717-724. [DOI] [PubMed] [Google Scholar]
- Zervas CG, Gregory SL, Brown NH (2001) Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J Cell Biol 152: 1007-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD (2004) NGF-induced axon growth is mediated by localized inactivation of Gsk-3beta and functions of the microtubule plus end binding protein APC. Neuron 42: 897-912. [DOI] [PubMed] [Google Scholar]